

Abstract
Although envelope glycoprotein M (gM) is highly conserved among herpesviruses, the varicella-zoster virus (VZV) gM homolog has never been investigated. Here we characterized the VZV gM homolog and analyzed its function in VZV-infected cells. The VZV gM homolog was expressed on virions as a glycoprotein modified with a complex N-linked oligosaccharide and localized mainly to the Golgi apparatus and the trans-Golgi network in infected cells. To analyze its function, a gM deletion mutant was generated using the bacterial artificial chromosome system in Escherichia coli, and the virus was reconstituted in MRC-5 cells. VZV is highly cell associated, and infection proceeds mostly by cell-to-cell spread. Compared with wild-type VZV, the gM deletion mutant showed a 90% reduction in plaque size and 50% of the cell-to-cell spread in MRC-5 cells. The analysis of infected cells by electron microscopy revealed numerous aberrant vacuoles containing electron-dense materials in cells infected with the deletion mutant virus but not in those infected with wild-type virus. However, enveloped immature particles termed L particles were found at the same level on the surfaces of cells infected with either type of virus, indicating that envelopment without a capsid might not be impaired. These results showed that VZV gM is important for efficient cell-to-cell virus spread in cell culture, although it is not essential for virus growth.
Varicella-zoster virus (VZV), which causes varicella (chicken pox) and herpes zoster (shingles), is a member of the genus Varicellovirus within the Alphaherpesvirinae subfamily of Herpesviridae (7). Although the diseases caused by this agent are endemic in the human population and it has one of the most significant morbidities among human herpesviruses, research on the biology of VZV has lagged behind that of other alphaherpesviruses, because VZV is highly cell associated and its growth is slower than that of other herpesviruses. The genomic organization of VZV is similar to that of herpes simplex virus type 1 (HSV-1), so the roles of many VZV genes have been predicted from their HSV-1 homologs (9). Despite this similarity, the human infections elicited by these viruses have different properties, implying differences in at least some of the viral gene functions.
The genome of herpesviruses encodes many glycoproteins, which are involved in various aspects of the viral life cycle, such as entry into the host cell (43) and the envelopment and maturation of progeny virions (29, 30), and are major immunological determinants for host defense mechanisms. VZV encodes at least seven proteins that are glycosylated—glycoproteins B, C, E, H, I, K, and L (gB, gC, gE, gH, gI, gK, and gL)—and contains the sequences for gM and gN, although it is not certain whether VZV gM and gN are glycosylated (7).
Although herpesviruses encode several glycoproteins, the genes for only five are conserved among all herpesviruses: gB, gH, gL, gM, and gN. The conservation of these glycoproteins in herpesvirus evolution suggests that they have important roles in virus life cycles. While gB and gH/gL are essential for viral replication in cell culture (43), disruption of gM in HSV-1 (2, 26), pseudorabies virus (PRV) (10), or equine herpesvirus 1 (EHV-1) (36) shows 1 to 10% of the wild-type growth. The gM genes encoded by human cytomegalovirus (HCMV) (16) and Marek's disease virus serotype 1 (48) are reported to be essential for viral replication. Therefore, the functional roles of the gM protein during viral replication may differ among herpesviruses. The gM homologs in PRV (17), bovine herpesvirus 1 (24), infectious laryngotracheitis virus (13), and HCMV (25) all form a disulfide-linked complex with a gN homolog.
Although the gM/gN complex is dispensable for alphaherpesvirus replication in cell culture, a null mutation of the gM gene or null mutations of both the gM and gN genes give rise to some interesting phenotypes. The deletion of gM in PRV (10), EHV-1 (36), or EHV-4 (51) impairs not only the cell-to-cell spread of infection but also the penetration of cell-free virus into cells. The gM of HSV-1 or PRV expressed in cell culture inhibits the cell-cell fusion induced by the herpesvirus glycoproteins gB, gD, and the gH-gL complex (8, 21) and by another viral protein, human respiratory syncytial virus F protein (8). The expression of gM causes other viral membrane proteins to relocate to the trans-Golgi network (TGN), suggesting that gM functions in the maturation of viral particles and the cell-to-cell spread of infection (8). The gM/gN complex is found in the viral particles of PRV (17) or HCMV (25). Recently, antibodies against the gM/gN complex were shown to have a neutralizing function in HCMV infection (42). These findings suggest that the gM/gN complex is involved in virus entry into host cells.
In contrast to the information available for other herpesviruses, the gN homolog encoded by VZV has only been reported to be nonessential for viral growth in cell culture (38), and the VZV homolog of gM has not been investigated at all. Therefore, the aim of this study was to identify and characterize the VZV gM homolog and to investigate its function in cell culture. For these analyses, we produced monospecific antibodies (Abs) against VZV gM and constructed a gM-deficient mutant virus. We found that the VZV gM homolog is a membrane glycoprotein expressed on virions and that the gM-deficient mutant exhibited impaired cell-to-cell spread compared with the parental virus.
MATERIALS AND METHODS
Cells and viruses.
MRC-5 cells were cultured in minimum essential medium with 10% fetal bovine serum at 37°C under a 5% CO2 atmosphere. Human melanoma cells (MeWo cells) were cultured in Dulbecco's modified Eagle's medium with 10% fetal bovine serum at 37°C under a 5% CO2 atmosphere. To generate MeWo cells stably expressing VZV gM (designated MeWo-gM cells), a plasmid encoding hemagglutinin (HA)-tagged VZV gM was constructed. A nucleotide fragment containing VZV gM with the HA tag was constructed by PCR with primers VZ50HAF (5′-GTCGACATGTACCCATACGACGTACCAGATTACGCTGGAACTCCAAAG-3′) and VZ50HAR (5′-CTCGAGCTACTCCCACCCACTGTT-3′) using pOka-BAC as the template. The establishment of pOka-BAC is described elsewhere (33). The resulting nucleotide fragment was ligated into SalI- and XhoI-digested pCAGGS (35). The pCAGGS plasmid was kindly provided by J. Miyazaki, Osaka University, Osaka, Japan. Nucleotide fragments containing the neomycin resistance gene were extracted from pMC1neopolyA (Stratagene) (47) by SalI and XhoI digestion. This neomycin resistance gene was inserted into the SalI cloning site of pCAGGS-MCS containing HA-tagged VZV gM, generating pCAGGS-neoHA-gM. The resultant pCAGGS-neoHA-gM plasmid contained an HA-tagged VZV gM gene, regulated by the HCMV immediate-early enhancer and the chicken β-actin promoter, and a neomycin resistance gene. MeWo-gM cells were constructed by transfecting MeWo cells with pCAGGS-neoHA-gM using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. The MeWo-gM cells were selected and propagated in the presence of G418 (Nacalai Tesque). Transduction of the gM gene into MeWo-gM cells was confirmed by PCR using primers VZ50F80 (5′-TGATCGCTCGTATAACAGCTCAT-3′) and VZ50R1230 (5′-GATGTTCACTTTGGGTGCGG-3′), and the expression of gM in MeWo-gM cells was verified by an immunofluorescence assay (data not shown) and Western blotting using an anti-gM Ab. The parental Oka (pOka) strain was used in this study and propagated in MRC-5 cells. Cell-free virus was prepared as follows. The infected cells were first treated with 0.1% EDTA in phosphate-buffered saline and then sonicated, and the lysates were spun at 500 × g for 5 min at 4°C. The supernatant was transferred to new tubes for use as cell-free virus.
Construction of rpOka and gM deletion mutant viruses.
The recombinant pOka virus (rpOka) was constructed as previously reported (33). In brief, rpOka was reconstituted by transfecting MRC-5 cells with the pOka bacterial artificial chromosome (BAC) genome (pOka-BAC) and then infecting them with a recombinant adenovirus harboring the Cre recombinase, AxCANCre, to delete the BAC sequence from the reconstituted virus. AxCANCre was kindly provided by Y. Kawaguchi, Tokyo University (18, 46). The gM deletion mutant virus (rpOkaΔgM) was constructed using pOka-BAC. To generate the gM-deficient mutant virus, a kanamycin resistance (Kmr) gene flanked by partial sequences of gM was constructed. The DNA fragments containing the Kmr gene were amplified from pCR2.1-TOPO (Invitrogen) by PCR with primers VZ50KMF200 (5′-TGCGTTTCTGTCGATGATACACATATGCACGCATCAATCTGAGAAGCAACGACAGCAAGCGAACCGGAATTGC-3′) and VZ50KMR980 (5′-CCATGGTATATACACTGATCACGGCCGCGATTATACACCGAGAAACGCCTTTTTCAATTCAGAAGAACTC-3′). The DNA fragments were introduced by electroporation into Escherichia coli DH10B containing pOka-BAC and pGETrec, which was a kind gift from P. A. Ioannou (34), and the transformants were incubated and restreaked onto plates containing chloramphenicol and kanamycin. The presence of the expected recombination in the resulting colonies was confirmed by PCR amplification of the appropriate regions and restriction enzyme analysis.
Purification of virus particles.
The cell-free virus samples were loaded onto a Histodenz (Sigma-Aldrich) gradient and subjected to ultracentrifugation at 27,000 rpm for 1 h at 4°C (P40ST-1689 rotor; Hitachi High Technologies). Fractions were collected from the top. The presence of virions in each fraction was analyzed by PCR with primers VZ50F80 and VZ50R1230. The presence of gM in each fraction was examined by Western blotting. The fractions containing both the viral genome and the gM protein were used as suspensions of purified virions (data not shown).
Endoglycosidase digestion.
For endoglycosidase digestion, endoglycosidase H (endo H) and peptide N-glycosidase F (PNGase F) were purchased from New England Biolabs. The rpOka-infected cells were harvested 48 h after infection. rpOka- or mock-infected MRC-5 cells were lysed with radioimmunoprecipitation assay buffer (0.01 M Tris-HCl [pH 7.4]), 0.15 M NaCl, 0.1% sodium deoxycholate, 1% Nonidet P-40, 0.1% sodium dodecyl sulfate [SDS], 1 mM EDTA) and spun at 70,000 rpm for 1 h in a TLA110 rotor (Beckman Coulter). The supernatants were then resuspended in digestion buffer (New England Biolabs), and the samples were digested with endo H or PNGase F in accordance with the manufacturer's protocol.
Abs.
Monospecific Abs were produced against synthetic peptides [(C)EDELLYERSNGWE] of the gM protein (Sigma-Aldrich, Japan). These peptides were used to immunize rabbits, and antisera were obtained (Sigma-Aldrich). Polyclonal Abs against open reading frame 16 (ORF16), ORF49, and gB were produced as described previously (40). Mouse monoclonal antibodies (MAbs) against α-tubulin (clone B-5-1-2; Sigma-Aldrich), GM130 (clone 35; BD Biosciences), p230-TGN (clone 15; BD Biosciences), EEA1 (clone 14; BD Biosciences), cathepsin D (clone BC011; EMD Biosciences), and nucleoporin p62 (clone 53; BD Biosciences) were purchased. Tetramethyl rhodamine isocyanate (TRITC)-conjugated swine anti-rabbit immunoglobulin G (IgG) (Dako) and an Alexa Fluor 488-conjugated F′(ab′)2 fragment of goat anti-mouse IgG (Invitrogen) were also obtained.
Western blotting.
MRC-5 cells were infected with VZV at a multiplicity of infection (MOI) of 0.01 and incubated for 48 h at 37°C. MeWo and MeWo-gM cells were inoculated with virus-infected cells and incubated for 72 h at 37°C. Samples were separated by SDS-polyacrylamide gel electrophoresis (PAGE) and electrotransferred to polyvinylidene difluoride membranes. The membranes were incubated first with primary Abs and then with peroxidase-conjugated secondary Abs. The bound Abs were detected by chemiluminescence using Western blotting detection reagents (GE Healthcare Biosciences) and were visualized on X-ray film.
Immunofluorescence microscopy.
Indirect immunofluorescence assays were performed as described previously (1, 40). All samples were analyzed on a model DMIRE2 Leica confocal microscope (Leica Microsystems).
RNA extraction and cDNA preparation for RT-PCR.
Total RNA was extracted from virus-infected MRC-5 cells by using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. cDNA was synthesized with the SuperScript II First Strand cDNA synthesis kit (Invitrogen) in accordance with the manufacturer's instructions. PCR amplification was performed with the primer pairs described below, using the cDNA as the template. The primer pairs were as follows: for ORF51, ORF51F2041 (5′-ACCTTAAACCGCCCGGTTTGGCGAACAACC-3′) and ORF51R (5′-ACCTTATAAACTTTCAAAATTTACCGC-3′); for cellular elongation factor, EF-F (5′-GCTCCAGCATGTTGTCACCATTC-3′) and EF-R (5′-GGTGAATTTGAAGCTGGTATCTC-3′). PCR of the extracted RNA was performed without the reverse transcription (RT) reaction in order to rule out DNA contamination.
Southern blotting.
pOka-BAC DNA, pOka-BACΔgM DNA, or DNA extracted from rpOka- or rpOkaΔgM-infected cells (viral DNA) was digested with BamHI and separated by electrophoresis in a 0.5% agarose gel. Viral DNA was extracted from infected cells as previously described (33). After transfer to nylon membranes, the blots were probed with horseradish peroxidase-bound nucleotide probes using the ECL direct nucleic acid labeling and detection system (GE Healthcare Biosciences) in accordance with the manufacturer's instructions. The probes for the gM gene, ORF62 derived from pOka-BAC, and the Kmr gene derived from pCR2.1-TOPO (Invitrogen) were made by PCR with primers VZ50F80 and VZ50R1280, IE62fWA (5′-GCTTTTGGTAGAACTGGTTC-3′) and IE62RvA (5′-TTCAACCAGAACCCAGAACG-3′), and KMF (5′-ATGATTGAACAAGATGGATTG-3′) and KMR (5′-AAGAAGGCGATAGAAGGCGATG-3′), respectively. Bound probes were detected by chemiluminescence using ECL detection reagents and were visualized on X-ray film.
Determination of plaque size.
Cells growing on six-well plates were infected in duplicate with cell-free virus at an MOI of 0.005 and incubated for 1 h to permit virus adsorption. The medium was then replaced with fresh medium, and the infected cells were incubated at 37°C for 10 days. The cells were then stained with 1% crystal violet-70% ethanol. The plaques were counted and their areas measured by ImageJ software (http://rsb.info.nih.gov/ij/).
Analysis of virus spread.
To analyze the cell-to-cell spread of the recombinant virus, an infectious center assay was performed as described previously (15). Briefly, 24 h after seeding, cells were infected at an MOI of 0.005 with cell-free viruses as described above. The infected cells were harvested at the indicated time points by trypsinization. The cells were serially diluted, added to uninfected cells on six-well plates, incubated at 37°C for 7 days, and stained with 1% crystal violet-70% ethanol. The plaques were counted, and the number of infectious centers was evaluated as the increase over the value on day zero.
Electron microscopy.
rpOka- and rpOkaΔgM-infected MRC-5 cell cultures in culture flasks with a 75-cm2 growth surface (Greiner) were scraped off the plate and pelleted by low-speed centrifugation 36 h after cell-to-cell infection. The cells were immediately fixed for 120 min at 4°C with 2.5% glutaraldehyde and 4% paraformaldehyde in 0.05 M sodium cacodylate buffer (pH 7.4). Small pieces were postfixed in sodium cacodylate-buffered 1.5% osmium tetroxide for 60 min at 4°C, block stained in 0.5% uranyl acetate, dehydrated through a series of ethanol concentrations, and embedded in Epon resin (TAAB). Ultrathin sections were stained with uranyl acetate and lead citrate. Viral particles, purified by Histodenz gradients as described above, were negatively stained using 3% uranyl acetate. The samples were examined under a Hitachi (Tokyo, Japan) electron microscope (H-7650).
RESULTS
VZV gM protein is expressed in both infected cells and virions as a glycoprotein modified with a complex N-linked oligosaccharide.
In previous reports of other herpesviruses, gM was identified as one of the glycoproteins expressed on the viral envelope (3, 10, 19, 27, 37, 50, 51). Therefore, first, to determine whether VZV gM is present in viral particles and whether it is glycosylated, purified virions and infected-cell lysates were digested with the endoglycosidase endo H or PNGase F and subjected to Western blot analysis with an anti-gM Ab. gM was detected in infected-cell lysates as a broad band with a molecular mass of 42 to 48 kDa and a faint band with a molecular mass of 37 kDa (Fig. 1Aa), while no 37-kDa gM was detected in virions. Because VZV is a highly cell associated virus, the presence of viral particles in the purified fraction was confirmed with negative staining by electron microscopy (Fig. 1Ab). The expression of gB was also examined in order to evaluate the purity of the virion preparation. gB is synthesized as a type I membrane glycoprotein (uncleaved) in infected cells. It is then cleaved into 68-kDa and 66-kDa proteins and incorporated into the virion as a heterodimer (20). As shown in Fig. 1Aa, only the cleaved form of gB was detected in the virion preparation, indicating that the virions were purified without contamination by uncleaved gB, while both forms were present in infected cells. Furthermore, ORF16 protein, which is a homolog of the processivity subunit of DNA polymerase and a nonstructural protein, was not detected in the purified virion lysates (Fig. 1Aa), indicating that the virion preparation was not contaminated with nonstructural viral proteins. In addition, EEA1, a 180-kDa hydrophilic peripheral membrane protein that localizes to early endosomes, was not detected in the virion lysates (Fig. 1Aa), indicating that the preparation was not contaminated with cytoplasmic membranes.
FIG. 1.

Western blotting of VZV- and mock-infected MRC-5 cells and purified virions. (A) (a) Cells and purified virions were lysed, separated by SDS-PAGE, and subjected to Western blotting with an anti-gM monospecific Ab, an anti-gB Ab, an anti-ORF16 Ab, or an anti-EEA1 MAb. Molecular masses are given to the left of Western blots. Infected cell, rpOka-infected MRC-5 cells; virion, rpOka virions; mock, mock-infected MRC-5 cells. (b) The purified virions used for Western blotting were confirmed by electron microscopy and negative staining. Scale bar, 200 nm. (B) Lysates were digested with endo H or PNGase F, resolved by SDS-PAGE under reducing conditions, and electrotransferred to polyvinylidene difluoride membranes. The blots were reacted with an anti-gM or anti-gB Ab. Open arrowheads indicate the precursor of gM.
Following PNGase F digestion, the molecular mass of the 42- to 48-kDa gM in both lysates was shifted to approximately 35 kDa, while it was not changed by endo H digestion (Fig. 1B). However, the 37-kDa gM detected only in infected-cell lysates was digested with both endo H and PNGase F. The results show that the VZV gM homolog is also a membrane glycoprotein modified with a complex N-linked oligosaccharide and is present in virions as a glycoprotein and that the 37-kDa form of gM is a precursor of the mature form. Endoglycosidase digestion of the lysates was also confirmed by Western blotting of gB. As shown in Fig. 1B, gB was digested with both endo H and PNGase F, confirming that the lysates were digested with both endoglycosidases.
VZV gM proteins were localized mainly to the Golgi apparatus and TGN in VZV-infected cells.
In HSV-1, gM has been reported to localize with a marker of the Golgi apparatus and partially with a marker of the TGN in infected cells (4). To examine the localization of gM in VZV-infected cells, mock- or rpOka-infected MRC-5 cells were harvested at 48 h postinfection (hpi), fixed, and double stained with an anti-gM Ab and with Abs against several cellular proteins. Since gM was expressed abundantly at 48 hpi in VZV-infected cells and the localization was clearly observed, this time point was chosen for the study. As shown in Fig. 2, strong gM expression was observed in the perinuclear region. gM colocalized with GM130 (Fig. 2A), a marker of the Golgi matrix, and p230 (Fig. 2B), a marker of the TGN, and the localizations of GM130 and p230 were altered by VZV infection (Fig. 2A and B). gM also partially colocalized with LAMP1, a marker of late endosomes and lysosomes (Fig. 2C), but did not colocalize with cathepsin D, a marker of lysosomes (Fig. 2D), indicating that gM localized slightly to late endosomes but not to lysosomes. However, gM scarcely overlapped with EEA1, a marker of early endosomes (Fig. 2E), and did not overlap with nucleoporin p62, a marker of the nuclear membrane (Fig. 2F). These results suggest that VZV gM localized mainly to the Golgi apparatus and TGN.
FIG. 2.

Subcellular localization of gM in rpOka-infected MRC-5 cells. MRC-5 cells were either mock infected or infected with rpOka at an MOI of 0.01, fixed 48 h later, and stained with a rabbit anti-gM monospecific Ab and a mouse anti-GM130 (A), anti-p230 (B), anti-LAMP1 (C), anti-cathepsin D (D), anti-EEA1 (E), or anti-nucleoporin p62 (F) MAb. Nuclei were stained with Hoechst 33342. The anti-gM Ab was visualized with TRITC-conjugated anti-rabbit IgG, and the anti-GM130, anti-p230, anti-LAMP1, anti-cathepsin D, anti-EEA1, and anti-nucleoporin p62 mouse MAbs were visualized with an Alexa Fluor 488-conjugated F′(ab′)2 fragment of goat anti-mouse IgG. Localization was analyzed by confocal laser scanning microscopy. Costained areas appear yellow in the merged images. Bars, 10 μm.
Construction and characterization of the gM deletion mutant in VZV.
To examine the function of VZV gM in infected cells, a deletion mutant of the gM gene was generated in E. coli by mutagenesis of a BAC clone harboring the VZV genome (pOka-BAC) (Fig. 3). Nucleotides 408 to 1058 (encoding amino acids 136 to 353) of the gM gene in the pOka-BAC clone were replaced with the Kmr gene (Fig. 3B). This segment was chosen so as to avoid negative effects on the up- and downstream genes ORF48, ORF49, and ORF51. The deletion of gM and the insertion of the Kmr cassette at the correct site of the VZV BAC genome were verified by restriction enzyme digestion (BamHI) (Fig. 4A) and Southern blot analyses using the BamHI-digested BAC and viral genome probed with either the Kmr (Fig. 4B and D), the gM (Fig. 4C and E), or the ORF62 (Fig. 4C and E) gene. The restriction digests showed the predicted 6.9-kb fragment of the gM gene from pOka-BAC DNA or rpOka viral DNA and the predicted 7.2-kb fragment of the Kmr cassette from pOka-BACΔgM DNA or rpOkaΔgM viral DNA. The ORF62 probe hybridized with the predicted fragments (4.8 kb) in all samples (Fig. 4C and E).
FIG. 3.

Structure of the pOka-BAC genome and construction of the gM deletion mutant virus. (A) Schematic map of the pOka-BAC genome showing the long (UL) and short (US) unique regions, the inverted-repeat sequences (TRL, IRL, IRS, and TRS), the BAC sequence, the gM gene, and the vicinity of these sequences. (B) The relevant region of the genome is enlarged. Pointed rectangles represent ORFs in their transcriptional orientation. The indicated nucleotide fragment containing the kanamycin resistance gene (Kmr) was used for deletion of the gM gene by homologous recombination in E. coli. (C) The gM gene deletion mutant was constructed from the pOka-BAC genome (pOka-BACΔgM). (D) The BAC sequence was removed from the reconstituted recombinant virus by Cre recombinase (rpOkaΔgM). The resulting mutant virus contained a single loxP site. Black rectangles with a white letter L represent loxP sites.
FIG. 4.

Restriction enzyme digestion and Southern blot analyses. (A) pOka-BAC and pOka-BACΔgM were digested with BamHI and separated on a 0.8% agarose gel. Molecular size markers are shown at the left. (B through E) Southern blot analyses of BamHI-digested pOka-BACΔgM DNA and pOka-BAC DNA (B and C) and of rpOkaΔgM and rpOka viral DNA (D and E) were performed. The blots were treated with labeled probes for the Kmr gene (B and D) and the gM and ORF62 genes (C and E).
The absence of gM protein expression in rpOkaΔgM-infected cells was verified by Western blot analysis. As expected, gM protein was detected in lysates of rpOka-infected MRC-5 cells but not in those of mock- or rpOkaΔgM-infected cells (Fig. 5A), confirming that the gM gene had been deleted from the VZV genome. Levels of protein or mRNA expression from the neighboring ORF49 or ORF51 gene were similar in cells infected with the two types of virus, indicating that gM's neighboring genes were not affected by the mutagenesis (Fig. 5B and D). α-Tubulin or elongation factor was used as a positive control for protein or mRNA expression of the cell lysates and was detected in all the samples (Fig. 5C and E).
FIG. 5.
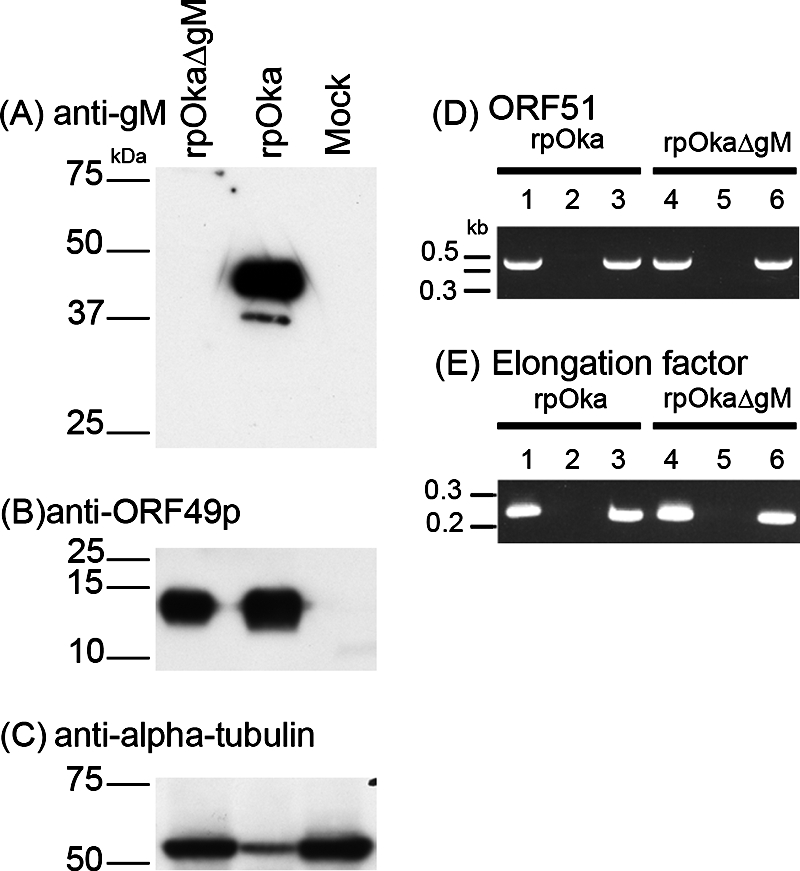
Absence of gM expression in rpOkaΔgM-infected cells. rpOka-, rpOkaΔgM-, or mock-infected MRC-5 cells were harvested at 48 hpi. (A, B, and C) Cells were lysed, separated by SDS-PAGE, and subjected to Western blotting with an anti-gM (A) or anti-ORF49 (B) Ab or with an anti-α-tubulin MAb (C). (D and E) RNA was extracted, and RT-PCRs of ORF51 (D, lanes 3 and 6) and elongation factor (E, lanes 3 and 6) were performed using specific primer pairs. Lanes 2 and 5, PCRs without RT, performed as a control for contamination with genomic DNA. Lanes 1 and 4, PCRs of viral DNA, performed as a positive control. Sizes of molecular mass markers are given on the left.
Plaque formation and cell-to-cell spread were impaired in the gM deletion mutant.
To analyze the growth properties of rpOkaΔgM, plaque formation and infectious center assays were performed. The infection of MRC-5 cells by rpOkaΔgM caused plaques (0.35 mm2 ± 0.01 mm2 [average ± standard error]) about 90% smaller than those caused by the wild-type virus, rpOka (4.09 mm2 ± 0.37 mm2) (Fig. 6), and the same result was obtained when MeWo cells were used (data not shown), suggesting that plaque formation was impaired in rpOkaΔgM-infected cells. The plaques in rpOkaΔgM-infected MeWo-gM cells, stably expressing gM protein, were approximately 33% larger than those in rpOkaΔgM-infected MeWo cells (Fig. 7A), indicating that the phenotype of the mutant was due to the deletion of the gM gene. The expression of gM in MeWo-gM cells was confirmed by Western blotting. As shown Fig. 7B, the 42- to 48-kDa form of gM was detected in rpOkaΔgM-infected MeWo-gM cells, while only the 37-kDa form, a precursor of gM, was detected in mock-infected MeWo-gM cells, indicating that gM expression was found in MeWo-gM cells; however, the formation of the mature form of gM is required for the expression of the other virus gene(s). In addition, the expression of gM was lower in rpOkaΔgM-infected MeWo-gM cells than in rpOka-infected MeWo cells. This may be due to the lower copy numbers of the gM gene in MeWo-gM cells.
FIG. 6.

Comparison of plaque sizes of rpOkaΔgM and rpOka. MRC-5 cells were infected with cell-free rpOka or rpOkaΔgM virus at an MOI of 0.005 and were cultured for 10 days. The cells were fixed and stained with 1% crystal violet-70% ethanol.
FIG. 7.

Comparison of plaque sizes of rpOkaΔgM in MeWo and MeWo-gM cells. (A) MeWo or MeWo-gM cells were infected with cell-free rOkaΔgM virus at an MOI of 0.005 and were cultured for 7 days. The cells were fixed and stained with 1% crystal violet-70% ethanol. Plaque sizes were scanned and measured using ImageJ. Error bars, standard errors. Statistical significance was determined by Student's t test. (B) MeWo cells infected with rpOka (lane 1) or rpOkaΔgM (lane 2) or mock infected (lane 3) and MeWo-gM cells infected with rpOkaΔgM (lane 4) or mock infected (lane 5) were harvested at 72 hpi. The cells were lysed, separated by SDS-PAGE, and subjected to Western blotting with an anti-gM Ab and an anti-tubulin MAb.
These phenomena indicated that the absence of gM expression in VZV infection caused a reduction in plaque formation. Thus, an infectious center assay was next performed to examine the cell-to-cell spreading of progeny viruses. MRC-5 cells were infected with rpOka or rpOkaΔgM, and the number of infected cells from 0 to 5 days was titrated. In this assay, rpOka exhibited an obviously faster spread than the mutant virus in MRC-5 cells. After 5 days of cell spread, the number of cells infected by rpOkaΔgM was about half the number infected by rpOka (Fig. 8). These results suggested that viral growth was marginally affected and that the cell-to-cell spread of viral progeny was impaired by the deletion of gM from the VZV genome.
FIG. 8.

Infectious center assay of rpOka and rpOkaΔgM. MRC-5 cells in six-well plates were infected independently with similar titers of cell-free rpOka or rpOkaΔgM virus and were then treated with trypsin from day 1 to day 5 postinfection. The treated cells were diluted and overlaid onto newly prepared, uninfected MRC-5 cells in six-well plates. Seven days later, the overlaid cells were fixed and stained with 1% crystal violet-70% ethanol, and the number of infected cells was assessed by counting the number of VZV plaques. The number of infected cells was normalized to the initial viral titer per dish; the increase (n-fold) is expressed as the number of infected cells from one initial infected cell on day zero. Error bars, standard errors. Statistical significance was determined by a paired Student t test.
The number of vacuoles containing relatively electron dense materials was increased in cells infected with the gM deletion mutant.
To assess the defect in cells infected with the gM deletion mutant, electron microscopic analyses were performed. To this end, MRC-5 cells were infected with rpOkaΔgM or rpOka by inoculating them with virus-infected MRC-5 cells. At 36 h after cell-to-cell infection, cells were fixed, stained, and analyzed. As shown in Fig. 9B, after infection with rpOkaΔgM, numerous aberrant vacuoles containing electron-dense materials were found in the cytoplasm. Enveloped particles termed L particles (28, 44, 45) lining the outer surfaces of the cells (Fig. 9C and F) and capsid assembly in the nucleus (Fig. 9A and D) were observed in cells infected with either type of virus. L particles are noninfectious virus-like particles that lack both capsids and virus DNA (28, 45).
FIG. 9.

Electron microscopy of rpOkaΔgM- or rpOka-infected cells. MRC-5 cells were infected with rpOkaΔgM (A, B, and C) or rpOka (D, E, and F) and analyzed at 36 h after cell-to-cell infection. (A and D) Nucleocapsids were observed in the nuclei of both rpOkaΔgM- and pOka-infected MRC-5 cells. (B and E) Aberrant vacuoles containing relatively electron dense materials (arrows) were found in the cytoplasm of rpOkaΔgM-infected cells but not in that of pOka-infected cells. (C and F) Numerous extracellular L particles (arrowheads) lining the surface of the plasma membrane were observed in both rpOkaΔgM- and rpOka-infected cells. Bars, 1 μm.
DISCUSSION
Although recent studies have greatly increased our understanding of viral glycoproteins such as gB and the gE/gI complex in VZV infection, this is the first report on the characterization and function of gM during VZV infection. gM is one of a few envelope glycoproteins conserved among alpha-, beta-, and gammaherpesviruses, but the gM gene has been reported to be nonessential for viral growth in herpesviruses HSV-1 (2, 26), PRV (10), EHV-1 (36), and EHV-4 (51).
In this study, abundant gM proteins with a molecular mass of 42 to 48 kDa or 37 kDa were detected in VZV-infected cells, and only the 42- to 48-kDa form was present in virions as a glycoprotein with a complex N-linked oligosaccharide. Generally, gM proteins encoded by herpesviruses are highly hydrophobic proteins that are predicted to contain six to eight transmembrane domains. They have a potential N-glycosylation site within the first extracellular domain and a conserved cysteine residue within the same loop that is predicted to form a disulfide bond with the relevant gN homolog (10, 17). VZV gM is also predicted to be an integral membrane protein with eight transmembrane domains, a putative N-glycosylation site, and a cysteine residue that could form a disulfide bond in the first ectodomain (Fig. 10).
FIG. 10.

Potential topology of VZV gM. The topology of VZV gM was predicted using the TMHMM transmembrane topology prediction server (http://www.cbs.dtu.dk/services/TMHMM-2.0/). The rectangle at the top is a schematic representation of the predicted gM protein. aa, amino acids. Dashed lines indicate the region of the gM gene replaced with the kanamycin resistance gene. The approximate locations of an N-linked glycosylation site (NAT, asparagine-alanine-threonine; branched structure, N-linked sugar chain) and a cysteine (C) that could form a disulfide bond (-S-S-) are indicated.
A putative N-linked glycosylation site in the first ectodomain is conserved among the gM proteins encoded by herpesviruses, which implies that the N-linked sugar chain in gM makes an important contribution to its function. However, despite this conservation, the gM of infectious laryngotracheitis virus is not glycosylated (12). The gM protein is a virion component in other herpesviruses (3, 10, 22, 27, 37, 50). In HCMV virions, gM is reported to be the most abundant glycoprotein (49). The deletion of gM impairs not only viral growth but also viral penetration of and entry into cells (10, 36). The gM encoded by VZV may also be involved in viral entry into and egress from host cells, because it is incorporated into virions.
Because VZV gM was found to be an envelope glycoprotein, we next examined its localization in infected cells. VZV gM was localized mainly to the Golgi apparatus and TGN of infected cells, indicating that gM together with other envelope glycoproteins entered the TGN for secondary envelopment, because VZV budding into TGN-derived vesicles has been reported (14). Furthermore, gM was localized partially to the late endosomes of infected cells, which might have reflected gM incorporation into virions, because mature or immature virions have been reported to be transported to late endosomes (14). In addition, gM was scarcely localized to early endosomes, indicating that gM may not traffic to early endosomes and may traffic directly to the TGN for secondary envelopment.
Recently, Baines et al. reported that HSV-1 gM was localized at the perinuclear region near the nuclear membrane and that gM was incorporated into virions during budding at the inner nuclear membrane (4). In the present study, no VZV gM expression was observed on the nuclear membrane, showing that there may be different biological functions of gM among alphaherpesviruses. However, the detection of gM on the nuclear membrane may depend on the stage of infection or the recognition site of the antibody used for detection. Further study would be required to analyze this issue more.
To analyze the function of VZV gM more precisely, we performed an electron microscopic examination, and the morphological features of rpOkaΔgM- and rpOka-infected MRC-5 cells were compared. Whereas cell-to-cell spread and plaque formation were impaired in rpOkaΔgM infection, L particles lining the outer surfaces of the cells and capsid assembly in the nucleus were observed in mutant virus-infected cells. This showed that the single deletion of gM of VZV did not cause significant impairment of particle formation, although the formation of mature virions was thought to be partially impaired. Ultrastructural studies of PRV (5) and EHV-1 (41) indicate that the deletion of gM impairs secondary envelopment, and this phenotype is more severe when the gE/gI complex is deleted together with gM. Deletion of the C-terminal region of gE in a gM deletion mutant of PRV showed a phenotype similar to that of a gE/gI/gM triple-deletion mutant (6). gM may have supplemental roles in secondary envelopment, which were not affected in the gM single-deletion mutant. rpOkaΔgM showed impairment of cell-to-cell spread of progeny but no impairment of L particle formation. Aberrant vacuoles containing electron-dense materials were found in rpOkaΔgM-infected MRC-5 cells, while they were scarcely found in rpOka-infected cells. In light of these data, the deletion of gM is thought to partially impair virion maturation and the related protein sorting, although the detailed mechanism is unknown. In PRV-infected cells, gM interacted with UL49, and a gE/gI/gM deletion mutant failed to incorporate UL49 into viral particles (11). As well, VZV gM may be more important for reciprocal functions with other gene products.
Here, by deleting VZV gM, we found that, although VZV gM was nonessential for virus growth, its loss caused impairments in plaque size and cell-to-cell spread, indicating that VZV gM may play an important role in virus spread to neighboring cells. In contrast, a gM gp2 double-negative mutant of EHV-1 (39) shows a defect in viral egress from infected cells, but only a slight impairment in cell-to-cell spread, compared with the parental virus and a gp2 single-deletion mutant. Therefore, the roles of gM may be redundant and/or have variable importance among the herpesviruses.
VZV gE is the most abundant viral glycoprotein in VZV-infected cells (32), and its deletion from VZV abolishes the virus's ability to reproduce (31). Recently, insulin-degrading enzyme was found to be a cellular receptor for VZV, and gE was shown to be the viral ligand for the receptor (23). In PRV, triple deletion of the gM, gE, and gI genes significantly impairs viral growth and secondary envelopment (5), indicating a redundancy between gM and the gE/gI complex. The gE/gI complex has an essential role in the cell-to-cell spread of VZV in vitro, and VZV gM may assist the gE/gI complex in this function.
Complementation of the growth of the rpOkaΔgM virus in MeWo-gM cells was partial. It appeared that not all of the virus particles produced in rpOkaΔgM-infected MeWo-gM cells were complemented with gM, in view of the lower expression of gM in MeWo-gM cells than in wild-type virus-infected MeWo cells (Fig. 7B).
In summary, we showed that VZV gM is a complex-type N-linked glycoprotein, that it is present in viral particles, and that the gM deletion mutant of VZV shows impaired viral growth and cell-to-cell spread. This is the first report to demonstrate that VZV gM is an envelope glycoprotein and plays a role in viral cell-to-cell spread.
Acknowledgments
We thank Panayiotis A. Ioannou (Cell and Gene Therapy Research Group, Murdoch Children's Research Institute, Royal Children's Hospital, Australia) for providing pGETrec, Yasushi Kawaguchi (Division of Viral Infection, Department of Infectious Disease Control, International Research Center for Infectious Diseases, Institute of Medical Science, University of Tokyo, Tokyo, Japan) for AxCANCre, Jun-ichi Miyazaki (Division of Stem Cell Regulation Research, Osaka University Graduate School of Medicine, Osaka, Japan) for pCAGGS, Ulrich H. Koszinowski (Max von Pettenkofer Institut fur Virologie, Ludwig-Maximilians-Universität München, Munich, Germany) for pHA2, and Junji Sashihara (Osaka University Graduate School of Medicine, Osaka, Japan) for technical assistance.
This study was supported in part by a grant-in-aid for Scientific Research on Priority Areas from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan.
Footnotes
Published ahead of print on 31 October 2007.
REFERENCES
- 1.Akkapaiboon, P., Y. Mori, T. Sadaoka, S. Yonemoto, and K. Yamanishi. 2004. Intracellular processing of human herpesvirus 6 glycoproteins Q1 and Q2 into tetrameric complexes expressed on the viral envelope. J. Virol. 787969-7983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baines, J. D., and B. Roizman. 1991. The open reading frames UL3, UL4, UL10, and UL16 are dispensable for the replication of herpes simplex virus 1 in cell culture. J. Virol. 65938-944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baines, J. D., and B. Roizman. 1993. The UL10 gene of herpes simplex virus 1 encodes a novel viral glycoprotein, gM, which is present in the virion and in the plasma membrane of infected cells. J. Virol. 671441-1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baines, J. D., E. Wills, R. J. Jacob, J. Pennington, and B. Roizman. 2007. Glycoprotein M of herpes simplex virus 1 is incorporated into virions during budding at the inner nuclear membrane. J. Virol. 81800-812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brack, A. R., J. M. Dijkstra, H. Granzow, B. G. Klupp, and T. C. Mettenleiter. 1999. Inhibition of virion maturation by simultaneous deletion of glycoproteins E, I, and M of pseudorabies virus. J. Virol. 735364-5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brack, A. R., B. G. Klupp, H. Granzow, R. Tirabassi, L. W. Enquist, and T. C. Mettenleiter. 2000. Role of the cytoplasmic tail of pseudorabies virus glycoprotein E in virion formation. J. Virol. 744004-4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen, J. I., S. E. Straus, and A. M. Arvin. 2006. Varicella-zoster virus: replication, pathogenesis, and management, p. 2774-2818. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 5th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 8.Crump, C. M., B. Bruun, S. Bell, L. E. Pomeranz, T. Minson, and H. M. Browne. 2004. Alphaherpesvirus glycoprotein M causes the relocalization of plasma membrane proteins. J. Gen. Virol. 853517-3527. [DOI] [PubMed] [Google Scholar]
- 9.Davison, A. J., and N. M. Wilkie. 1983. Location and orientation of homologous sequences in the genomes of five herpesviruses. J. Gen. Virol. 641927-1942. [DOI] [PubMed] [Google Scholar]
- 10.Dijkstra, J. M., N. Visser, T. C. Mettenleiter, and B. G. Klupp. 1996. Identification and characterization of pseudorabies virus glycoprotein gM as a nonessential virion component. J. Virol. 705684-5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fuchs, W., B. G. Klupp, H. Granzow, C. Hengartner, A. Brack, A. Mundt, L. W. Enquist, and T. C. Mettenleiter. 2002. Physical interaction between envelope glycoproteins E and M of pseudorabies virus and the major tegument protein UL49. J. Virol. 768208-8217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuchs, W., and T. C. Mettenleiter. 1999. DNA sequence of the UL6 to UL20 genes of infectious laryngotracheitis virus and characterization of the UL10 gene product as a nonglycosylated and nonessential virion protein. J. Gen. Virol. 802173-2182. [DOI] [PubMed] [Google Scholar]
- 13.Fuchs, W., and T. C. Mettenleiter. 2005. The nonessential UL49.5 gene of infectious laryngotracheitis virus encodes an O-glycosylated protein which forms a complex with the non-glycosylated UL10 gene product. Virus Res. 112108-114. [DOI] [PubMed] [Google Scholar]
- 14.Gershon, A. A., D. L. Sherman, Z. Zhu, C. A. Gabel, R. T. Ambron, and M. D. Gershon. 1994. Intracellular transport of newly synthesized varicella-zoster virus: final envelopment in the trans-Golgi network. J. Virol. 686372-6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gomi, Y., H. Sunamachi, Y. Mori, K. Nagaike, M. Takahashi, and K. Yamanishi. 2002. Comparison of the complete DNA sequences of the Oka varicella vaccine and its parental virus. J. Virol. 7611447-11459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hobom, U., W. Brune, M. Messerle, G. Hahn, and U. H. Koszinowski. 2000. Fast screening procedures for random transposon libraries of cloned herpesvirus genomes: mutational analysis of human cytomegalovirus envelope glycoprotein genes. J. Virol. 747720-7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jöns, A., J. M. Dijkstra, and T. C. Mettenleiter. 1998. Glycoproteins M and N of pseudorabies virus form a disulfide-linked complex. J. Virol. 72550-557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanegae, Y., G. Lee, Y. Sato, M. Tanaka, M. Nakal, T. Sakaki, S. Sugano, and I. Saito. 1995. Efficient gene activation in mammalian cells by using recombinant adenovirus expressing site-specific Cre recombinase. Nucleic Acids Res. 233816-3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kari, B., W. Li, J. Cooper, R. Goertz, and B. Radeke. 1994. The human cytomegalovirus UL100 gene encodes the gC-II glycoproteins recognized by group 2 monoclonal antibodies. J. Gen. Virol. 753081-3086. [DOI] [PubMed] [Google Scholar]
- 20.Keller, P. M., A. J. Davison, R. S. Lowe, C. D. Bennett, and R. W. Ellis. 1986. Identification and structure of the gene encoding gpII, a major glycoprotein of varicella-zoster virus. Virology 152181-191. [DOI] [PubMed] [Google Scholar]
- 21.Klupp, B. G., R. Nixdorf, and T. C. Mettenleiter. 2000. Pseudorabies virus glycoprotein M inhibits membrane fusion. J. Virol. 746760-6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lehner, R., H. Meyer, and M. Mach. 1989. Identification and characterization of a human cytomegalovirus gene coding for a membrane protein that is conserved among human herpesviruses. J. Virol. 633792-3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li, Q., M. A. Ali, and J. I. Cohen. 2006. Insulin degrading enzyme is a cellular receptor mediating varicella-zoster virus infection and cell-to-cell spread. Cell 127305-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liang, X., B. Chow, C. Raggo, and L. A. Babiuk. 1996. Bovine herpesvirus 1 UL49.5 homolog gene encodes a novel viral envelope protein that forms a disulfide-linked complex with a second virion structural protein. J. Virol. 701448-1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mach, M., B. Kropff, P. Dal Monte, and W. Britt. 2000. Complex formation by human cytomegalovirus glycoproteins M (gpUL100) and N (gpUL73). J. Virol. 7411881-11892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.MacLean, C. A., S. Efstathiou, M. L. Elliott, F. E. Jamieson, and D. J. McGeoch. 1991. Investigation of herpes simplex virus type 1 genes encoding multiply inserted membrane proteins. J. Gen. Virol. 72897-906. [DOI] [PubMed] [Google Scholar]
- 27.MacLean, C. A., L. M. Robertson, and F. E. Jamieson. 1993. Characterization of the UL10 gene product of herpes simplex virus type 1 and investigation of its role in vivo. J. Gen. Virol. 74975-983. [DOI] [PubMed] [Google Scholar]
- 28.McLauchlan, J., and F. J. Rixon. 1992. Characterization of enveloped tegument structures (L particles) produced by alphaherpesviruses: integrity of the tegument does not depend on the presence of capsid or envelope. J. Gen. Virol. 73269-276. [DOI] [PubMed] [Google Scholar]
- 29.Mettenleiter, T. C. 2004. Budding events in herpesvirus morphogenesis. Virus Res. 106167-180. [DOI] [PubMed] [Google Scholar]
- 30.Mettenleiter, T. C. 2002. Herpesvirus assembly and egress. J. Virol. 761537-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mo, C., J. Lee, M. Sommer, C. Grose, and A. M. Arvin. 2002. The requirement of varicella zoster virus glycoprotein E (gE) for viral replication and effects of glycoprotein I on gE in melanoma cells. Virology 304176-186. [DOI] [PubMed] [Google Scholar]
- 32.Montalvo, E. A., R. T. Parmley, and C. Grose. 1985. Structural analysis of the varicella-zoster virus gp98-gp62 complex: posttranslational addition of N-linked and O-linked oligosaccharide moieties. J. Virol. 53761-770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagaike, K., Y. Mori, Y. Gomi, H. Yoshii, M. Takahashi, M. Wagner, U. Koszinowski, and K. Yamanishi. 2004. Cloning of the varicella-zoster virus genome as an infectious bacterial artificial chromosome in Escherichia coli. Vaccine 224069-4074. [DOI] [PubMed] [Google Scholar]
- 34.Narayanan, K., R. Williamson, Y. Zhang, A. F. Stewart, and P. A. Ioannou. 1999. Efficient and precise engineering of a 200 kb β-globin human/bacterial artificial chromosome in E. coli DH10B using an inducible homologous recombination system. Gene Ther. 6442-447. [DOI] [PubMed] [Google Scholar]
- 35.Niwa, H., K. Yamamura, and J. Miyazaki. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108193-199. [DOI] [PubMed] [Google Scholar]
- 36.Osterrieder, N., A. Neubauer, C. Brandmuller, B. Braun, O. R. Kaaden, and J. D. Baines. 1996. The equine herpesvirus 1 glycoprotein gp21/22a, the herpes simplex virus type 1 gM homolog, is involved in virus penetration and cell-to-cell spread of virions. J. Virol. 704110-4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pilling, A., A. J. Davison, E. A. R. Telford, and D. M. Meredith. 1994. The equine herpesvirus type 1 glycoprotein homologous to herpes simplex virus type 1 glycoprotein M is a major constituent of the virus particle. J. Gen. Virol. 75439-442. [DOI] [PubMed] [Google Scholar]
- 38.Ross, J., M. Williams, and J. I. Cohen. 1997. Disruption of the varicella-zoster virus dUTPase and the adjacent ORF9A gene results in impaired growth and reduced syncytia formation in vitro. Virology 234186-195. [DOI] [PubMed] [Google Scholar]
- 39.Rudolph, J., and N. Osterrieder. 2002. Equine herpesvirus type 1 devoid of gM and gp2 is severely impaired in virus egress but not direct cell-to-cell spread. Virology 293356-367. [DOI] [PubMed] [Google Scholar]
- 40.Sadaoka, T., H. Yoshii, T. Imazawa, K. Yamanishi, and Y. Mori. 12 September 2007. Deletion mutant of varicella-zoster virus gene open reading frame 49 shows reduced virus growth in human malignant melanoma cells, but not in human embryonic fibroblasts. J. Virol. doi: 10.1128/JVI.01183-07. (Subsequently published, J. Virol. 81:12654-12665, 2007.) [DOI] [PMC free article] [PubMed]
- 41.Seyboldt, C., H. Granzow, and N. Osterrieder. 2000. Equine herpesvirus 1 (EHV-1) glycoprotein M: effect of deletions of transmembrane domains. Virology 278477-489. [DOI] [PubMed] [Google Scholar]
- 42.Shimamura, M., M. Mach, and W. J. Britt. 2006. Human cytomegalovirus infection elicits a glycoprotein M (gM)/gN-specific virus-neutralizing antibody response. J. Virol. 804591-4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spear, P. G., and R. Longnecker. 2003. Herpesvirus entry: an update. J. Virol. 7710179-10185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Szilágyi, J. F., and J. Berriman. 1994. Herpes simplex virus L particles contain spherical membrane-enclosed inclusion vesicles. J. Gen. Virol. 751749-1753. [DOI] [PubMed] [Google Scholar]
- 45.Szilágyi, J. F., and C. Cunningham. 1991. Identification and characterization of a novel non-infectious herpes simplex virus-related particle. J. Gen. Virol. 72661-668. [DOI] [PubMed] [Google Scholar]
- 46.Tanaka, M., H. Kagawa, Y. Yamanashi, T. Sata, and Y. Kawaguchi. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J. Virol. 771382-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thomas, K. R., and M. R. Capecchi. 1987. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 51503-512. [DOI] [PubMed] [Google Scholar]
- 48.Tischer, B. K., D. Schumacher, M. Messerle, M. Wagner, and N. Osterrieder. 2002. The products of the UL10 (gM) and the UL49.5 genes of Marek's disease virus serotype 1 are essential for virus growth in cultured cells. J. Gen. Virol. 83997-1003. [DOI] [PubMed] [Google Scholar]
- 49.Varnum, S. M., D. N. Streblow, M. E. Monroe, P. Smith, K. J. Auberry, L. Pasa-Tolic, D. Wang, D. G. Camp II, K. Rodland, S. Wiley, W. Britt, T. Shenk, R. D. Smith, and J. A. Nelson. 2004. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J. Virol. 7810960-10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu, S. X., X. P. Zhu, and G. J. Letchworth. 1998. Bovine herpesvirus 1 glycoprotein M forms a disulfide-linked heterodimer with the UL49.5 protein. J. Virol. 723029-3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ziegler, C., F. T. Just, A. Lischewski, K. Elbers, and A. Neubauer. 2005. A glycoprotein M-deleted equid herpesvirus 4 is severely impaired in virus egress and cell-to-cell spread. J. Gen. Virol. 8611-21. [DOI] [PubMed] [Google Scholar]