

Abstract
The human immunodeficiency virus type 1 (HIV-1) V3 loop is critical for coreceptor binding and principally determines tropism for the CCR5 and CXCR4 coreceptors. The recent crystallographic resolution of V3 shows that its base is closely associated with the conserved coreceptor binding site on the gp120 core, whereas more distal regions protrude toward the cell surface, likely mediating interactions with coreceptor extracellular loops. However, these V3-coreceptor interactions and the structural basis for CCR5 or CXCR4 specificity are poorly understood. Using the dual-tropic virus HIV-1R3A, which uses both CCR5 and CXCR4, we sought to identify subdomains within V3 that selectively mediate R5 or X4 tropism. An extensive panel of V3 mutants was evaluated for effects on tropism and sensitivity to coreceptor antagonists. Mutations on either side of the V3 base (residues 3 to 8 and 26 to 33) ablated R5 tropism and made the resulting X4-tropic Envs more sensitive to the CXCR4 inhibitor AMD3100. When mutations were introduced within the V3 stem, only a deletion of residues 9 to 12 on the N-terminal side ablated X4 tropism. Remarkably, this R5-tropic Δ9-12 mutant was completely resistant to several small-molecule inhibitors of CCR5. Envs with mutations in the V3 crown (residues 13 to 20) remained dual tropic. Similar observations were made for a second dual-tropic isolate, HIV-189.6. These findings suggest that V3 subdomains can be identified that differentially affect R5 and X4 tropism and modulate sensitivity to CCR5 and CXCR4 inhibitors. These studies provide a novel approach for probing V3-coreceptor interactions and mechanisms by which these interactions can be inhibited.
Human immunodeficiency virus (HIV) entry requires a coordinated interaction between envelope glycoprotein (Env) trimers on the virion surface with CD4 and a chemokine receptor, typically CCR5 (2, 11, 18, 20, 22) or CXCR4 (24), on the target cell. Whereas binding of gp120 to CD4 is required for the initial conformational changes that facilitate coreceptor interactions (9, 35), binding to CCR5 or CXCR4 is required to release gp41 to interact with the cell membrane and to form the six-helix bundle that provides the energy for membrane fusion (8, 19, 60). The gp120-coreceptor interactions that are required for these events likely involve (i) the bridging sheet (a four-stranded β-sheet on the gp120 core) and the base of the V3 loop with the coreceptor N terminus and (ii) more distal regions of V3 with coreceptor extracellular loops (ECLs) (14, 15, 29, 33, 48, 49, 56).
The V3 loop is the primary determinant for R5 or X4 tropism (28). However, the mechanism and structural basis that underlie the specificity of V3-coreceptor interactions are poorly understood. During HIV infection, viruses that utilize CCR5 are characteristically transmitted (16, 38, 51, 53, 58), whereas viruses that utilize CXCR4 can evolve during the progression to disease (13, 47). The evolution of X4 tropism in vivo and in vitro has been associated with an increase in the net positive charge in V3 (26, 42, 44, 54), particularly with the acquisition of positively charged residues at amino acid positions 11, 24, and 25 (6, 17, 25, 26, 32, 46), and with the loss of the conserved 301N glycosylation site in the V3 base (45). While these features suggest direct interactions between V3 and negatively charged residues on CXCR4 (5, 7, 21, 39, 63), direct contact sites for these interactions have not been delineated (27). Moreover, the interaction of V3 with CCR5 is even less well understood, although molecular-dynamics modeling approaches have predicted interactions with both the N terminus and ECL2 (3, 4, 30, 43). Interestingly, although the R5-to-X4 coreceptor switch is well described, many CXCR4-utilizing viruses that evolve in vivo retain R5 tropism (13, 52, 55), suggesting that dual-tropic Envs retain structural features within V3 that permit both R5 and X4 to be engaged.
The recent crystallographic resolution of V3 on a CD4-bound gp120 provides new opportunities to characterize coreceptor interactions and structural determinants for virus tropism. The structure shows three regions of the V3 loop: a conserved base that is closely associated with the bridging sheet on the gp120 core, a flexible stem that extends away from the core, and a conserved β-hairpin tip (31). In the present study, we hypothesized that dual-tropic HIV type 1 (HIV-1) Envs could provide an opportunity to identify domains within V3 that differentially affect CCR5 and CXCR4 utilization. This possibility was suggested by the report of Yang et al., who noted for the dual-tropic Env HIV-189.6P that a symmetrical deletion of residues 5 to 7 and 27 to 29 within the V3 base abrogated R5 tropism but did not affect entry on CXCR4-positive (CXCR4+) cells (65). Using the dual-tropic HIV-1 clade B isolate R3A, previously described for its ability to deplete thymocytes in ex vivo organ cultures (40), we made an extensive panel of small deletion and alanine substitution mutants within V3. We report that residues 3 to 8 and 26 to 33 within the V3 base are critical for maintaining R5 tropism, whereas a deletion of residues 9 to 12 (Δ9-12) on the N-terminal side of the V3 stem selectively ablates X4 tropism. In contrast, Envs with mutations in the V3 crown and C-terminal stem remained dual tropic. We also found that the X4-tropic V3 base mutants became more sensitive to the CXCR4 antagonist AMD3100, whereas the R5-tropic Δ9-12 mutant became resistant to several CCR5 antagonists. Similar findings were observed for HIV-189.6, another dual-tropic clade B Env. These findings suggest that, for dual-tropic isolates, subdomains within V3 can be identified that differentially mediate R5 and X4 tropism, as well as sensitivity to coreceptor antagonists. This approach may be useful in developing models for V3-coreceptor interactions and understanding mechanisms of sensitivity or resistance to X4 and R5 antagonists.
MATERIALS AND METHODS
Plasmid construction.
Plasmid pHSPG-R3A has been described previously (40). All V3 mutants were made using the PCR-based Quickchange site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. Each mutant was confirmed by sequencing. To prepare NL4-3 recombinant viruses containing the mutant Envs, the envelope expression vectors were digested with EcoRI and XhoI and ligated to the similarly digested NL4-3 backbone. The ligations were transformed into Stbl2 competent cells (Invitrogen), and recombinant-clone identity was confirmed by restriction digestion and sequencing.
Cells.
The Japanese quail fibrosarcoma cell line QT6 and the human embryonic kidney cell line 293T were maintained in Dulbecco's modified Eagle medium (DMEM) (high glucose) supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, and 2 mM penicillin-streptomycin. The human astroglioma cell line U87 stably expressing CD4 and CXCR4 (U87.CD4.CXCR4) or CD4 and CCR5 (U87.CD4.CCR5) were maintained in DMEM (high glucose) supplemented with 20% FBS, 2 mM glutamine, and 2 mM penicillin-streptomycin.
Cell-cell fusion assay.
To quantify cell-cell fusion events, we used the gene reporter fusion assay previously described (23, 50). Briefly, to generate effector cells, QT6 cells in six-well plates were infected with the recombinant vaccinia virus strain VTF1.1 expressing T7 polymerase (1) at a multiplicity of infection of 10 for 1 h at 37°C and then transfected for 4 h by the standard calcium phosphate method with 2 μg of the desired env-expressing plasmid. Following transfection, the cells were incubated overnight at 32°C with rifampin at a concentration of 100 μg/ml. To generate target cells, QT6 cells in 48-well plates were transfected with the desired receptors and the T7 luciferase reporter plasmid in a total of 2 μg by the standard calcium phosphate method for 4 h and expressed overnight at 37°C. Effector cells were mixed with target cells in the presence of 100 μg/ml rifampin and 100 nM cytosine arabinoside, and cell-cell fusion was assessed 7 to 8 h later by lysing with 0.5% Triton X-100-phosphate-buffered saline. After addition of luciferase substrate (Promega), luciferase activity was quantified with a Thermo Labsystems Luminoskan Ascent luminometer. For inhibition experiments, various concentrations of coreceptor inhibitor were added to the target cells at the time of mixing with the effector cells, and fusion inhibition was measured as the percent reduction in luciferase activity. Relative fusion values less than 5% were considered nonfunctional.
Luciferase reporter viruses.
Luciferase reporter viruses were prepared by transfecting 293T cells for 6 h by the standard calcium phosphate method with a plasmid encoding the NL4-3 luciferase virus backbone (pNL-Luc-E−R−) (10, 12) and the desired env-expressing plasmid. Supernatants were harvested 48 h posttransfection and stored at −80°C. Virus concentrations were determined by an enzyme-linked immunosorbent assay for the viral p24 antigen (Coulter). Equivalent amounts of virus were used to spin infect U87.CD4.CXCR4 and U87.CD4.CCR5 cells for 1 h at 1,300 rpm. Following spin infections, cells were incubated at 37°C and lysed 72 h postinfection with 0.5% Triton X-100-phosphate-buffered saline. After the addition of luciferase substrate (Promega), luciferase activity was quantified with a Thermo Labsystems Luminoskan Ascent luminometer. For inhibition experiments, various concentrations of coreceptor inhibitor were added to the target cells 30 min prior to the addition of pseudotyped virus. Pseudotype inhibition was measured as the percent reduction in luciferase activity. Relative infection values of less than 5% were considered nonfunctional.
Viral growth curves.
Plasmids encoding recombinant NL4-3 viruses containing R3A Envs were transfected into 293T cells for 6 h by the standard calcium phosphate method. Virus was collected 48 h posttransfection and stored at −80°C. Virus concentrations were determined by an enzyme-linked immunosorbent assay for the viral p24 antigen (Coulter), and equivalent amounts of virus were added to U87.CD4.CXCR4 and U87.CD4.CCR5 cells. After 18 h at 37°C, excess virus was removed by washing the cells twice in DMEM (high glucose) supplemented with 2.5% FBS. Viral replication was monitored over 2 weeks by measuring viral reverse transcriptase (RT) activity in culture supernatants.
RESULTS
Mutations within the V3 base selectively ablate R5 tropism.
As previously noted, a deletion of residues 5 to 7 and 27 to 29 within the HIV-189.6P V3 base resulted in selective loss of R5 tropism while X4 tropism was retained (65). These findings suggested that determinants within the V3 base were critical for CCR5, but not CXCR4, usage. To determine if this finding could be extended to another dual-tropic virus and to more clearly identify the residues involved, we introduced a series of 2- and 4-amino-acid deletions and alanine substitutions individually and in combination into the V3 base of the HIV-1R3A Env (Table 1). Western blot analysis confirmed that all of these constructs were expressed and processed (data not shown). Envs were first evaluated in cell-cell fusion assays on QT6 cells (Fig. 1A). Whereas parental R3A mediated fusion on both CXCR4+ and CCR5+ cells, deletion mutations on either side of the V3 base resulted in a striking and selective ablation of CCR5-dependent fusion to levels <5% of those of R3A, while CXCR4-dependent fusion was retained at 24 to 170% of that of R3A. Similarly, the alanine substitutions 5-6AA, 28-29AA, and 5-6/28-29AA had no effect on CXCR4-mediated fusion but reduced fusion on CCR5+ cells to levels 13 to 40% of those of R3A.
TABLE 1.
Mutations in the V3 loop of R3A
| Mutation | Sequencea | |
|---|---|---|
| V3 base | ||
| R3A | CTRPGNNTRKRVTLGPGRVYYTTGQIIGDIRKAHC | |
| Δ3-4 | CT GNNTRKRVTLGPGRVYYTTGQIIGDIRKAHC | |
| Δ5-6 | CTRPNTRKRVTLGPGRVYYTTGQIIGDIRKAHC | |
| Δ5-8 | CTRPRKRVTLGPGRVYYTTGQIIGDIRKAHC | |
| Δ26-29 | CTRPGNNTRKRVTLGPGRVYYTTGQIRKAHC | |
| Δ28-29 | CTRPGNNTRKRVTLGPGRVYYTTGQIIIRKAHC | |
| Δ30-31 | CTRPGNNTRKRVTLGPGRVYYTTGQIIGDKAHC | |
| Δ32-33 | CTRPGNNTRKRVTLGPGRVYYTTGQIIGDIRHC | |
| Δ5-6/28-29 | CTRP NTRKRVTLGPGRVYYTTGQIIIRKAHC | |
| Δ5-8/26-29 | CTRP RKRVTLGPGRVYYTTGQIRKAHC | |
| 5-6AA | CTRPAANTRKRVTLGPGRVYYTTGQIIGDIRKAHC | |
| 28-29AA | CTRPGNNTRKRVTLGPGRVYYTTGQIIAAIRKAHC | |
| 5-6/28-29AA | CTRPAANTRKRVTLGPGRVYYTTGQIIAAIRKAHC | |
| V3 crown and stem | ||
| R3A | CTRPGNNTRKRVTLGPGRVYYTTGQIIGDIRKAHC | |
| ΔV3(9,9) | CTRPGNNTRK GAGIIGDIRKAHC | |
| Δ13-14 | CTRPGNNTRKRV GPGRVYYTTGQIIGDIRKAHC | |
| Δ15-18 | CTRPGNNTRKRVTL VYYTTGQIIGDIRKAHC | |
| Δ19-20 | CTRPGNNTRKRVTLGPGR YTTGQIIGDIRKAHC | |
| Δ11-12 | CTRPGNNTRK TLGPGRVYYTTGQIIGDIRKAHC | |
| Δ21-22 | CTRPGNNTRKRVTLGPGRVY TGQIIGDIRKAHC | |
| Δ11-12/21-22 | CTRPGNNTRK TLGPGRVY TGQIIGDIRKAHC | |
| Δ9-12 | CTRPGNNT TLGPGRVYYTTGQIIGDIRKAHC | |
| Δ21-24 | CTRPGNNTRKRVTLGPGRVY QIIGDIRKAHC | |
| Δ9-12/21-24 | CTRPGNNT TLGPGRVY QIIGDIRKAHC | |
| Δ24-25 | CTRPGNNTRKRVTLGPGRVYYTT IIGDIRKAHC | |
| 9-12AAAA | CTRPGNNTAAAATLGPGRVYYTTGQIIGDIRKAHC | |
| Δ9-10 | CTRPGNNT RVTLGPGRVYYTTGQIIGDIRKAHC | |
| 9-10AA | CTRPGNNTAARVTLGPGRVYYTTGQIIGDIRKAHC | |
| Δ10-11 | CTRPGNNTR VTLGPGRVYYTTGQIIGDIRKAHC | |
| 10-11AA | CTRPGNNTRAAVTLGPGRVYYTTGQIIGDIRKAHC | |
Amino acids in boldface are insertions.
FIG. 1.

Effects of mutations in the HIV-1R3A V3 base on tropism. (A) Fusion activity in a cell-cell fusion assay is shown for Envs with small deletions and alanine substitutions (Table 1). Percent fusion was calculated by using luciferase activity normalized to R3A fusion on CD4+ CXCR4+ or CD4+ CCR5+ QT6 cells. The values are means plus standard errors of the mean (SEM). The data shown are the averages of three experiments. (B) Infectivities of the indicated Envs are shown using a single-cycle luciferase reporter virus on U87.CD4.CXCR4 and U87.CD4.CCR5 cells. Percent infection was calculated by using luciferase activity normalized to R3A infection on U87.CD4.CXCR4 cells or U87.CD4.CCR5 cells. The values are means plus SEM. The data shown are the averages of three experiments. (C) Growth curves for viruses containing the Δ5-6 and Δ28-29 Envs in comparison with parental R3A are shown on U87.CD4.CXCR4 cells (left) and U87.CD4.CCR5 cells (right). RT activity in culture supernatants was measured at the indicated time points. The results from one of two independent experiments are shown.
To validate the effects of these mutations in the context of an infectious particle, we analyzed a subset of the V3 base mutants for the ability to mediate entry using a single-cycle HIV reporter virus carrying a luciferase reporter gene (Fig. 1B). Pseudovirions bearing the parental R3A Env were able to infect both U87.CD4.CXCR4 and U87.CD4.CCR5 cells. However, the V3 base deletion mutants (Δ5-6, Δ28-29, and Δ5-6/28-29) and alanine substitution mutants (5-6AA, 28-29AA, and 5-6/28-29AA) could infect U87.CD4.CXCR4 cells but not U87.CD4.CCR5 cells. Thus, in the more physiologic context of an infection assay, the V3 base alanine substitution mutants are strictly X4 tropic.
To assess the effects of V3 base mutations on replication-competent viruses, Δ5-6 and Δ28-29 Envs were introduced into an NL4-3 infectious provirus, and RT activity was monitored over time in U87.CD4.CXCR4 and U87.CD4.CCR5 target cells (Fig. 1C). Whereas both V3 base mutants replicated in U87.CD4.CXCR4 cells at levels comparable to or slightly slower than those of R3A, these mutants were unable to replicate in U87.CD4.CCR5 cells up to 12 days postinfection. Collectively, these data show that mutations on either side of the V3 base can selectively ablate R5 tropism while having minimal to no effect on X4 tropism.
Mutations in the V3 base confer increased sensitivity to AMD3100.
We next evaluated the effects of the X4-tropic V3 base mutants on sensitivity to the CXCR4-specific antagonist AMD3100. Envs containing deletions or alanine substitutions of residues 5 and 6 or 28 and 29, alone and in combination, were evaluated in both cell-cell fusion (Fig. 2A) and pseudotype infection (Fig. 2B) assays on CXCR4-expressing cells in the presence of AMD3100. Parental R3A was sensitive to AMD3100 inhibition, with a 50% inhibitory concentration (IC50) of 40 nM in both cell-cell fusion assays and pseudotype infections. Interestingly, while all V3 base mutant Envs remained sensitive to AMD3100, several exhibited increased sensitivity. In cell-cell fusion assays, deletion mutants Δ5-6, Δ28-29, and Δ5-6/28-29 and alanine substitution mutants 28-29AA and 5-6/28-29AA exhibited 1.8- to 5-fold-lower IC50s, while in pseudotype infection assays, Δ5-6 and Δ5-6/28-29 exhibited 6.2-fold and 5.3-fold-lower IC50s, respectively, although little to no effect was seen for the Δ28-29 or alanine mutants. Thus, V3 base mutations not only altered the coreceptor tropism of R3A, but also rendered some of these strictly X4-tropic Envs more sensitive to a CXCR4 inhibitor. As described in Discussion, this finding is consistent with the view that V3 base mutations may confer increased dependence of the X4-tropic Envs on CXCR4 regions targeted by AMD3100 (i.e., the ECLs).
FIG. 2.
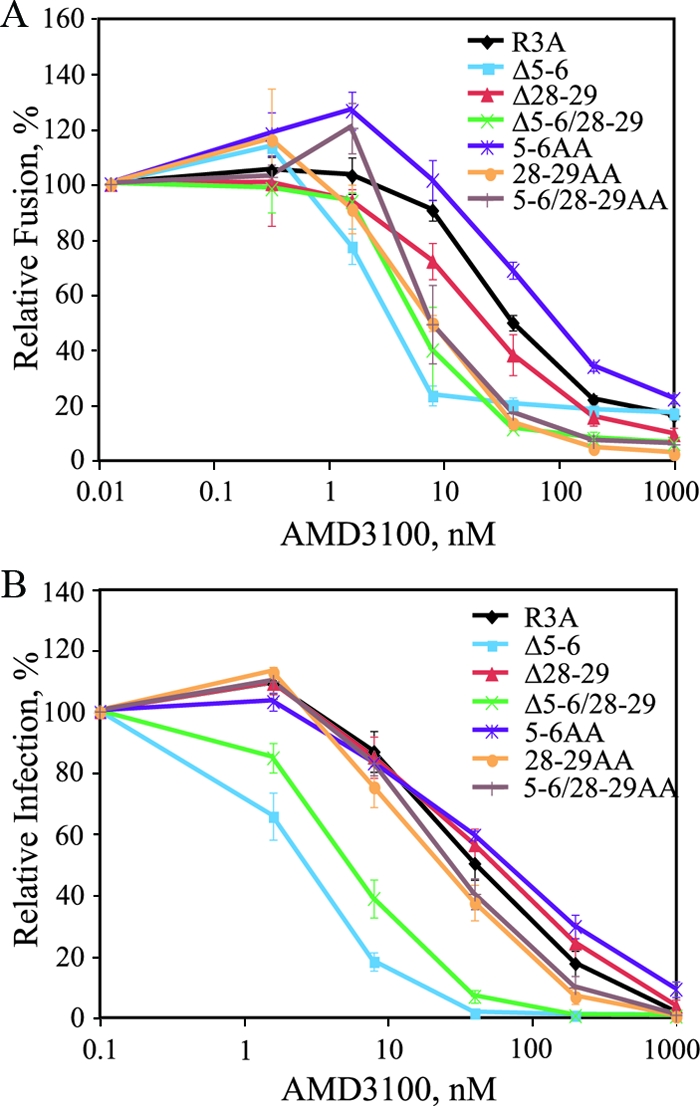
Sensitivities of V3 base mutants to AMD3100. (A) Sensitivities to AMD3100 of mutants containing two amino acid deletions or alanine substitutions in the V3 base are shown using a cell-cell fusion assay with CXCR4+ QT6 cells. Percent fusion was calculated by using luciferase activity normalized to fusion in the absence of inhibitor for each Env. The values are means ± standard errors of the mean (SEM). The data shown are the averages of three experiments. (B) Sensitivities of V3 base mutants to AMD3100 are shown using a pseudotype infection assay on U87.CD4.CXCR4 cells. Percent fusion was calculated by using luciferase activity normalized to infection in the absence of inhibitor for each virus. The values are means ± SEM. The data shown are the averages of three experiments.
A mutation within the V3 stem selectively ablates X4 tropism.
We recently reported the derivation of a replication-competent variant of R3A with 15 amino acids deleted from the V3 crown and stem (36). This ΔV3(9,9) Env contains only the first and last 9 amino acids of V3, excluding the paired cysteines, and a Gly-Ala-Gly linker introduced to bridge the N- and C-terminal V3 stems. In contrast to the V3 base mutations described above, the ΔV3(9,9) Env lost the ability to use CXCR4 but retained R5 tropism. We next sought to determine the minimal domain within the region deleted on the ΔV3(9,9) Env that could selectively ablate X4 tropism. A series of 2- and 4-amino-acid deletions and alanine substitutions in the V3 crown and stem were introduced into the R3A Env (Table 1). Western blot analysis again confirmed that all of these constructs were expressed and processed (data not shown). In cell-cell fusion assays, small deletions within the V3 crown (Δ13-14, Δ15-18, and Δ19-20), as well as within the V3 stem (Δ11-12, Δ21-22, Δ11-12/21-22, Δ21-24, and Δ24-25), remained dual tropic to various degrees, with R5-dependent fusion ranging from 13 to 140% and X4-dependent fusion ranging from 8 to 83% of that of parental R3A (Fig. 3A). The finding that the Δ11-12, Δ21-24, and Δ24-25 mutants remained dual tropic was interesting, since positively charged residues at positions 11, 24, and 25 have been implicated as determinants of X4 tropism (6, 17, 25, 26, 46). Remarkably, the Δ9-12 Env became exclusively R5 tropic, as did the Δ9-12/21-24 Env that also contained this deletion, suggesting that this region on the N-terminal side of the V3 stem plays a critical role in maintaining X4 tropism for R3A.
FIG. 3.

Effects of mutations in the V3 crown and stem on tropism. (A) Effects of 2- and 4-amino-acid deletions in the V3 crown (residues 13 to 20) and stem (residues 9 to 12 and 21 to 25) on cell-cell fusion are shown. Percent fusion was calculated using luciferase activity normalized to R3A fusion on CD4+ CXCR4+ or CD4+ CCR5+ cells. The values are means plus standard errors of the mean (SEM). The data shown are the averages of three experiments. (B) Effects of deletions and alanine substitutions within the N-terminal side of the V3 stem (residues 9 to 12) on cell-cell fusion are shown. Percent fusion was calculated by using luciferase activity normalized to R3A fusion on CD4+ CXCR4+ or CD4+ CCR5+ cells. The values are means plus SEM. The data shown are the averages of three experiments. (C) Growth curves for infectious viruses containing the parental R3A and Δ9-12 Envs on SupT1 cells (CXCR4+ CCR5−) and SupCCR5 cells (CXCR4+ CCR5+) are shown. RT activity in culture supernatants was measured at the indicated time points. The results from one of two independent experiments are shown.
V3 residues 9 to 12 comprise the minimal domain for maintaining X4 tropism.
The Δ9-12 mutation, which ablated X4 tropism in the R3A Env, removes residues Arg-Lys-Arg-Val from the N-terminal side of the V3 stem. When residues 9 to 12 were replaced by four alanines, a reduction in X4 tropism to 14% of that of parental R3A was also seen, with no effect on the level of R5-mediated fusion (Fig. 3B). Since this region contains three positively charged residues, we determined if a smaller domain necessary for maintaining X4 tropism could be more finely mapped. A deletion of the Arg-Val residues at positions 11 and 12 (i.e., Δ11-12) had been shown to reduce but not ablate X4 tropism (Fig. 3A). When deletion or alanine substitution mutants involving residues 9 and 10 or 10 and 11 (i.e., Δ9-10, 9-10AA, Δ10-11, and 10-11AA) were evaluated, all of these Envs could mediate fusion on CXCR4+ and CCR5+ cells (Fig. 3B). Although the V3 stem and crown mutants functioned poorly when pseudotyped onto an HIV reporter virus (data not shown), when the Δ9-12 Env was incorporated into an NL4-3 provirus, this virus could infect CXCR4+ SupT1 cells engineered to express high levels of CCR5 but was unable to infect SupT1 cells lacking CCR5 (Fig. 3C). These data suggest that residues 9 to 12 on the N-terminal side of the V3 stem collectively comprise a critical domain for conferring X4 tropism for R3A.
V3 mutations that ablate X4 tropism confer resistance to CCR5 inhibitors.
We reported that the R3A ΔV3(9,9) Env containing a deletion of the distal half of V3 became completely resistant to small-molecule inhibitors of CCR5 (36). Because these inhibitors bind to regions of CCR5 that are likely targeted by V3, we proposed that this resistance reflected a decreased dependence on V3 for entry. Given this finding and the observation that mutations in the V3 base yielded strictly X4-tropic viruses with increased sensitivity to AMD3100, we assessed the effects of the Δ9-12 deletion on sensitivity to several CCR5 inhibitors (Fig. 4). As expected, parental R3A was sensitive to inhibition by CMPD-167, AD101, Schering D, and Aplaviroc, with IC50s of 4 nM, 60 nM, 23 nM, and 50 pM, respectively. However, the Δ9-12 Env was completely resistant to these CCR5 antagonists at drug concentrations up to 10,000 nM. Similar results were observed for the ΔV3(9,9) Env, consistent with our reported results (36).
FIG. 4.

Sensitivities of the R5-tropic mutant Δ9-12 Env to CCR5 inhibitors. The sensitivities of R3A, ΔV3(9,9), and Δ9-12 to CMPD-167 (A), AD101 (B), Schering D (C), and Aplaviroc (D) are shown using a cell-cell fusion assay on CCR5+ cells. Percent fusion was calculated by using luciferase activity normalized to fusion in the absence of inhibitor for each Env. The values are means ± standard errors of the mean. The data shown are the averages of three experiments.
To determine if mutations elsewhere in V3 affected sensitivity to R5 antagonists, the full panel of V3 crown and stem mutants shown in Table 1 was assessed for sensitivity to AD101 (Fig. 5). With the exception of Δ9-12 and a combination deletion mutant that also contained this mutation (Δ9-12/21-24), all deletion mutants were sensitive to AD101, with IC50s similar to or less than those of R3A (Fig. 5A). Interestingly, when residues 9 to 12 were replaced with alanines, this Env remained sensitive to AD101 (Fig. 5B). Similar results were seen when smaller deletions and alanine substitutions within the 9-to-12 region were assessed (i.e., Δ9-10, Δ10-11, Δ11-12, 9-10AA, and 10-11AA). Thus, the Δ9-12 mutation, shown to be the minimum determinant for ablating X4 tropism, was also the minimum determinant for conferring complete resistance to CCR5 antagonists.
FIG. 5.
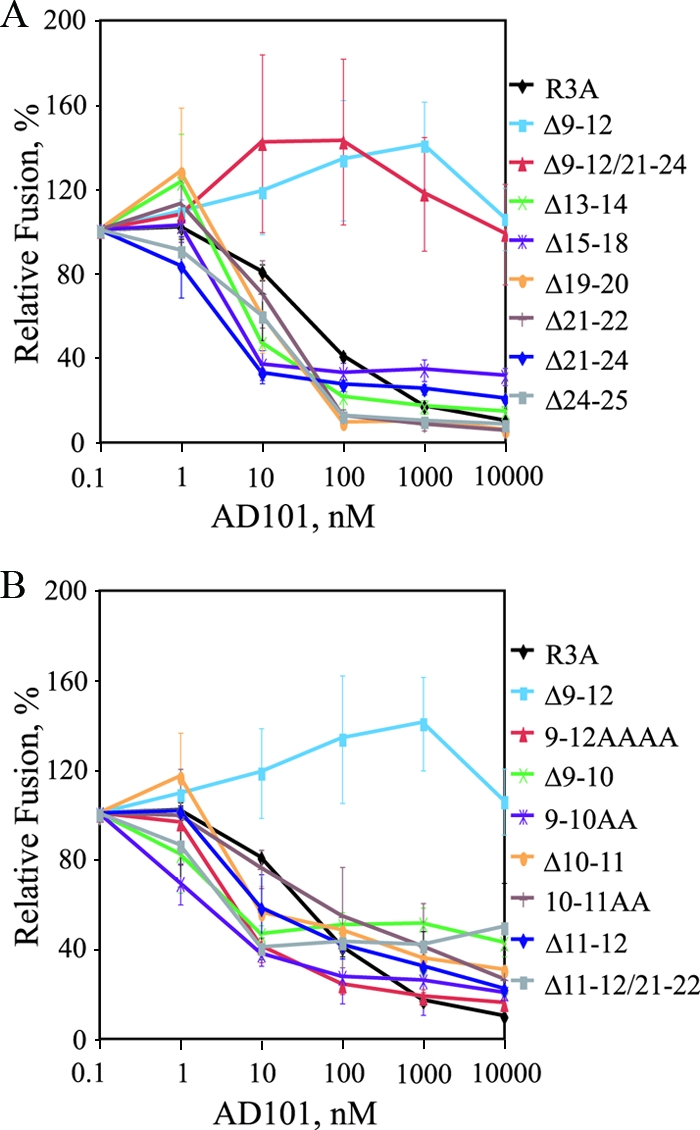
Sensitivities of HIV-1R3A V3 crown and stem mutants to AD101. (A) Sensitivities to AD101 of mutants containing 2- or 4- amino-acid deletions in the V3 crown and stem are shown using a cell-cell fusion assay with CCR5+ cells. Percent fusion was calculated by using luciferase activity normalized to fusion in the absence of inhibitor for each Env. The values are means ± standard errors of the mean (SEM). The data shown are the averages of three experiments. (B) Sensitivities to AD101 of mutants containing small deletions or alanine substitutions within V3 stem residues 9 to 12 are shown using a cell-cell fusion assay with CCR5+ cells. Percent fusion was calculated by using luciferase activity normalized to fusion in the absence of inhibitor for each Env. The values are means ± SEM. The data shown are the averages of three experiments.
V3 mutations in HIV-189.6 have similar effects on tropism and drug sensitivity.
To determine if the V3 mutations that selectively modulated R5 and X4 tropism for R3A could confer a similar phenotype on a different HIV-1 genetic background, analogous mutations were introduced into the dual-tropic Env 89.6. Similar to R3A, deletion of V3 base residues 5 and 6 or 28 and 29, ablated R5-dependent fusion to <5% of that of parental 89.6, while X4-dependent fusion was retained at 20 to 27%. Moreover, the Δ9-12 deletion reduced X4-dependent fusion to background levels, while R5-dependent fusion was retained at 18% of parental 89.6 (Fig. 6A). When the sensitivities of these Envs to CXCR4 and CCR5 inhibitors were assessed in cell-cell fusion assays, the V3 base mutations conferred increased sensitivity to AMD3100 (IC50, 105 nM for parental 89.6 compared to 20 nM for Δ5-6 and 30 nM for Δ28-29), while the Δ9-12 mutation conferred complete resistance to Schering D (IC50, 300 nM for parental 89.6 compared to >10,000 nM for Δ9-12) (Fig. 6B and C). Thus, for both R3A and 89.6, small deletions in the V3 base selectively ablated R5 tropism and conferred increased sensitivity to a CXCR4 inhibitor, while the Δ9-12 deletion in the V3 stem selectively ablated X4 tropism and conferred complete resistance to a CCR5 inhibitor.
FIG. 6.

Effects of HIV-189.6 V3 deletion mutants on coreceptor tropism and sensitivity to coreceptor inhibitors. (A) Fusion activities of Envs containing deletions in the V3 base (Δ5-6 and Δ28-29) and within the N-terminal side of the V3 stem (Δ9-12) are shown in a cell-cell fusion assay. Percent fusion was calculated by using luciferase activity normalized to 89.6 fusion on CD4+ CXCR4+ or CD4+ CCR5+ cells. The values are means plus standard errors of the mean (SEM). The data shown are the averages of three experiments. (B) Sensitivities of V3 base deletion mutants (Δ5-6 and Δ28-29) to AMD3100 are shown using a cell-cell fusion assay with CXCR4+ cells. Percent fusion was calculated by using luciferase activity normalized to fusion in the absence of inhibitor for each Env. The values are means ± SEM. The data shown are the averages of three experiments. (C) Sensitivity of the V3 stem deletion mutant Δ9-12 to Schering D is shown using a cell-cell fusion assay on CCR5+ cells. Percent fusion was calculated by using luciferase activity normalized to fusion in the absence of inhibitor for each Env. The values are means ± SEM. The data shown are the averages of three experiments.
DISCUSSION
Although the V3 loop has long been recognized as the principle determinant for coreceptor tropism, the structural basis for CCR5 or CXCR4 specificity is poorly understood. In this study, using the dual-tropic HIV-1 Env R3A, we identified distinct regions within the V3 loop that could selectively mediate R5 or X4 tropism. We found that deletions of two or four residues from either the N-terminal (Δ3-4, Δ5-6, and Δ5-8) or C-terminal (Δ26-29, Δ28-29, Δ30-31, and Δ32-33) side of the V3 base, alone or in combination, selectively ablated R5 tropism with little or only modest effects on X4 usage. Similar results were seen for HIV-189.6 when the Δ5-6 or Δ28-29 mutation was introduced. These results extend the findings of Yang et al., who reported that a deletion of 6 amino acids, 3 on each side of the V3 base of dual-tropic HIV-189.6P (residues 5 to 7 and 27 to 29), ablated R5 but not X4 tropism (65). Moreover, using a panel of alanine substitution mutants within the V3 base that preserved the 35-amino-acid length of V3, a similar selective loss of R5 tropism was observed in virus infection assays. Huang et al. noted that, for R5-tropic isolates, residues in the V3 base are more conserved than residues in the stem and crown, whereas for X4 isolates, variation in V3 is more generally distributed (31). These findings suggest that the base of V3, perhaps in association with the highly conserved bridging sheet, contributes to a structure that is indispensable for CCR5 utilization while it is either not required or less important for CXCR4 use (Fig. 7A).
FIG. 7.

Model for coreceptor utilization and sensitivity to entry inhibitors for the dual-tropic Envs R3A and 89.6. (A) Although Env interactions with both the coreceptor N terminus and the ECLs occur, the interaction of the bridging sheet and V3 base with the coreceptor N terminus is critical for R5 tropism, while the interaction between more distal regions of V3 and the coreceptor ECLs is critical for X4 tropism. (B) Deletions at either side of the V3 base (residues 3 to 8 and 26 to 33, indicated by the white boxes) ablate R5 tropism, resulting in X4-tropic Envs with an increased reliance on the V3-ECL interaction. Because AMD3100 binds within the ECLs of CXCR4 and blocks this interaction, V3 base mutations increase sensitivity to this inhibitor. (C) A deletion of residues 9 to 12 on the N-terminal side of the V3 stem (white box) directly or indirectly disrupts a region that is critical for X4 tropism, resulting in a pure R5-tropic Env. However, this mutation also perturbs the V3-ECL interaction for CCR5, resulting in an increased reliance on the bridging-sheet-V3 base-N terminus interaction. Because CCR5 inhibitors also bind within the transmembrane helices of CCR5 to block the V3-ECL interaction, the Δ9-12 mutant is resistant to these compounds.
In contrast to the R3A V3 base, the stem region, in particular residues 9 to 12, played a critical role in CXCR4 utilization. We previously reported that an R3A Env containing only the first and last 10 amino acids of V3 (i.e., a deletion of the distal 15 amino acids) became exclusively R5 tropic (36). Several studies have proposed that the more distal regions of V3, specifically the V3 crown and its β-hairpin tip, are critical for interactions with coreceptor ECLs (6, 14, 15, 29, 33). However, when we deleted 2 or 4 amino acids within the V3 crown (i.e., residues 13 to 20), these Envs remained functional and dual tropic to various degrees in cell-cell fusion assays (Fig. 3). Indeed, the Δ15-18 mutant, which removed the Gly-Pro-Gly-Arg tip, retained fusion on CCR5+ and CXCR4+ cells. In contrast, among Envs with deletions of 2 and 4 amino acids on either side of the V3 stem, only the Δ9-12 mutant completely lost function on CXCR4-expressing cells, while other stem deletion mutants remained dual tropic. Although alanine substitutions and smaller deletions within the 9-to-12 region reduced X4 tropism, a deletion of all four residues was required to fully ablate X4-mediated fusion. A similar result was seen for HIV-189.6 containing a Δ9-12 mutation, indicating that this model is not restricted to a single HIV-1 Env. This finding suggests residues 9 to 12 collectively comprise or are essential to the formation of a minimal domain for CXCR4 engagement (Fig. 7A).
The Δ9-12 mutation removes three of the seven positively charged residues within the R3A V3 loop. Given that an increase in the net positive charge of V3 has been associated with X4 tropism (26, 32, 41, 42, 54), our findings support the importance of a positively charged V3 for this phenotype. However, the clustering of these residues raises interesting issues in view of recently proposed models for CXCR4 utilization. R3A satisfies the 11/25 rule, which states that a positively charged residue at the 11th or 25th position in V3 predicts X4 tropism (17, 26, 41, 64). Although this rule accurately predicts X4 tropism for only ∼50% of Envs, Cardozo et al. have shown on a panel of well-characterized HIV-1 Envs that if a positively charged residue is present at the 11th, 24th, or 25th position, the overall predictive value increases to 94%, with a positive predictive value for X4- and R5-tropic viruses of 86% and 96%, respectively (6). These authors also utilized three-dimensional molecular modeling to propose a model in which residues 11, 24, and 25 associate to form a “patch” that when positively charged interacts with CXCR4 and when negatively charged or neutral interacts with CCR5. Notably, several of our mutants removed the Arg at position 11 (Δ11-12, Δ10-11, 10-11AA, and Δ11-12/21-22), yet these Envs retained function on CCR5+ and CXCR4+ cells (Fig. 3). Moreover, an Env with a deletion of residues 24 and 25 mediated fusion on CXCR4 and CCR5 at levels approximating those of the parental R3A, indicating that, at least in this context, these residues do not form a structure required for CXCR4 (or CCR5) interactions. Rather, our results support a model in which positively charged residues at positions 9, 10, and 11 collectively contribute to a CXCR4-interactive site, possibly with one or more negatively charged residues on the CXCR4 ECLs.
For both R3A and 89.6, a loss of R5 tropism for the V3 base mutants correlated with increased sensitivity to AMD3100, while a loss of X4 tropism for the Δ9-12 stem mutant correlated with complete resistance to several CCR5 antagonists. CXCR4 and CCR5 antagonists are thought to bind to membrane-proximal residues in close association with the ECLs. Rather then directly inhibiting gp120 binding, they have been proposed to act by an allosteric mechanism, altering the conformational repertoire of the ECLs to render drug bound receptors unable to interact with gp120 (59). Resistance of HIV-1 and HIV-2 mutants containing more extensive deletions in V3 to CCR5 and/or CXCR4 antagonists has suggested that all of these compounds act by disrupting a V3-ECL interaction (36, 37). Given evidence that the bridging sheet and V3 base collectively comprise a surface that interacts with the coreceptor N terminus, while the more distal V3 regions engage the coreceptor ECLs (22, 27, 31) (Fig. 7A), the increased sensitivity of our V3 base mutants to AMD3100 likely results from a reduction in the contribution of the CXCR4 N terminus to gp120 binding and a resulting increased dependence on the V3-ECL interaction (Fig. 7B). Conversely, the Δ9-12 stem mutation may disrupt an ECL interaction that, while essential for X4 tropism, is less important for R5 tropism. This Env becomes more reliant on the CCR5 N terminus and therefore resistant to CCR5 antagonists (Fig. 7C). Resistance to CCR5 inhibitors has been observed in vitro and in vivo (34, 57). While the genetic determinants are complex, one mechanism could be an increased dependence of these Envs on the CCR5 N terminus with a reduced requirement for the V3-ECL interaction. Alternatively, mutations in V3 that confer resistance to CCR5 antagonists could also enable V3 to bind to ECL conformations presented by drug bound receptor (61, 62). Whatever the mechanism, our findings suggest that the 9-to-12 region contributes directly or indirectly to a CCR5 interaction and plays a pivotal role in this process.
In summary, our findings for two dual-tropic HIV-1 Envs show that determinants for R5 and X4 tropism can be dissociated, with residues from the V3 base contributing principally to R5 tropism and a positively charged cluster of residues in the N-terminal V3 stem contributing to X4 tropism. This approach has revealed a novel method to dissect Env structure/function and gp120-coreceptor interactions and provides strong evidence for direct V3 interactions with coreceptors. Ongoing studies to address this model with other dual-tropic Envs are in progress.
Acknowledgments
This work was supported by grant AI-045378 (to J.A.H.) and grant T32-AI-007632 from the National Institutes of Health.
We thank James Nolan, Jr., for his help with the figures and illustrations. We thank Robert Doms, Ronald Collman, and members of the J. A. Hoxie laboratory for helpful discussions. p24 assays were performed by the Viral/Molecular Core of the Penn Center for AIDS Research. We also thank Merck for CPMD-167, Schering-Plough for AD101, and Glaxo-Smith-Kline for Aplaviroc.
Footnotes
Published ahead of print on 31 October 2007.
REFERENCES
- 1.Alexander, W. A., B. Moss, and T. R. Fuerst. 1992. Regulated expression of foreign genes in vaccinia virus under the control of bacteriophage T7 RNA polymerase and the Escherichia coli lac repressor. J. Virol. 662934-2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alkhatib, G., C. Combadiere, C. C. Broder, Y. Feng, P. E. Kennedy, P. M. Murphy, and E. A. Berger. 1996. CC CKR5: a RANTES, MIP-1α, MIP-1β receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 2721955-1958. [DOI] [PubMed] [Google Scholar]
- 3.Atchison, R. E., J. Gosling, F. S. Monteclaro, C. Franci, L. Digilio, I. F. Charo, and M. A. Goldsmith. 1996. Multiple extracellular elements of CCR5 and HIV-1 entry: dissociation from response to chemokines. Science 2741924-1926. [DOI] [PubMed] [Google Scholar]
- 4.Bieniasz, P. D., R. A. Fridell, I. Aramori, S. S. Ferguson, M. G. Caron, and B. R. Cullen. 1997. HIV-1-induced cell fusion is mediated by multiple regions within both the viral envelope and the CCR-5 co-receptor. EMBO J. 162599-2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brelot, A., N. Heveker, M. Montes, and M. Alizon. 2000. Identification of residues of CXCR4 critical for human immunodeficiency virus coreceptor and chemokine receptor activities. J. Biol. Chem. 27523736-23744. [DOI] [PubMed] [Google Scholar]
- 6.Cardozo, T., T. Kimura, S. Philpott, B. Weiser, H. Burger, and S. Zolla-Pazner. 2007. Structural basis for coreceptor selectivity by the HIV type 1 V3 loop. AIDS Res. Hum. Retrovir. 23415-426. [DOI] [PubMed] [Google Scholar]
- 7.Chabot, D. J., P. F. Zhang, G. V. Quinnan, and C. C. Broder. 1999. Mutagenesis of CXCR4 identifies important domains for human immunodeficiency virus type 1 X4 isolate envelope-mediated membrane fusion and virus entry and reveals cryptic coreceptor activity for R5 isolates. J. Virol. 736598-6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan, D. C., D. Fass, J. M. Berger, and P. S. Kim. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89263-273. [DOI] [PubMed] [Google Scholar]
- 9.Chen, B., E. M. Vogan, H. Gong, J. J. Skehel, D. C. Wiley, and S. C. Harrison. 2005. Structure of an unliganded simian immunodeficiency virus gp120 core. Nature 433834-841. [DOI] [PubMed] [Google Scholar]
- 10.Chen, B. K., K. Saksela, R. Andino, and D. Baltimore. 1994. Distinct modes of human immunodeficiency virus type 1 proviral latency revealed by superinfection of nonproductively infected cell lines with recombinant luciferase-encoding viruses. J. Virol. 68654-660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choe, H., M. Farzan, Y. Sun, N. Sullivan, B. Rollins, P. D. Ponath, L. Wu, C. R. Mackay, G. LaRosa, W. Newman, N. Gerard, C. Gerard, and J. Sodroski. 1996. The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 851135-1148. [DOI] [PubMed] [Google Scholar]
- 12.Connor, R. I., B. K. Chen, S. Choe, and N. R. Landau. 1995. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 206935-944. [DOI] [PubMed] [Google Scholar]
- 13.Connor, R. I., K. E. Sheridan, D. Ceradini, S. Choe, and N. R. Landau. 1997. Change in coreceptor use correlates with disease progression in HIV-1-infected individuals. J. Exp. Med. 185621-628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cormier, E. G., and T. Dragic. 2002. The crown and stem of the V3 loop play distinct roles in human immunodeficiency virus type 1 envelope glycoprotein interactions with the CCR5 coreceptor. J. Virol. 768953-8957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cormier, E. G., D. N. Tran, L. Yukhayeva, W. C. Olson, and T. Dragic. 2001. Mapping the determinants of the CCR5 amino-terminal sulfopeptide interaction with soluble human immunodeficiency virus type 1 gp120-CD4 complexes. J. Virol. 755541-5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dean, M., M. Carrington, C. Winkler, G. A. Huttley, M. W. Smith, R. Allikmets, J. J. Goedert, S. P. Buchbinder, E. Vittinghoff, E. Gomperts, S. J. Donfield, D. Vlahov, R. Kaslow, A. Saah, C. Rinaldo, R. Detels, and S. O'Brien. 1996. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Science 2731856-1862. [DOI] [PubMed] [Google Scholar]
- 17.De Jong, J. J., A. De Ronde, W. Keulen, M. Tersmette, and J. Goudsmit. 1992. Minimal requirements for the human immunodeficiency virus type 1 V3 domain to support the syncytium-inducing phenotype: analysis by single amino acid substitution. J. Virol. 666777-6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deng, H., R. Liu, W. Ellmeier, S. Choe, D. Unutmaz, M. Burkhart, P. Di Marzio, S. Marmon, R. E. Sutton, C. M. Hill, C. B. Davis, S. C. Peiper, T. J. Schall, D. R. Littman, and N. R. Landau. 1996. Identification of a major co-receptor for primary isolates of HIV-1. Nature 381661-666. [DOI] [PubMed] [Google Scholar]
- 19.Doms, R. W., and J. P. Moore. 2000. HIV-1 membrane fusion: targets of opportunity. J. Cell Biol. 151F9-F14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doranz, B. J., J. Rucker, Y. Yi, R. J. Smyth, M. Samson, S. C. Peiper, M. Parmentier, R. G. Collman, and R. W. Doms. 1996. A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell 851149-1158. [DOI] [PubMed] [Google Scholar]
- 21.Doranz, B. J., M. J. Orsini, J. D. Turner, T. L. Hoffman, J. F. Berson, J. A. Hoxie, S. C. Peiper, L. F. Brass, and R. W. Doms. 1999. Identification of CXCR4 domains that support coreceptor and chemokine receptor functions. J. Virol. 732752-2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dragic, T., V. Litwin, G. P. Allaway, S. R. Martin, Y. Huang, K. A. Nagashima, C. Cayanan, P. J. Maddon, R. A. Koup, J. P. Moore, and W. A. Paxton. 1996. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 381667-673. [DOI] [PubMed] [Google Scholar]
- 23.Edinger, A. L., and R. W. Doms. 1999. A cell-cell fusion assay to monitor HIV-1 Env interactions with chemokine receptors. Methods Mol. Med. 1741-49. [DOI] [PubMed] [Google Scholar]
- 24.Feng, Y., C. C. Broder, P. E. Kennedy, and E. A. Berger. 1996. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272872-877. [DOI] [PubMed] [Google Scholar]
- 25.Fouchier, R. A., M. Brouwer, S. M. Broersen, and H. Schuitemaker. 1995. Simple determination of human immunodeficiency virus type 1 syncytium-inducing V3 genotype by PCR. J. Clin. Microbiol. 33906-911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fouchier, R. A., M. Groenink, N. A. Kootstra, M. Tersmette, H. G. Huisman, F. Miedema, and H. Schuitemaker. 1992. Phenotype-associated sequence variation in the third variable domain of the human immunodeficiency virus type 1 gp120 molecule. J. Virol. 663183-3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hartley, O., P. J. Klasse, Q. J. Sattentau, and J. P. Moore. 2005. V3: HIV's switch-hitter. AIDS Res. Hum. Retrovir. 21171-189. [DOI] [PubMed] [Google Scholar]
- 28.Hoffman, T. L., and R. W. Doms. 1999. HIV-1 envelope determinants for cell tropism and chemokine receptor use. Mol. Membr. Biol. 1657-65. [DOI] [PubMed] [Google Scholar]
- 29.Hu, Q., J. O. Trent, G. D. Tomaras, Z. Wang, J. L. Murray, S. M. Conolly, J. M. Navenot, A. P. Barry, M. L. Greenberg, and S. C. Peiper. 2000. Identification of ENV determinants in V3 that influence the molecular anatomy of CCR5 utilization. J. Mol. Biol. 302359-375. [DOI] [PubMed] [Google Scholar]
- 30.Hu, Q., K. B. Napier, J. O. Trent, Z. Wang, S. Taylor, G. E. Griffin, S. C. Peiper, and R. J. Shattock. 2005. Restricted variable residues in the C-terminal segment of HIV-1 V3 loop regulate the molecular anatomy of CCR5 utilization. J. Mol. Biol. 350699-712. [DOI] [PubMed] [Google Scholar]
- 31.Huang, C. C., M. Tang, M. Y. Zhang, S. Majeed, E. Montabana, R. L. Stanfield, D. S. Dimitrov, B. Korber, J. Sodroski, I. A. Wilson, R. Wyatt, and P. D. Kwong. 2005. Structure of a V3-containing HIV-1 gp120 core. Science 3101025-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jensen, M. A., F. S. Li, A. B. van't Wout, D. C. Nickle, D. Shriner, H. X. He, S. McLaughlin, R. Shankarappa, J. B. Margolick, and J. I. Mullins. 2003. Improved coreceptor usage prediction and genotypic monitoring of R5-to-X4 transition by motif analysis of human immunodeficiency virus type 1 env V3 loop sequences. J. Virol. 7713376-13388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kajumo, F., D. A. Thompson, Y. Guo, and T. Dragic. 2000. Entry of R5X4 and X4 human immunodeficiency virus type 1 strains is mediated by negatively charged and tyrosine residues in the amino-terminal domain and the second extracellular loop of CXCR4. Virology 271240-247. [DOI] [PubMed] [Google Scholar]
- 34.Kuhmann, S. E., P. Pugach, K. J. Kunstman, J. Taylor, R. L. Stanfield, A. Snyder, J. M. Strizki, J. Riley, B. M. Baroudy, I. A. Wilson, B. T. Korber, S. M. Wolinsky, and J. P. Moore. 2004. Genetic and phenotypic analyses of human immunodeficiency virus type 1 escape from a small-molecule CCR5 inhibitor. J. Virol. 782790-2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kwong, P. D., R. Wyatt, J. Robinson, R. W. Sweet, J. Sodroski, and W. A. Hendrickson. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393648-659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laakso, M. M., F. H. Lee, B. Haggarty, C. Agrawal, K. M. Nolan, M. J. Biscone, J. Romano, A. P. Jordan, G. L. Leslie, E. G. Meissner, L. Su, J. A. Hoxie, and R. W. Doms. 2007. V3 loop truncations in HIV-1 envelope impart resistance to coreceptor inhibitors and enhanced sensitivity to neutralizing antibodies. PLoS Pathog. 3e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin, G., A. Bertolotti-Ciarlet, B. Haggarty, J. Romano, K. M. Nolan, G. L. Leslie, A. P. Jordan, C. C. Huang, P. D. Kwong, R. W. Doms, and J. A. Hoxie. 2007. Replication-competent variants of human immunodeficiency virus type 2 lacking the V3 loop exhibit resistance to chemokine receptor antgonists. J. Virol. 819956-9966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu, R., W. A. Paxton, S. Choe, D. Ceradini, S. R. Martin, R. Horuk, M. E. MacDonald, H. Stuhlmann, R. A. Koup, and N. R. Landau. 1996. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 86367-377. [DOI] [PubMed] [Google Scholar]
- 39.Lu, Z., J. F. Berson, Y. Chen, J. D. Turner, T. Zhang, M. Sharron, M. H. Jenks, Z. Wang, J. Kim, J. Rucker, J. A. Hoxie, S. C. Peiper, and R. W. Doms. 1997. Evolution of HIV-1 coreceptor usage through interactions with distinct CCR5 and CXCR4 domains. Proc. Natl. Acad. Sci. USA 946426-6431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meissner, E. G., K. M. Duus, F. Gao, X. F. Yu, and L. Su. 2004. Characterization of a thymus-tropic HIV-1 isolate from a rapid progressor: role of the envelope. Virology 32874-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Milich, L., B. Margolin, and R. Swanstrom. 1993. V3 loop of the human immunodeficiency virus type 1 Env protein: interpreting sequence variability. J. Virol. 675623-5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moulard, M., H. Lortat-Jacob, I. Mondor, G. Roca, R. Wyatt, J. Sodroski, L. Zhao, W. Olson, P. D. Kwong, and Q. J. Sattentau. 2000. Selective interactions of polyanions with basic surfaces on human immunodeficiency virus type 1 gp120. J. Virol. 741948-1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Napier, K. B., Z. X. Wang, S. C. Peiper, and J. O. Trent. 2007. CCR5 interactions with the variable 3 loop of gp120. J. Mol. Model 1329-41. [DOI] [PubMed] [Google Scholar]
- 44.Pastore, C., R. Nedellec, A. Ramos, S. Pontow, L. Ratner, and D. E. Mosier. 2006. Human immunodeficiency virus type 1 coreceptor switching: V1/V2 gain-of-fitness mutations compensate for V3 loss-of-fitness mutations. J. Virol. 80750-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pollakis, G., S. Kang, A. Kliphuis, M. I. Chalaby, J. Goudsmit, and W. A. Paxton. 2001. N-linked glycosylation of the HIV type-1 gp120 envelope glycoprotein as a major determinant of CCR5 and CXCR4 coreceptor utilization. J. Biol. Chem. 27613433-13441. [DOI] [PubMed] [Google Scholar]
- 46.Resch, W., N. Hoffman, and R. Swanstrom. 2001. Improved success of phenotype prediction of the human immunodeficiency virus type 1 from envelope variable loop 3 sequence using neural networks. Virology 28851-62. [DOI] [PubMed] [Google Scholar]
- 47.Richman, D. D., and S. A. Bozzette. 1994. The impact of the syncytium-inducing phenotype of human immunodeficiency virus on disease progression. J. Infect. Dis. 169968-974. [DOI] [PubMed] [Google Scholar]
- 48.Rizzuto, C., and J. Sodroski. 2000. Fine definition of a conserved CCR5-binding region on the human immunodeficiency virus type 1 glycoprotein 120. AIDS Res. Hum. Retrovir. 16741-749. [DOI] [PubMed] [Google Scholar]
- 49.Rizzuto, C. D., R. Wyatt, N. Hernandez-Ramos, Y. Sun, P. D. Kwong, W. A. Hendrickson, and J. Sodroski. 1998. A conserved HIV gp120 glycoprotein structure involved in chemokine receptor binding. Science 2801949-1953. [DOI] [PubMed] [Google Scholar]
- 50.Rucker, J., B. J. Doranz, A. L. Edinger, D. Long, J. F. Berson, and R. W. Doms. 1997. Cell-cell fusion assay to study role of chemokine receptors in human immunodeficiency virus type 1 entry. Methods Enzymol. 288118-133. [DOI] [PubMed] [Google Scholar]
- 51.Samson, M., F. Libert, B. J. Doranz, J. Rucker, C. Liesnard, C. M. Farber, S. Saragosti, C. Lapoumeroulie, J. Cognaux, C. Forceille, G. Muyldermans, C. Verhofstede, G. Burtonboy, M. Georges, T. Imai, S. Rana, Y. Yi, R. J. Smyth, R. G. Collman, R. W. Doms, G. Vassart, and M. Parmentier. 1996. Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 382722-725. [DOI] [PubMed] [Google Scholar]
- 52.Scarlatti, G., E. Tresoldi, A. Bjorndal, R. Fredriksson, C. Colognesi, H. K. Deng, M. S. Malnati, A. Plebani, A. G. Siccardi, D. R. Littman, E. M. Fenyo, and P. Lusso. 1997. In vivo evolution of HIV-1 co-receptor usage and sensitivity to chemokine-mediated suppression. Nat. Med. 31259-1265. [DOI] [PubMed] [Google Scholar]
- 53.Schuitemaker, H., M. Koot, N. A. Kootstra, M. W. Dercksen, R. E. de Goede, R. P. van Steenwijk, J. M. Lange, J. K. Schattenkerk, F. Miedema, and M. Tersmette. 1992. Biological phenotype of human immunodeficiency virus type 1 clones at different stages of infection: progression of disease is associated with a shift from monocytotropic to T-cell-tropic virus population. J. Virol. 661354-1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shioda, T., S. Oka, S. Ida, K. Nokihara, H. Toriyoshi, S. Mori, Y. Takebe, S. Kimura, K. Shimada, and Y. Nagai. 1994. A naturally occurring single basic amino acid substitution in the V3 region of the human immunodeficiency virus type 1 env protein alters the cellular host range and antigenic structure of the virus. J. Virol. 687689-7696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Simmons, G., D. Wilkinson, J. D. Reeves, M. T. Dittmar, S. Beddows, J. Weber, G. Carnegie, U. Desselberger, P. W. Gray, R. A. Weiss, and P. R. Clapham. 1996. Primary, syncytium-inducing human immunodeficiency virus type 1 isolates are dual-tropic and most can use either Lestr or CCR5 as coreceptors for virus entry. J. Virol. 708355-8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suphaphiphat, P., A. Thitithanyanont, S. Paca-Uccaralertkun, M. Essex, and T. H. Lee. 2003. Effect of amino acid substitution of the V3 and bridging sheet residues in human immunodeficiency virus type 1 subtype C gp120 on CCR5 utilization. J. Virol. 773832-3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsamis, F., S. Gavrilov, F. Kajumo, C. Seibert, S. Kuhmann, T. Ketas, A. Trkola, A. Palani, J. W. Clader, J. R. Tagat, S. McCombie, B. Baroudy, J. P. Moore, T. P. Sakmar, and T. Dragic. 2003. Analysis of the mechanism by which the small-molecule CCR5 antagonists SCH-351125 and SCH-350581 inhibit human immunodeficiency virus type 1 entry. J. Virol. 775201-5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van't Wout, A. B., N. A. Kootstra, G. A. Mulder-Kampinga, N. Albrecht-van Lent, H. J. Scherpbier, J. Veenstra, K. Boer, R. A. Coutinho, F. Miedema, and H. Schuitemaker. 1994. Macrophage-tropic variants initiate human immunodeficiency virus type 1 infection after sexual, parenteral, and vertical transmission. J. Clin. Investig. 942060-2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Watson, C., S. Jenkinson, W. Kazmierski, and T. Kenakin. 2005. The CCR5 receptor-based mechanism of action of 873140, a potent allosteric noncompetitive HIV entry inhibitor. Mol. Pharmacol. 671268-1282. [DOI] [PubMed] [Google Scholar]
- 60.Weissenhorn, W., A. Dessen, S. C. Harrison, J. J. Skehel, and D. C. Wiley. 1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature 387426-430. [DOI] [PubMed] [Google Scholar]
- 61.Westby, M., M. Lewis, J. Whitcomb, M. Youle, A. L. Pozniak, I. T. James, T. M. Jenkins, M. Perros, and E. van der Ryst. 2006. Emergence of CXCR4-using human immunodeficiency virus type 1 (HIV-1) variants in a minority of HIV-1-infected patients following treatment with the CCR5 antagonist maraviroc is from a pretreatment CXCR4-using virus reservoir. J. Virol. 804909-4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Westby, M., C. Smith-Burchnell, J. Mori, M. Lewis, M. Mosley, M. Stockdale, P. Dorr, G. Ciaramella, and M. Perros. 2007. Reduced maximal inhibition in phenotypic susceptibility assays indicates that viral strains resistant to the CCR5 antagonist maraviroc utilize inhibitor-bound receptor for entry. J. Virol. 812359-2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Willett, B. J., K. Adema, N. Heveker, A. Brelot, L. Picard, M. Alizon, J. D. Turner, J. A. Hoxie, S. Peiper, J. C. Neil, and M. J. Hosie. 1998. The second extracellular loop of CXCR4 determines its function as a receptor for feline immunodeficiency virus J. Virol. 726475-6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xiao, L., S. M. Owen, I. Goldman, A. A. Lal, J. J. deJong, J. Goudsmit, and R. B. Lal. 1998. CCR5 coreceptor usage of non-syncytium-inducing primary HIV-1 is independent of phylogenetically distinct global HIV-1 isolates: delineation of consensus motif in the V3 domain that predicts CCR-5 usage. Virology 24083-92. [DOI] [PubMed] [Google Scholar]
- 65.Yang, Z. Y., B. K. Chakrabarti, L. Xu, B. Welcher, W. P. Kong, K. Leung, A. Panet, J. R. Mascola, and G. J. Nabel. 2004. Selective modification of variable loops alters tropism and enhances immunogenicity of human immunodeficiency virus type 1 envelope. J. Virol. 784029-4036. [DOI] [PMC free article] [PubMed] [Google Scholar]