

Abstract
The E3L proteins encoded by vaccinia virus bind double-stranded RNA and mediate interferon resistance, promote virus growth, and impair virus-mediated apoptosis. Among the cellular proteins implicated as targets of E3L is the protein kinase regulated by RNA (PKR). To test in human cells the role of PKR in conferring the E3L mutant phenotype, HeLa cells stably deficient in PKR generated by an RNA interference-silencing strategy were compared to parental and control knockdown cells following infection with either an E3L deletion mutant (ΔE3L) or wild-type (WT) virus. The growth yields of WT virus were comparable in PKR-sufficient and -deficient cells. By contrast, the single-cycle yield of ΔE3L virus was increased by nearly 2 log10 in PKR-deficient cells over the impaired growth in PKR-sufficient cells. Furthermore, virus-induced apoptosis characteristic of the ΔE3L mutant in PKR-sufficient cells was effectively abolished in PKR-deficient HeLa cells. The viral protein synthesis pattern was altered in ΔE3L-infected PKR-sufficient cells, characterized by an inhibition of late viral protein expression, whereas in PKR-deficient cells, late protein accumulation was restored. Phosphorylation of both PKR and the α subunit of protein synthesis initiation factor 2 (eIF-2α) was elevated severalfold in ΔE3L-infected PKR-sufficient, but not PKR-deficient, cells. WT virus did not significantly increase PKR or eIF-2α phosphorylation in either PKR-sufficient or -deficient cells, both of which supported efficient WT viral protein production. Finally, apoptosis induced by infection of PKR-sufficient HeLa cells with ΔE3L virus was blocked by a caspase antagonist, but mutant virus growth was not rescued, suggesting that translation inhibition rather than apoptosis activation is a principal factor limiting virus growth.
Among the antiviral defenses of mammalian cells are the production and action of interferons (IFN) and, subsequently, the programmed death of virus-infected cells. Viruses have evolved mechanisms to counter apoptosis and to antagonize the actions of IFN (16, 40). Poxviruses, including vaccinia virus, are large DNA viruses that possess >200 genes and replicate in the cytoplasm of infected host cells (35). Several poxvirus genes specify proteins that act to impair host responses to infection, including antagonism of IFN signaling, IFN action, and apoptosis (40, 44, 54). One such vaccinia virus gene is E3L.
The vaccinia virus E3L gene encodes two proteins, p25 and p20, that are synthesized early during virus infection, likely by a leaky scanning mechanism of translation initiation (10). The E3L proteins possess two functional domains: an N-terminal Z-DNA binding domain discovered by homology to a repeated domain found in the A-to-I RNA-editing enzyme ADAR1 (22, 36, 51) and a C-terminal double-stranded RNA (dsRNA) binding domain discovered by homology to the protein kinase regulated by RNA (PKR) (8, 18, 33) that is now known in several cellular dsRNA binding proteins (50). Vaccinia virus with the E3L gene deleted (ΔE3L) displays a different growth phenotype and mediates a different apoptotic response from wild-type (WT) virus in cell culture. The host range of ΔE3L mutant virus is restricted, as exemplified by the replication of ΔE3L virus in rabbit RK13 cells but not in human HeLa cells (3, 9). In HeLa cells, where ΔE3L virus growth is restricted, multiplication of WT virus is resistant to IFN pretreatment, whereas in ΔE3L permissive cells the mutant virus growth is sensitive to the antiviral actions of IFN (2, 3, 9, 47). Furthermore, ΔE3L mutant-virus-infected HeLa cells undergo apoptosis, whereas WT virus-infected HeLa cells do not (21, 28). E3L mutants that retain dsRNA binding activity complement the ΔE3L deletion and rescue mutant virus growth in cultured cells (9, 47), whereas the N-terminal and the C-terminal domains of E3L are required for pathogenesis in the mouse model (6).
The key cellular target and the molecular mechanism by which E3L antagonizes host responses in vaccinia virus-infected cells have remained elusive. Among the RNA binding proteins implicated as physiologic targets of E3L are cellular proteins activated by dsRNA, including the PKR kinase (10), the family of 2′,5′-oligoadenylate synthetases (OAS) that act through RNase L (37), and the IFN regulatory factors IRF3 and IRF7 (48). Studies with null mouse embryonic fibroblasts (MEFs) and ΔE3L mutant virus showed that the yield of ΔE3L mutant virus was about 20-fold higher in both RNase L single-null MEFs and combined RNase L and PKR double-null MEFs than in WT MEFs (57), whereas other studies indicated that the basal level of PKR was the critical determinant for the ability of ΔE3L mutant virus to replicate in different cell lines (26, 32).
PKR, the protein kinase regulated by dsRNA, is a well-characterized antiviral protein in the IFN defense system(16, 40). The Pkr gene, often seen to be constitutively expressed at variable but low levels in untreated and uninfected cells, is transcriptionally activated by IFN treatment and virus infection (40, 49, 56). In response to physiologic stimuli, including virus infection, inactive PKR protein is activated by a dsRNA-mediated dimerization and subsequent autophosphorylation process (33, 34). Following RNA-dependent autoactivation, among the best-characterized substrates of PKR is the α subunit of protein synthesis initiation factor 2 (eIF-2α) (41). Phosphorylation of eIF-2α on serine 51 leads to inhibition of translation. PKR also is reported to modulate signal transduction responses, including the NF-κB activation pathway (5, 24) and the p38 mitogen-activated protein kinase pathway (15).
Vaccinia virus encodes two protein antagonists of PKR, E3L and K3L (10). Two mechanisms have been advanced to account for the PKR antagonism of E3L, one by sequestering the activator RNA (20, 47) and the other by direct protein-protein interaction involving the substrate binding region of PKR to form inactive heterodimers (39, 45). K3L is thought to impair PKR activity through a fundamentally different mechanism than E3L (40). K3L is a homologue of eIF-2α and acts as a pseudosubstrate to block the phosphorylation of eIF-2α (7). Unlike the E3L deletion virus, mutant vaccinia virus possessing the K3L deletion replicates in HeLa cells and remains IFN insensitive (3, 26).
In addition to impairment of the antiviral activity of IFN and effects on host range, the vaccinia virus E3L protein also mediates resistance to apoptosis (14). HeLa cells infected with vaccinia virus with the E3L gene deleted have been shown to undergo apoptosis, whereas HeLa cells infected with WT vaccinia virus do not (21, 28). Studies with vaccinia virus recombinants expressing either mutant E3L proteins or alternative dsRNA binding proteins established that suppression of apoptosis induction in infected HeLa cells correlates with dsRNA binding activity (21). We recently established HeLa cell clones stably deficient in PKR and showed that apoptosis in these HeLa cells induced by dsRNA is PKR dependent (58).
To test whether the cellular PKR protein is a primary target of the vaccinia virus E3L protein in infected human cells and to assess the extent to which the PKR protein is required for virus-induced apoptotic responses in cells infected with ΔE3L mutant virus, we studied HeLa cells either stably deficient or sufficient in PKR. We examined the role of PKR in virus replication and E3L mutant virus-induced apoptosis by comparing WT and mutant virus strains (ΔE3L, lacking E3L; Δ83N, possessing an N-terminal deletion of E3L; and Δ26C, possessing a C-terminal deletion of E3L) in parental HeLa cells (PKR+) compared to a PKR-deficient HeLa cell clone (PKR knockdown [PKRkd]) in which >95% of PKR protein expression is stably silenced and a PKR knockdown control cell clone (PKRkd-con) with parental levels of PKR. The growth of ΔE3L virus and Δ26C virus expressing a truncated E3L protein lacking dsRNA binding activity was restricted in the PKR-sufficient parental PKR+ and control PKRkd-con HeLa cells, but yields were increased by nearly 2 log10 in PKR-deficient HeLa cells. Yields of the WT and the Δ83N mutant were comparable in the PKR-sufficient and -deficient HeLa cells and remained resistant to both IFN-α and -β. Vaccinia virus-induced apoptosis seen in ΔE3L- and Δ26C-infected PKR-sufficient cells was absent in infected PKR-deficient cells. In ΔE3L-infected PKR-deficient cells, the phosphorylation of PKR and eIF-2α was abolished, resulting in a normal expression pattern of viral proteins. Although ΔE3L-induced apoptosis could be inhibited by treatment of PKR-sufficient cells with a caspase inhibitor, the growth of ΔE3L virus was not rescued.
MATERIALS AND METHODS
Cell maintenance and viruses.
HeLa, BHK, and RK13 cells were maintained in Dulbecco's modified Eagle's medium complemented with 5% or 10% (vol/vol) fetal bovine serum (HyClone), respectively; 100 μg/ml of penicillin; and 100 units/ml streptomycin (Invitrogen) as previously described (58). HeLa stable transfectant clones, PKRkd and PKRkd-con (58), were maintained in the above-mentioned medium containing 1 μg/ml puromycin (Sigma). The Copenhagen strain (VC-2) of WT vaccinia virus, ΔE3L virus mutants, and mutants with the first 83 N-terminal amino acids of E3L (Δ83N) or last 26 C-terminal amino acids of E3L (Δ26C) deleted were previously described (8, 9). IFN treatment was with 1,000 units/ml of recombinant IFN-αA/D (PBL InterferonSource) or 1,000 units/ml of IFN-β (Toray Industries) for 24 h. Caspase inhibitor treatment, either 1% dimethyl sulfoxide (DMSO) mock control or 100 μM z-VAD-fmk (EMD Biosciences no. 627609), was initiated 2 h before virus infection and was maintained throughout the 24-h single-cycle incubation period.
Single-cycle virus growth assay.
HeLa parental cells and stably transformed cell clones were seeded into six-well plates (6 × 105 cells per well) and either pretreated with recombinant IFN-αA/D (1,000 units/ml) or IFN-β (1,000 units/ml) for 24 h or left untreated. Virus infection was at a multiplicity of infection (MOI) of 5 with WT virus or the indicated deletion mutant virus (ΔE3L, Δ83N, or Δ26C). Virus absorption was carried out for 1 h in 0.2 ml Puck's saline A modified to contain 20 mM MgCl2 and 1% fetal bovine serum. The inoculum was then removed, the monolayers were rinsed twice with phosphate-buffered saline, and the cultures were incubated in 1.5 ml of maintenance medium. Infected cultures were harvested at 24 h postinfection (p.i.) by scraping the infected cells into the medium. Virus yields were determined by plaque titration on rabbit kidney RK13 cells.
Western blot analysis.
Whole-cell extracts were prepared in the presence of 1 mM phenylmethylsulfonyl fluoride and 1% (vol/vol) protease inhibitor mixture (Sigma) as described previously (11, 58). For analysis of eIF-2α and PKR phosphorylation, 50 mM NaF and 2 mM Na2VO3 were included in the extract buffer. Proteins were fractionated by sodium dodecyl sulfate-10% polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, blocked, and then probed with an appropriate dilution of primary antibody in phosphate-buffered saline containing 3% (wt/vol) skim milk. Rabbit polyclonal antibodies were used to detect human PKR (Santa Cruz Biotechnology Inc.), phospho-PKR(thr446) (Santa Cruz Biotechnology Inc.), eIF-2α (Cell Signaling Technology), and phospho-eIF-2α(ser51) (Cell Signaling Technology); mouse monoclonal antibodies were used to detect human poly(ADP-ribose) polymerase (PARP) (BD Pharmingen), β-actin (Sigma), and α-tubulin (Sigma). Vaccinia virus proteins were detected using monospecific antibodies against the B5, D5, E3L, and I3 proteins. D5 and I3 antibodies were generously provided by P. Tracktman (Medical College of Wisconsin, Milwaukee), the B5 monoclonal antibody was provided by B. Moss (NIH, Bethesda, MD), and the E3L antibody and polyclonal antibody prepared against purified virus virions were as previously described (53). Western blot detection was done with IRDye 800CW-conjugated anti-rabbit immunoglobulin G or IRDye 680-conjugated anti-mouse immunoglobulin G secondary antibody according to the manufacturer's protocols (Li-Cor). Immunoreactive bands were visualized using an Odyssey infrared imaging system, and quantitation was carried out using the Quantity One software program.
Caspase 3/7 activation assay.
Effector caspase 3/7 activation was measured by the Caspase Glo 3/7 assay (Promega) as previously described (58). HeLa cells were seeded into 96-well plates at a density of 1.5 × 104 per well. After overnight culture, the cells were infected with virus at an MOI of 5. At the specified time, usually 24 h p.i., 100 μl of Caspase-Glo 3/7 reagent was added to each well and mixed gently. After 1 h of incubation at room temperature in the dark, the luciferase reporter activity was measured using a Perkin-Elmer Victor 3V luminometer plate reader model 1420.
RESULTS
Growth of the vaccinia virus ΔE3L mutant is rescued in PKR-deficient HeLa cells.
Prior studies showed that vaccinia virus ΔE3L has a limited host range and replicates poorly in HeLa cells compared to WT virus (3, 9). To test whether PKR plays an obligatory role in limiting the growth of the ΔE3L mutant in human cells, a HeLa clonal line was examined in which PKR protein expression is stably knocked down by RNA interference (58). These PKRkd cells possess less than 5% of the PKR protein found in either their parental PKR+ cells or PKRkd-con cells (Fig. 1A). While both IFN-α and IFN-β induced PKR in the PKR+ and PKRkd-con HeLa cells, the level of PKR measured by Western immunoblot assay in the PKRkd clone was nearly undetectable even after treatment with type I IFN (Fig. 1A, lanes 4 to 6). These three HeLa cell clonal lines were then examined for the ability to support the growth of the ΔE3L mutant compared to WT virus and also to mount an IFN-induced antiviral state against these viruses.
FIG. 1.

Single-cycle multiplication of ΔE3L mutant compared to WT vaccinia virus in PKR+, PKRkd, and PKRkd-con HeLa cells. WT parental (PKR+), PKR-deficient knockdown (PKRkd), and PKR-sufficient control knockdown (PKRkd-con) HeLa cells were mock treated or treated with 1,000 IU/ml of IFN-αA/D (α) or IFN-β (β) for 24 h. (A) Western blot analysis comparing PKR expression levels. Whole-cell extract protein (10 μg) was analyzed in each lane; the membrane was probed with antibody against human PKR and antibody against β-actin as a loading control. (B) Virus yields. Cells were infected with either WT or ΔE3L virus at an MOI of 5. At 24 h p.i., cells were harvested and virus yields were determined by plaque titration on RK13 cells. The results shown are the means ± standard deviations determined from a minimum of three experiments.
As shown in Fig. 1B, the single-cycle yields of WT vaccinia virus were comparable in the PKR-sufficient (PKR+ and PKRkd-con) and PKR-deficient (PKRkd) HeLa cells. Furthermore, neither IFN-α nor IFN-β pretreatment significantly reduced the yield of WT virus in any of the HeLa cells, PKRkd, PKRkd-con, or parental PKR+ (Fig. 1B). Consistent with prior findings, the yield of the ΔE3L mutant was about 100-fold lower than that of WT virus in PKR-sufficient cells, both the parental PKR+ cells and the PKRkd-con cells. By contrast, the yield of the ΔE3L mutant in PKRkd cells was significantly increased (Fig. 1B, center), with a minimum of 30- to 50-fold-higher yields of ΔE3L in the PKRkd cells than in the PKR+ and PKRkd-con cells. Even though the growth of the ΔE3L mutant was rescued in the absence of PKR, pretreatment with IFN-α or IFN-β only modestly decreased the yield of ΔE3L virus in PKRkd cells, about three- to fivefold (Fig. 1B, center).
Phosphorylation of initiation factor eIF-2α is impaired in ΔE3L mutant-infected PKR-deficient HeLa cells.
One mechanism by which the protein synthesis pattern is regulated in virus-infected cells is through the phosphorylation of eIF-2α at serine 51, which causes an inhibition of translation (12, 41). eIF-2α, the best-characterized substrate of PKR, is phosphorylated following dsRNA-mediated autophosphorylation and activation of PKR that includes PKR phosphorylation at threonine 446 (4, 40, 42, 43). However, kinases in addition to PKR are known in eukaryotic cells that also phosphorylate eIF-2α at serine 51, including HRI, PERK, and GCN2 (12, 41). dsRNA, a PKR activator, has been described in vaccinia virus-infected HeLa cells (55), and the E3L gene product is a dsRNA binding protein (26, 33). Furthermore, antagonism of eIF-2α phosphorylation in poxvirus-infected cells has been attributed to two viral proteins: E3L, which binds dsRNA, and K3L, which is an eIF-2α homologue (7, 26). Therefore, to test directly the roles of PKR and E3L in determining the phosphorylation status of eIF-2α in virus-infected cells, we carried out a time course Western assay to measure the phosphorylation states of both PKR (Thr446) and eIF-2α (Ser51) in PKR-deficient and PKR-sufficient HeLa cell clones following infection with either WT or ΔE3L vaccinia virus.
As shown in Fig. 2, the amounts of phospho-PKR and phospho-eIF-2α were greatly increased in both PKR-sufficient cells, PKR+ parental and PKRkd-con control, following infection with ΔE3L mutant virus (Fig. 2, right, lanes 18 to 20 and 28 to 30). By contrast, little increase in phosphorylation of either PKR or eIF-2α was seen in WT virus-infected PKR-sufficient cells at any time after infection (Fig. 2, lanes 1 to 5 and 11 to 15). In the ΔE3L mutant-virus-infected cells, the increase in phosphorylation of eIF-2α was seen at times after infection when PKR phosphorylation was increased, beginning at 6 h (Fig. 2, lanes 18 to 20 and 28 to 30). By contrast, phosphorylation of eIF-2α in the PKR-deficient PKRkd cells was not detectably increased at any time following infection with either ΔE3L mutant or WT virus (Fig. 2, lanes 6 to 10 and 21 to 25). The amounts of both PKR protein and phospho-PKR were extremely low and marginally detectable in the PKRkd cells (Fig. 2, lanes 6 to 10 and 21 to 25).
FIG. 2.

Time courses of PKR and eIF-2α phosphorylation following infection of PKR+, PKRkd, and PKRkd-con cells with ΔE3L mutant compared to WT vaccinia virus. Whole-cell extracts were prepared from uninfected cells (0) or infected cells at 3, 6, 9, and 12 h p.i. with either WT or ΔE3L virus. Whole-cell extract protein (30 μg) was analyzed in each lane. The membranes were probed with antibodies against PKR, phospho-T446-PKR, eIF-2α, phospho-eIF-2α, vaccinia virus E3L, and β-actin as a loading control.
In the WT virus-infected PKR-sufficient HeLa cells, two size forms of E3L protein (p25 and p20) were detected. Their expression levels were comparable in PKR-sufficient (lanes 3 to 5 and 13 to 15) and -deficient (lanes 8 to 10) HeLa cells (Fig. 2, left). The E3L proteins were not detected in cells infected with ΔE3L mutant virus (Fig. 2, right) or in uninfected cells (Fig. 2). Finally, as an internal gel-loading control, the levels of β-actin were similar for all lanes. Failure to express the E3L protein in the mutant-infected PKR-sufficient HeLa cells correlated with the rapid and abundant phosphorylation of PKR and eIF-2α, whereas neither PKR nor eIF-2α phosphorylation was seen in the PKR-deficient cells. These results suggest that the eIF-2α phosphorylation seen in vaccinia virus-infected cells is predominantly, if not exclusively, mediated by the PKR kinase and that the lack of expression of E3L proteins in infected cells is sufficient to greatly elevate the level of eIF-2α phosphorylation.
Expression of vaccinia virus late proteins in ΔE3L mutant-infected cells is inhibited in PKR-sufficient cells and rescued in PKR-deficient cells.
To examine the possible functional consequences of the difference in eIF-2α phosphorylation seen in ΔE3L mutant-infected PKR-deficient cells compared to PKR-sufficient HeLa cells for the production of vaccinia virus proteins, three viral proteins (D5, I3, and B5) known to be expressed at different infection stages of the virus life cycle were analyzed (Fig. 3), in addition to the E3L protein (Fig. 2). The D5 gene encodes a 90-kDa protein, an intrinsic nucleoside triphosphatase, that is expressed at early times after virus infection (13); the I3 gene encodes a 34-kDa single-stranded DNA binding protein that is expressed at early and intermediate times p.i. (38); and the B5 gene encodes a 42-kDa membrane glycoprotein expressed at late times p.i. that is required for efficient envelopment of intracellular virions (19).
FIG. 3.

Time course of viral gene expression following infection of PKR+, PKRkd, and PKRkd-con cells with WT compared to ΔE3L mutant vaccinia virus. Whole-cell extracts were prepared at increasing times (h) after infection with either WT or ΔE3L vaccinia virus. Ten micrograms of protein was analyzed in each lane. The membranes were probed with antibodies against the vaccinia virus early gene product I3, the early and intermediate gene product D5, and the late gene product B5. β-Actin was included as a loading control.
In ΔE3L mutant-virus-infected HeLa cells, viral protein expression varied depending on the presence or absence of PKR (Fig. 3, right). Expression of the late protein B5 was inhibited in the two PKR-sufficient lines (PKR+, lanes 17 to 20, and PKRkd-con, lanes 27 to 30) infected with ΔE3L virus. The absence of PKR (PKRkd, lanes 22 to 25) rescued the expression of the B5 protein in ΔE3L mutant-infected cells (Fig. 3, right). By contrast, in WT virus-infected HeLa cells, the expression patterns of the three viral proteins, D5, I3, and B5, were not affected by the presence (PKR+ and PKRkd-con) or absence (PKRkd) of PKR protein (Fig. 3, left). The production of the three vaccinia proteins in ΔE3L-infected PKRkd cells, but not PKR+ or PKRkd-con cells, resembled that of WT virus-infected PKR+, PKRkd-con, and PKRkd cells. However, in ΔE3L mutant-infected PKR-sufficient cells, the early gene product D5, while detected from 6 h p.i., did not increase at the later time points, 9 and 12 h p.i. (Fig. 3, lanes 18 to 20 and 28 to 30). These results suggest that the reduced expression of virus proteins seen in ΔE3L mutant-infected PKR-sufficient cells, especially the late B5 protein (Fig. 3, right), correlated with the increased phosphorylation of eIF-2α detected from 6 h p.i (Fig. 2), whereas the increased protein expression seen in the PKR-deficient ΔE3L-infected cells (Fig. 3, right) correlated with the absence of eIF-2α phosphorylation (Fig. 2).
Effects of PKR on the growth of vaccinia virus mutants lacking either the N- or C-terminal domain of E3L.
The single-cycle growth rates of domain-selective E3L deletion mutants, Δ83N and Δ26C, were compared in PKR-deficient and PKR-sufficient HeLa cells (Fig. 4). For reference, the ΔE3L virus lacking the entire E3L gene grew poorly in PKR-sufficient PKR+ parental and PKRkd-con control cells compared to WT virus but nearly as well as the WT in the PKR-deficient PKRkd cells (Fig. 1B and 4). The presence or absence of PKR did not significantly affect the growth of the Δ83N mutant with the Z-DNA binding region deleted from E3L; Δ83N virus growth was comparable to that of WT virus in all three HeLa lines. Deletion of the dsRNA binding region from E3L reduced Δ26C mutant virus yields about 5- to 10-fold in PKR-sufficient cells, but not in PKR-deficient PKRkd cells, compared to WT virus (Fig. 4).
FIG. 4.

Single-cycle multiplication of vaccinia virus mutants lacking the N- and C-terminal domains of E3L in PKR+, PKRkd, and PKRkd-con cells. Cells were infected at an MOI of 5 with either WT, ΔE3L, Δ26C, or Δ83N virus. At 24 h p.i., cells were harvested and virus yields were determined by plaque titration on RK13 cells. The results shown are the means ± standard deviations determined from a minimum of three experiments.
Apoptosis induced by infection with ΔE3L mutant vaccinia virus is mediated by PKR.
It is known that infection of HeLa cells with the ΔE3L mutant induces apoptosis in HeLa cells (21), and our previous work established that PKR plays a proapoptotic role in HeLa cells transfected with a synthetic dsRNA (58). To examine whether PKR also plays a role in vaccinia virus-induced apoptosis in HeLa cells, PKR-sufficient and PKR-deficient cells were examined for cleavage of PARP and for caspase 3/7 activation, two indicators of apoptosis.
As an initial measure of apoptosis, cleavage of the 116-kDa PARP protein was analyzed by Western blot analysis at 24 h after infection with either WT virus or the ΔE3L mutant. As shown in Fig. 5A, the 85-kDa cleavage fragment of PARP was readily detected in extracts of PKR-sufficient parental PKR+ (lane 3) and control PKRkd-con (lane 9) HeLa cells infected with the ΔE3L mutant. By contrast, PARP cleavage was not detectable in the PKRkd cells infected with either the ΔE3L or WT virus (lanes 5 and 6). Substantially less PARP cleavage was seen in WT virus-infected PKR-sufficient HeLa cells.
FIG. 5.

Vaccinia virus-mediated apoptosis is impaired in PKR-deficient HeLa cells. (A) Western immunoblot measurement of PARP cleavage with WT or ΔE3L mutant vaccinia virus at 24 h p.i. Whole-cell extract protein (10 μg) was analyzed in each lane. (B) Caspase 3/7 activation assay. Apoptosis induction by vaccinia virus infection of PKR+, PKRkd, and PKRkd-con cells was measured in 96-well plates, either mock infected or infected with the indicated deletion mutant virus at an MOI of 5. Apoptosis was measured using a Caspase-Glo 3/7 kit at 24 h p.i. Each experiment was repeated a minimum of three times. The error bars indicate standard deviations.
A second measure of apoptosis, activation of the effector caspases 3 and 7, was utilized. Caspase activation, monitored by caspase 3/7-dependent generation of the substrate for luciferase, was significantly reduced in the PKR-deficient PKRkd cells following infection with WT or E3L mutant vaccinia virus (Fig. 5B, center) compared to either the parental PKR+ or the negative control PKRkd-con line. The ΔE3L and Δ26C mutants were the most efficient activators of apoptosis in infected PKR-sufficient PKR+ and PKRkd-con cells. Caspase activation by the Δ83N mutant was comparable to that by WT virus in PKR-sufficient cells (Fig. 5B).
The ΔE3L mutant host range phenotype is apoptosis independent in HeLa cells.
We have demonstrated that depletion of PKR can both rescue ΔE3L-infected HeLa cells from apoptosis (Fig. 5) and prevent activation of eIF-2α phosphorylation (Fig. 2), thereby restoring the synthesis of the beacon late viral protein B5 (Fig. 3). However, from these results, it is unclear whether apoptosis or translation inhibition mediated by PKR is primarily responsible for the host range phenotype of reduced yield of the ΔE3L mutant in PKR-sufficient cells. To further assess the role of apoptosis in reducing ΔE3L virus yields, we tested whether ΔE3L virus growth in PKR-sufficient cells could be rescued by a pancaspase pharmacological inhibitor, z-VAD-fmk.
Treatment of the PKR-sufficient PKR+ and PKRkd-con cell lines with z-VAD-fmk completely inhibited apoptosis induced by ΔE3L infection, as demonstrated by two measurements, cell morphology (Fig. 6A) and PARP cleavage (Fig. 6B). In PKR-sufficient cells treated with the caspase inhibitor, either no or very few apoptotic bodies were observed by microscopy (Fig. 6A), and the formation of the 85-kDa PARP cleavage fragment was blocked (Fig. 6B, compare lanes 3 and 9 to lanes 2 and 8). Somewhat surprisingly, however, inhibition of apoptosis by z-VAD-fmk did not rescue the growth of the ΔE3L virus in PKR-sufficient cells (Fig. 6C, center). Single-cycle yields of ΔE3L mutant virus remained low in the PKR+ and the negative control PKRkd-con lines and were comparable in the presence and absence of z-VAD-fmk. About 1- to 2-log10-higher yields of ΔE3L virus were obtained in the PKR-deficient PKRkd cells, either with or without caspase inhibitor treatment (Fig. 6C).
FIG. 6.

The apoptosis of PKR-sufficient HeLa cells induced by ΔE3L vaccinia virus can be inhibited by caspase inhibitor. PKR+, PKRkd, and PKRkd-con HeLa cells were infected with ΔE3L mutant virus in the presence (+) or absence (−) of the caspase inhibitors z-VAD-fmk (100 μM) and DMSO (1%) or left uninfected. (A) Cytopathic effect as revealed by microscopy. Images were taken at 24 h p.i. (B) Western blot analysis. Whole-cell extracts were prepared 24 h p.i., and 10 μg protein was loaded in each lane. The membranes were probed with antibodies against PARP and with β-actin as a loading control. (C) Virus yields. Infected HeLa cells were harvested at 24 h p.i., and yields of ΔE3L mutant virus were determined by plaque titration on RK13 cells. The results shown are the means ± standard deviations determined from a minimum of three experiments.
The effect of inhibiting apoptosis on the production of viral proteins in ΔE3L mutant-infected HeLa cells was also assessed (Fig. 7). Blockage of apoptosis with the z-VAD-fmk caspase inhibitor did not rescue B5 synthesis in PKR-sufficient HeLa cells (Fig. 7A, lanes 3 and 9). Similar to the observation with the late B5 glycoprotein, when antisera against vaccinia virion proteins was used to measure viral protein accumulation, efficient production of virion proteins was observed in the PKR-deficient PKRkd cells infected with ΔE3L mutant virus (lanes 5 and 6) compared to the two PKR-sufficient lines (Fig. 7B, lanes 2 and 3 and lanes 8 and 9). Addition of the z-VAD-fmk inhibitor did not increase virion protein production in the parental PKR+ and control PKRkd-con lines, and the production of virion structural proteins remained high in the PKRkd cells treated with the caspase inhibitor (lane 7B).
FIG. 7.
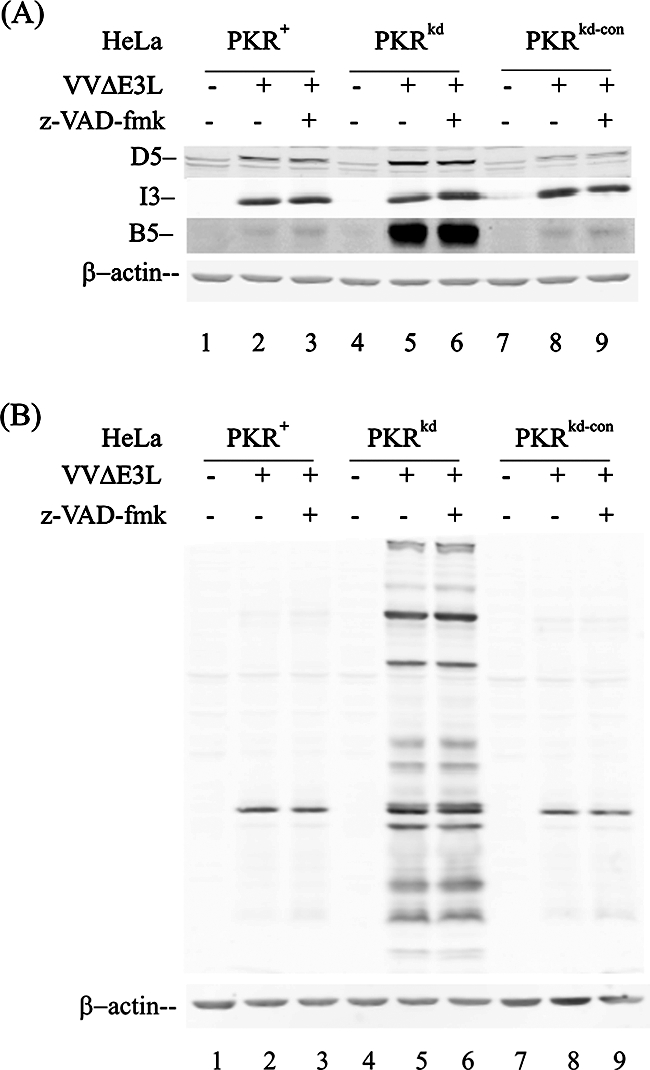
Inhibition of apoptosis does not rescue the production of late viral proteins in ΔE3L mutant-infected PKR-sufficient HeLa cells. PKR+, PKRkd, and PKRkd-con HeLa cells were infected with ΔE3L mutant virus in the presence (+) or absence (−) of the caspase inhibitors z-VAD-fmk (100 μM) and DMSO (1%) or left uninfected. Whole-cell extracts were prepared at 12 h p.i., and 10 μg protein was analyzed by Western blot analysis. The filter membranes were probed as follows: (A) monospecific antibodies against vaccinia virus proteins I3, D5, and B5 as indicated; (B) rabbit polyclonal antibody prepared against whole virions. β-Actin was monitored as a loading control.
DISCUSSION
The vaccinia virus E3L gene product is known as an IFN resistance protein, as a determinant of host range and viral pathogenesis, and as a modulator of cellular apoptotic and signal transduction pathways (26, 27, 31). Mutant vaccinia virus strains with either the entire E3L gene deleted or expressing a truncated E3L protein that lacks the C-terminal dsRNA binding domain display a limited host range for replication (26) and enhanced apoptosis (14, 21). In this study, we used human cells in which >95% of the PKR protein was stably depleted (58) to further investigate the role of PKR in determining the E3L mutant phenotype. Several important points emerge from our findings. In addition to showing that the PKR protein plays an obligatory and major role in restricting growth in HeLa cells infected with vaccinia virus lacking E3L, our findings also establish that the virus-induced apoptosis in parental HeLa cells infected with E3L deletion mutant virus is mediated by PKR. Cell death itself does not appear to be the direct determinant of restricted ΔE3L mutant virus growth and likely is not responsible for the ΔE3L virus restricted host range. Rather, activation of eIF-2α by PKR and the subsequent inhibition of late viral protein production appears responsible for limiting ΔE3L replication in HeLa cells.
The substantial reduction in the PKR protein level in the PKRkd HeLa cells largely rescued the growth of ΔE3L in these cells and greatly impaired the apoptotic response triggered by ΔE3L infection. These results indicate that PKR has an essential and primary role in the innate response of HeLa cells that is antagonized by the E3L protein. Vaccinia virus has been reported to utilize two mechanisms to block the function of PKR, mediated by the E3L and K3L genes, respectively (7, 8, 26, 40, 45-47). Our results that show that the depletion of PKR in the PKRkd HeLa cells can complement the ΔE3L mutant and largely restore viral growth are consistent with the notion that, at least in HeLa cells (26), it is E3L rather than K3L that is the dominant vaccinia virus antagonist of PKR and that PKR is the primary cellular protein targeted by E3L. Our findings would also favor a mechanism of E3L antagonism in HeLa cells that involves a selective functional effect of E3L on PKR. Such a mechanism could include direct interaction of E3L with PKR involving the C-terminal region of E3L to down-regulate the autoactivation of PKR and subsequent phosphorylation of eIF-2α (39, 47). While E3L protein is a dsRNA binding protein that sequesters dsRNA activators of cellular proteins, such a mechanism seemingly would necessitate selective RNA structure or cellular localization in order to discriminate among potential targets in addition to PKR. Our data do not permit us to distinguish between the two possible mechanisms of E3L antagonism, direct protein-protein interaction or sequestering of RNA.
Combined biological and biochemical studies have identified three different IFN-regulated dsRNA binding proteins whose activities are antagonized by E3L (10, 30, 37). These include, in addition to PKR (8, 20), the dsRNA-dependent OAS and the dsRNA-selective adenosine deaminase (ADAR1) (30, 37). In addition, the activation of the IFN regulatory factors IRF3 and IRF7 is impaired by E3L (48). dsRNA is either an effector (IRF, OAS, and PKR) or a substrate (ADAR1) of these proteins (17, 40), and dsRNA is known to be produced during vaccinia virus infection (20, 55). Both OAS and ADAR1, like PKR, could affect the translational patterns in cells (40). Rapid activation of OAS and RNase L has been seen in HeLa cells infected with modified Ankara ΔE3L mutant virus (31), whereas in studies with the Copenhagen strain ΔE3L mutant, the amount of PKR was found to be an especially important parameter conferring the ΔE3L mutant phenotype (26). Studies with knockout mice and derived MEFs have not given a clear and consistent indication of a singular cellular-protein function primarily disrupted by E3L. For example, the ΔE3L vaccinia virus did not replicate to high yield or cause significant disease in mice triply deficient for RNase L, PKR, and a third IFN-regulated antiviral protein, Mx1 (57). We found that the growth of the ΔE3L vaccinia virus lacking the entire E3L gene was largely recovered by depletion of PKR protein from human HeLa cells. Conceivably, in WT virus-infected mouse cells there may not be a singular limiting cellular-protein function that is antagonized by E3L that conversely impairs virus growth in ΔE3L mutant-infected mouse cells. In the HeLa cells stably knocked down for PKR, at least two other IFN-inducible gene products, ADAR1 RNA-specific deaminase and STAT1 transcription factor, are unaffected. While the PKR-deficient HeLa cells have less than 5% of the PKR protein of the PKR-sufficient parent and control cells, the basal and the IFN-inducible levels of ADAR1 and STAT1 are unaffected by the PKR knockdown (58).
The yield of ΔE3L virus in the PKRkd cells was only about 3-fold less than that of WT virus in the PKR-deficient HeLa cells and much higher (30- to 50-fold) than the yield of ΔE3L virus in PKR-sufficient cells, either the parental HeLa PKR+ or the negative knockdown control HeLa clone PKRkd-con. This finding is in contrast to our earlier observation with adenovirus, where the growth of virus depleted of the VAI gene was not rescued in the PKRkd cells (58), suggesting the importance of cellular factors in addition to PKR in conferring the VA RNA phenotype (23, 29). Depletion of PKR rescued the ΔE3L mutant vaccinia virus phenotype, as measured by viral growth, by viral protein synthesis patterns, and by virus-mediated apoptosis. Furthermore, when the Δ83N and Δ26C E3L deletion mutants were examined, virus expressing Δ83N, with the N-terminal 83 amino acids deleted, showed a phenotype similar to that of WT virus in the PKR-deficient cells, whereas virus expressing Δ26C, with the C-terminal 26 amino acids deleted, showed a phenotype similar to that of ΔE3L virus. These findings suggest that the C-terminal region of E3L that includes the dsRNA binding domain is of fundamental importance in conferring the PKR-dependent ΔE3L mutant phenotype.
The E3L deletion virus, while substantially rescued in the absence of PKR, did not fully achieve the yield of WT virus in the PKR-deficient cells, indicating that E3L likely has targets in addition to PKR. Two additional candidates are IRF3 and NF-κB. Deletions of E3L have been shown to cause the activation of signal transduction cascades leading to up-regulation of host gene expression (27). The Δ83N mutant displayed a gene expression profile similar to that of WT virus, and the ΔE3L mutant profile was similar to that of the Δ26C mutant (27), which, interestingly, is conceptually similar to the rescue pattern that we observed for virus growth in PKR-deficient HeLa cells. Prior analyses suggested that activation of IRF3 and NF-κB signal transduction cascades by vaccinia virus infection (27) or dsRNA treatment (58) occur through a PKR-independent mechanism.
Vaccinia virus protein expression levels in WT- and ΔE3L mutant-infected PKRkd cells were comparably high, consistent with the impaired phosphorylation and activation of PKR and subsequent downstream phosphorylation of eIF-2α. By contrast, when the increased phosphorylation of PKR and eIF-2α was observed, expression of early and intermediate viral gene products (D5 and I3) was detected in both ΔE3L and WT virus-infected PKR-sufficient cells. While no increase of D5 was seen at later time points in ΔE3L-infected PKR-sufficient cells, expression of D5 and I3 was further increased with increasing time after infection in WT-infected cells. Late gene product (B5) expression was greatly reduced to a marginally detectable level in ΔE3L-infected PKR-sufficient cells, but in PKR-deficient cells, B5 was expressed to a high level. Thus, the inhibition of late vaccinia virus protein production seen in ΔE3L-infected cells, as measured by B5 or major virion capsid proteins, can be explained by a PKR-mediated translational inhibition and phosphorylation of eIF-2α.
Our findings also indicate that PKR is a major cellular determinant of the caspase-dependent HeLa apoptosis induced following infection with the ΔE3L mutant. We previously established that in PKR-sufficient HeLa cells PKR is a key mediator of the apoptotic response triggered by transfection of dsRNA into PKR-sufficient HeLa cells (58). dsRNA has been identified in situ within vaccinia virus-infected cells (1, 55), and dsRNA has been reported to be the initiating signal of cell death in ΔE3L mutant-infected HeLa cells (21). Cellular dsRNA binding proteins whose activities have been shown to be affected by E3L and which have been implicated from mouse gene disruption studies in apoptotic responses include RNase L, which is activated by the products of OAS (37) and ADAR1 (30, 52), in addition to PKR (21, 25, 53). Our findings from ΔE3L-infected PKR-deficient PKRkd cells and the two PKR-sufficient controls demonstrate that PKR is the key mediator of apoptosis in the vaccinia virus-infected cells. In the PKR-deficient HeLa cell clone, ΔE3L infection did not cause either caspase 3/7 activation or enhanced PARP cleavage, both of which were seen in the PKR-sufficient cell controls. Furthermore, the apoptotic response seen in PKR-sufficient HeLa cells following ΔE3L infection could be blocked by treatment with the caspase inhibitor z-VAD-fmk, consistent with a caspase-dependent virus-induced cell death. However, we observed that apoptosis of PKR-sufficient HeLa cells induced by infection with ΔE3L virus did not play an antiviral role as earlier hypothesized (21). The pharmacologic inhibitor of caspase function impaired the ΔE3L virus-induced apoptosis of PKR-sufficient cells but did not rescue ΔE3L mutant virus growth or restore the viral translation pattern in the nonpermissive HeLa cells.
Acknowledgments
This work was supported in part by research grants AI-12520 and AI-20611 (C.E.S.) and AI-52347 and AI-66326 (B.L.J.) from the National Institute of Allergy and Infectious Diseases, NIH, U.S. Public Health Service.
Footnotes
Published ahead of print on 24 October 2007.
REFERENCES
- 1.Bayliss, C. D., and R. C. Condit. 1993. Temperature-sensitive mutants in the vaccinia virus A18R gene increase double-stranded RNA synthesis as a result of aberrant viral transcription. Virology 194254-262. [DOI] [PubMed] [Google Scholar]
- 2.Beattie, E., K. L. Denzler, J. Tartaglia, M. E. Perkus, E. Paoletti, and B. L. Jacobs. 1995. Reversal of the interferon-sensitive phenotype of a vaccinia virus lacking E3L by expression of the reovirus S4 gene. J. Virol. 69499-505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beattie, E., E. Paoletti, and J. Tartaglia. 1995. Distinct patterns of IFN sensitivity observed in cells infected with vaccinia K3L- and E3L-mutant viruses. Virology 210254-263. [DOI] [PubMed] [Google Scholar]
- 4.Bevilacqua, P. C., C. X. George, C. E. Samuel, and T. R. Cech. 1998. Binding of the protein kinase PKR to RNAs with secondary structure defects: role of the tandem A-G mismatch and noncontiguous helixes. Biochemistry 376303-6316. [DOI] [PubMed] [Google Scholar]
- 5.Bonnet, M. C., C. Daurat, C. Ottone, and E. F. Meurs. 2006. The N-terminus of PKR is responsible for the activation of the NF-κB signaling pathway by interacting with the IKK complex. Cell Signal. 181865-1875. [DOI] [PubMed] [Google Scholar]
- 6.Brandt, T. A., and B. L. Jacobs. 2001. Both carboxy- and amino-terminal domains of the vaccinia virus interferon resistance gene, E3L, are required for pathogenesis in a mouse model. J. Virol. 75850-856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carroll, K., O. Elroy-Stein, B. Moss, and R. Jagus. 1993. Recombinant vaccinia virus K3L gene product prevents activation of double-stranded RNA-dependent, initiation factor 2 alpha-specific protein kinase. J. Biol. Chem. 26812837-12842. [PubMed] [Google Scholar]
- 8.Chang, H. W., and B. L. Jacobs. 1993. Identification of a conserved motif that is necessary for binding of the vaccinia virus E3L gene products to double-stranded RNA. Virology 194537-547. [DOI] [PubMed] [Google Scholar]
- 9.Chang, H. W., L. H. Uribe, and B. L. Jacobs. 1995. Rescue of vaccinia virus lacking the E3L gene by mutants of E3L. J. Virol. 696605-6608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang, H. W., J. C. Watson, and B. L. Jacobs. 1992. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc. Natl. Acad. Sci. USA 894825-4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Das, S., S. V. Ward, R. S. Tacke, G. Suske, and C. E. Samuel. 2006. Activation of the RNA-dependent protein kinase PKR promoter in the absence of interferon is dependent upon Sp proteins. J. Biol. Chem. 2813244-3253. [DOI] [PubMed] [Google Scholar]
- 12.Dey, M., C. Cao, A. C. Dar, T. Tamura, K. Ozato, F. Sicheri, and T. E. Dever. 2005. Mechanistic link between PKR dimerization, autophosphorylation, and eIF2α substrate recognition. Cell 122901-913. [DOI] [PubMed] [Google Scholar]
- 13.Evans, E., N. Klemperer, R. Ghosh, and P. Traktman. 1995. The vaccinia virus D5 protein, which is required for DNA replication, is a nucleic acid-independent nucleoside triphosphatase. J. Virol. 695353-5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garcia, M. A., S. Guerra, J. Gil, V. Jimenez, and M. Esteban. 2002. Anti-apoptotic and oncogenic properties of the dsRNA-binding protein of vaccinia virus, E3L. Oncogene 218379-8387. [DOI] [PubMed] [Google Scholar]
- 15.Goh, K. C., M. J. deVeer, and B. R. Williams. 2000. The protein kinase PKR is required for p38 MAPK activation and the innate immune response to bacterial endotoxin. EMBO J. 194292-4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haller, O., G. Kochs, and F. Weber. 2006. The interferon response circuit: induction and suppression by pathogenic viruses. Virology 344119-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hiscott, J. 2007. Triggering the innate antiviral response through IRF-3 activation. J. Biol. Chem. 28215325-15329. [DOI] [PubMed] [Google Scholar]
- 18.Ho, C. K., and S. Shuman. 1996. Mutational analysis of the vaccinia virus E3 protein defines amino acid residues involved in E3 binding to double-stranded RNA. J. Virol. 702611-2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Isaacs, S. N., E. J. Wolffe, L. G. Payne, and B. Moss. 1992. Characterization of a vaccinia virus-encoded 42-kilodalton class I membrane glycoprotein component of the extracellular virus envelope. J. Virol. 667217-7224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jacobs, B. L., and J. O. Langland. 1996. When two strands are better than one: the mediators and modulators of the cellular responses to double-stranded RNA. Virology 219339-349. [DOI] [PubMed] [Google Scholar]
- 21.Kibler, K. V., T. Shors, K. B. Perkins, C. C. Zeman, M. P. Banaszak, J. Biesterfeldt, J. O. Langland, and B. L. Jacobs. 1997. Double-stranded RNA is a trigger for apoptosis in vaccinia virus-infected cells. J. Virol. 711992-2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim, Y. G., M. Muralinath, T. Brandt, M. Pearcy, K. Hauns, K. Lowenhaupt, B. L. Jacobs, and A. Rich. 2003. A role for Z-DNA binding in vaccinia virus pathogenesis. Proc. Natl. Acad. Sci. USA 1006974-6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kitajewski, J., R. J. Schneider, B. Safer, S. M. Munemitsu, C. E. Samuel, B. Thimmappaya, and T. Shenk. 1986. Adenovirus VAI RNA antagonizes the antiviral action of interferon by preventing activation of the interferon-induced eIF-2 alpha kinase. Cell 45195-200. [DOI] [PubMed] [Google Scholar]
- 24.Kumar, A., Y. L. Yang, V. Flati, S. Der, S. Kadereit, A. Deb, J. Haque, L. Reis, C. Weissmann, and B. R. Williams. 1997. Deficient cytokine signaling in mouse embryo fibroblasts with a targeted deletion in the PKR gene: role of IRF-1 and NF-κB. EMBO J. 16406-416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langland, J. O., and B. L. Jacobs. 2004. Inhibition of PKR by vaccinia virus: role of the N- and C-terminal domains of E3L. Virology 324419-429. [DOI] [PubMed] [Google Scholar]
- 26.Langland, J. O., and B. L. Jacobs. 2002. The role of the PKR-inhibitory genes, E3L and K3L, in determining vaccinia virus host range. Virology 299133-141. [DOI] [PubMed] [Google Scholar]
- 27.Langland, J. O., J. C. Kash, V. Carter, M. J. Thomas, M. G. Katze, and B. L. Jacobs. 2006. Suppression of proinflammatory signal transduction and gene expression by the dual nucleic acid binding domains of the vaccinia virus E3L proteins. J. Virol. 8010083-10095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee, S. B., and M. Esteban. 1994. The interferon-induced double-stranded RNA-activated protein kinase induces apoptosis. Virology 199491-496. [DOI] [PubMed] [Google Scholar]
- 29.Lei, M., Y. Liu, and C. E. Samuel. 1998. Adenovirus VAI RNA antagonizes the RNA-editing activity of the ADAR adenosine deaminase. Virology 245188-196. [DOI] [PubMed] [Google Scholar]
- 30.Liu, Y., K. C. Wolff, B. L. Jacobs, and C. E. Samuel. 2001. Vaccinia virus E3L interferon resistance protein inhibits the interferon-induced adenosine deaminase A-to-I editing activity. Virology 289378-387. [DOI] [PubMed] [Google Scholar]
- 31.Ludwig, H., J. Mages, C. Staib, M. H. Lehmann, R. Lang, and G. Sutter. 2005. Role of viral factor E3L in modified vaccinia virus Ankara infection of human HeLa cells: regulation of the virus life cycle and identification of differentially expressed host genes. J. Virol. 792584-2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ludwig, H., Y. Suezer, Z. Waibler, U. Kalinke, B. S. Schnierle, and G. Sutter. 2006. Double-stranded RNA-binding protein E3 controls translation of viral intermediate RNA, marking an essential step in the life cycle of modified vaccinia virus Ankara. J. Gen. Virol. 871145-1155. [DOI] [PubMed] [Google Scholar]
- 33.McCormack, S. J., D. C. Thomis, and C. E. Samuel. 1992. Mechanism of interferon action: identification of a RNA binding domain within the N-terminal region of the human RNA-dependent P1/eIF-2 alpha protein kinase. Virology 18847-56. [DOI] [PubMed] [Google Scholar]
- 34.McKenna, S. A., D. A. Lindhout, I. Kim, C. W. Liu, V. M. Gelev, G. Wagner, and J. D. Puglisi. 2007. Molecular framework for the activation of RNA-dependent protein kinase. J. Biol. Chem. 28211474-11486. [DOI] [PubMed] [Google Scholar]
- 35.Moss, B. 2001. Poxviridae: the viruses and their replication, p. 2849-2883. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 36.Patterson, J. B., and C. E. Samuel. 1995. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol. Cell. Biol. 155376-5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rivas, C., J. Gil, Z. Melkova, M. Esteban, and M. Diaz-Guerra. 1998. Vaccinia virus E3L protein is an inhibitor of the interferon (IFN)-induced 2-5A synthetase enzyme. Virology 243406-414. [DOI] [PubMed] [Google Scholar]
- 38.Rochester, S. C., and P. Traktman. 1998. Characterization of the single-stranded DNA binding protein encoded by the vaccinia virus I3 gene. J. Virol. 722917-2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Romano, P. R., F. Zhang, S. L. Tan, M. T. Garcia-Barrio, M. G. Katze, T. E. Dever, and A. G. Hinnebusch. 1998. Inhibition of double-stranded RNA-dependent protein kinase PKR by vaccinia virus E3: role of complex formation and the E3 N-terminal domain. Mol. Cell. Biol. 187304-7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Samuel, C. E. 2001. Antiviral actions of interferons. Clin. Microbiol. Rev. 14778-809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Samuel, C. E. 1993. The eIF-2 alpha protein kinases, regulators of translation in eukaryotes from yeasts to humans. J. Biol. Chem. 2687603-7606. [PubMed] [Google Scholar]
- 42.Samuel, C. E. 1979. Mechanism of interferon action: phosphorylation of protein synthesis initiation factor eIF-2 in interferon-treated human cells by a ribosome-associated kinase possessing site specificity similar to hemin-regulated rabbit reticulocyte kinase. Proc. Natl. Acad. Sci. USA 76600-604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Samuel, C. E., R. Duncan, G. S. Knutson, and J. W. Hershey. 1984. Mechanism of interferon action. Increased phosphorylation of protein synthesis initiation factor eIF-2 alpha in interferon-treated, reovirus-infected mouse L929 fibroblasts in vitro and in vivo. J. Biol. Chem. 25913451-13457. [PubMed] [Google Scholar]
- 44.Seet, B. T., J. B. Johnston, C. R. Brunetti, J. W. Barrett, H. Everett, C. Cameron, J. Sypula, S. H. Nazarian, A. Lucas, and G. McFadden. 2003. Poxviruses and immune evasion. Annu. Rev. Immunol. 21377-423. [DOI] [PubMed] [Google Scholar]
- 45.Sharp, T. V., F. Moonan, A. Romashko, B. Joshi, G. N. Barber, and R. Jagus. 1998. The vaccinia virus E3L gene product interacts with both the regulatory and the substrate binding regions of PKR: implications for PKR autoregulation. Virology 250302-315. [DOI] [PubMed] [Google Scholar]
- 46.Shors, S. T., E. Beattie, E. Paoletti, J. Tartaglia, and B. L. Jacobs. 1998. Role of the vaccinia virus E3L and K3L gene products in rescue of VSV and EMCV from the effects of IFN-alpha. J. Interferon Cytokine Res. 18721-729. [DOI] [PubMed] [Google Scholar]
- 47.Shors, T., K. V. Kibler, K. B. Perkins, R. Seidler-Wulff, M. P. Banaszak, and B. L. Jacobs. 1997. Complementation of vaccinia virus deleted of the E3L gene by mutants of E3L. Virology 239269-276. [DOI] [PubMed] [Google Scholar]
- 48.Smith, E. J., I. Marie, A. Prakash, A. Garcia-Sastre, and D. E. Levy. 2001. IRF3 and IRF7 phosphorylation in virus-infected cells does not require double-stranded RNA-dependent protein kinase R or IκB kinase but is blocked by vaccinia virus E3L protein. J. Biol. Chem. 2768951-8957. [DOI] [PubMed] [Google Scholar]
- 49.Tanaka, H., and C. E. Samuel. 1994. Mechanism of interferon action: structure of the mouse PKR gene encoding the interferon-inducible RNA-dependent protein kinase. Proc. Natl. Acad. Sci. USA 917995-7999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tian, B., P. C. Bevilacqua, A. Diegelman-Parente, and M. B. Mathews. 2004. The double-stranded-RNA-binding motif: interference and much more. Nat. Rev. Mol. Cell Biol. 51013-1023. [DOI] [PubMed] [Google Scholar]
- 51.Toth, A. M., P. Zhang, S. Das, C. X. George, and C. E. Samuel. 2006. Interferon action and the double-stranded RNA-dependent enzymes ADAR1 adenosine deaminase and PKR protein kinase. Prog. Nucleic Acid Res. Mol. Biol. 81369-434. [DOI] [PubMed] [Google Scholar]
- 52.Wang, Q., M. Miyakoda, W. Yang, J. Khillan, D. L. Stachura, M. J. Weiss, and K. Nishikura. 2004. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J. Biol. Chem. 2794952-4961. [DOI] [PubMed] [Google Scholar]
- 53.Watson, J. C., H. W. Chang, and B. L. Jacobs. 1991. Characterization of a vaccinia virus-encoded double-stranded RNA-binding protein that may be involved in inhibition of the double-stranded RNA-dependent protein kinase. Virology 185206-216. [DOI] [PubMed] [Google Scholar]
- 54.Weber, F., G. Kochs, and O. Haller. 2004. Inverse interference: how viruses fight the interferon system. Viral Immunol. 17498-515. [DOI] [PubMed] [Google Scholar]
- 55.Weber, F., V. Wagner, S. B. Rasmussen, R. Hartmann, and S. R. Paludan. 2006. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 805059-5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Williams, B. R. 1999. PKR; a sentinel kinase for cellular stress. Oncogene 186112-6120. [DOI] [PubMed] [Google Scholar]
- 57.Xiang, Y., R. C. Condit, S. Vijaysri, B. Jacobs, B. R. Williams, and R. H. Silverman. 2002. Blockade of interferon induction and action by the E3L double-stranded RNA binding proteins of vaccinia virus. J. Virol. 765251-5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang, P., and C. E. Samuel. 2007. Protein kinase PKR plays a stimulus- and virus-dependent role in apoptotic death and virus multiplication in human cells. J. Virol. 818192-8200. [DOI] [PMC free article] [PubMed] [Google Scholar]