

The first structure of a microbial aspartokinase reveals details of its quaternary structure and the mode of substrate binding and provides insights into the catalytic mechanism.
Keywords: aspartokinase, amino-acid biosynthesis, enzyme catalysis, substrate binding, quaternary structure, archaea
Abstract
The activation of the β-carboxyl group of aspartate catalyzed by aspartokinase is the commitment step to amino-acid biosynthesis in the aspartate pathway. The first structure of a microbial aspartokinase, that from Methanococcus jannaschii, has been determined in the presence of the amino-acid substrate l-aspartic acid and the nucleotide product MgADP. The enzyme assembles into a dimer of dimers, with the interfaces mediated by both the N- and C-terminal domains. The active-site functional groups responsible for substrate binding and specificity have been identified and roles have been proposed for putative catalytic functional groups.
1. Introduction
The aspartokinase-catalyzed activation of the β-carboxyl group of aspartic acid is the commitment step in the pathway that ultimately leads to the synthesis of the amino acids lysine, methionine, threonine and isoleucine (Cohen, 1983 ▶; Viola, 2001 ▶). The aspartate pathway also produces several important metabolites, including diaminopimelic acid, a necessary component in bacterial cell-wall synthesis (Van Heijenoort, 2001 ▶), dipicolinic acid, important for sporulation in Gram-positive bacteria (Paidhungat et al., 2000 ▶), and the high-potential methyl-group donor S-adenosylmethionine (AdoMet; Roberts & Selker, 1995 ▶). The aspartate biosynthetic pathway is absent in mammals, but is present and essential in both plants and microorganisms, raising interest in the design of selective inhibitors of these enzymes as potential herbicides or antibiotics.
Gram-negative bacteria typically have three isofunctional aspartokinases (AK): two are bifunctional enzymes which also possess homoserine dehydrogenase activity, while the third AK is monofunctional. Most of the bifunctional AKs are composed of more than 800 amino acids, while the monofunctional enzymes lacking the dehydrogenase domain are smaller, with about 500 amino acids. The biochemistry of AKs in bacteria and plants is quite complex. The AKs from Escherichia coli include the bifunctional AK-HDH I which assembles into a 360 kDa tetrameric complex (Veron et al., 1985 ▶), while the second bifunctional enzyme (AK-HDH II) is found as a dimer in solution (Dautry-Varsat et al., 1977 ▶). The monofunctional AK III from E. coli assembles into a smaller dimer of only 96 kDa (Richaud et al., 1973 ▶). Additional complexity is evident in the regulation of the AKs, which are mediated by the end products of the pathway. Both activities of E. coli AK-HDH I and the activity of AK III are feedback-inhibited by threonine and lysine, respectively (Stadtman et al., 1961 ▶). AK-HDH II is not allosterically controlled, but is instead regulated at the genetic level by methionine, as are AK-HDH I by threonine and AK III by increased lysine levels (Cohen, 1983 ▶).
The first structure of an aspartokinase has very recently been determined for a lysine- and AdoMet-sensitive monofunctional isozyme from Arabidopsis thaliana (atAK) in a complex with both of its allosteric effectors (Mas-Droux et al., 2006 ▶). Native gel electrophoresis of atAK shows the enzyme to be a dimer in solution. A detailed analysis of the bound allosteric inhibitors, lysine and AdoMet, has revealed structural details of the feedback-inhibition mechanism (Mas-Droux et al., 2006 ▶). In this paper, we report the first structural study of an aspartokinase–substrate complex from the thermophilic archaeon Methanococcus jannaschii.
2. Experimental procedures
2.1. Preparation and purification of AK
The cDNA clone containing the open reading frame of the M. jannaschii gene encoding aspartokinase was obtained from ATCC (clone No. 624490). The gene was amplified from the template DNA using the polymerase chain reaction (PCR) method with primers that were designed to incorporate NdeI and BamHI restriction sites at the ends of the PCR product. Ligation of the digested PCR product into the pET-41a expression vector (NdeI/BamHI-digested) was performed with the Clonables ligation kit (Novagen). Positive inserts were verified by restriction-digest mapping and then by DNA sequencing at ACGT Inc. Protein expression in Rosetta (DE3) E. coli cells was induced during the growth cycle by the addition of 1 mM IPTG to the media. The cells were harvested by centrifugation after 6 h of growth at 303 K.
The cell paste was suspended in 50 mM Tris buffer pH 8.0, 50 mM KCl and 1 mM β-mercaptoethanol by ultrasonication. The nucleic acid component of the crude extract was removed by stirring with a 1% streptomycin sulfate solution. After centrifugation, the precipitated nucleic acids were removed and the soluble crude portion was placed in a water bath maintained at 358 K for 25 min. A significant portion of the soluble E. coli proteins denatured under these conditions and were discarded. The remaining soluble portion containing the heat-stable aspartokinase was subjected to successive anion-exchange chromatography: first on a high-capacity resin (Q Sepharose XL, Amersham Biosciences), followed by a high-resolution resin (Source 15 Q, Amersham Biosciences) for optimal separation. The protein was judged to be >99% pure at this point, as there were no other protein contaminants visualized by SDS–PAGE. The activity of aspartokinase was measured with a coupled enzyme assay using a heat-stable aspartate-β-semialdehyde dehydrogenase (M. jannaschii ASADH; Faehnle et al., 2005 ▶) in 100 mM HEPES pH 8.0 and 100 mM KCl at temperatures between 303 and 348 K. Feedback-inhibition studies of mjAK were carried out by including the amino-acid end products lysine, threonine, isoleucine and methionine in the reaction assay at varying concentrations up to 10 mM.
2.2. Crystallization of the enzyme–substrate complex
mjAK was dialyzed into 25 mM Tris pH 8.0, 100 mM KCl, 2 mM DTT and then concentrated to 30 mg ml−1. The ternary complex was formed by incubating mjAK (14 mg ml−1) in 5 mM MgCl2, 5 mM ADP and 30 mM l-aspartate for 1 h prior to crystallization experiments. The mjAK complex was screened against the Clear Strategy Screen (Brzozowski & Walton, 2001 ▶) with 100 mM buffer additives ranging from pH 5 to pH 9 in 0.5 pH-unit intervals using the hanging-drop vapor-diffusion method; 2 µl enzyme solution was added to 2 µl well solution and was suspended over well volumes of 600 µl. Preliminary crystals were identified in several conditions containing 800 mM sodium formate; single plate-shaped crystals were grown in Tris buffer pH 8.0 with 15% PEG 4000 as the precipitant. Diffraction-quality crystals were obtained through further optimization and reproducible crystals were obtained from 100 mM Tris buffer pH 8.5, 800 mM ammonium formate and 12–14% PEG 3350 at a final pH of 8.2. Cryoprotection of these crystals was achieved with 2 µl of an artificial solution composed of the well components with an additional 30% MPD added to a 1 µl aliquot of the mother liquor; crystals were transferred to this solution for 1 min prior to flash-cooling in liquid nitrogen.
2.3. Data collection and processing
A complete X-ray diffraction data set was collected for the native recombinant enzyme as a substrate–product complex (l-aspartate/MgADP) at 100 K at the Advanced Photon Source at Argonne National Laboratory on the BioCars-CAT beamline 14ID-B. Autoindexing of the initial diffraction images and the generation of a data-collection strategy were performed with HKL-2000 (Otwinowski & Minor, 1997 ▶), followed by integration and scaling of the diffraction data. The reduced data were 98.0% complete overall and 77.9% complete in the highest resolution shell (2.82–2.69 Å), with an R merge of 0.046 overall and 0.205 in the highest resolution shell. Data-collection statistics are given in Table 1 ▶.
Table 1. Data-collection statistics for M. jannaschii aspartokinase.
Values in parentheses are for the highest resolution shell (2.82–2.69 Å).
| Wavelength (Å) | 1.00 |
| Space group | P212121 |
| Molecules per ASU | 4 |
| Unit-cell parameters (Å) | a = 101.7, b = 104.5, c = 192.9 |
| Resolution limits (Å) | 30–2.7 |
| No. of observations | |
| Measured | 312893 |
| Unique | 54182 |
| Completeness (%) | 94.0 (77.9) |
| Rsym† (%) | 10.7 (24.8) |
| 〈I/σ(I)〉 | 24 (4.5) |
R
sym = 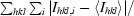
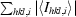 , where I
hkl,i is the intensity of an individual measurement of the reflection with Miller indices h, k and l and 〈Ihkl〉 is the mean intensity of that reflection.
, where I
hkl,i is the intensity of an individual measurement of the reflection with Miller indices h, k and l and 〈Ihkl〉 is the mean intensity of that reflection.
2.4. Structure solution and refinement
Maximum-likelihood molecular replacement (MR) as implemented in the program Phaser (McCoy et al., 2005 ▶) was used to carry out rotation and translation functions using all data between 30 and 2.7 Å resolution. The search model for MR was a homology model of mjAK constructed with the software CHAINSAW from the CCP4 program suite (Collaborative Computational Project, Number 4, 1994 ▶), where a sequence alignment was used as a guide to truncate non-conserved residues in the AK structure from A. thaliana (PDB code 2cdq; ∼30% sequence identity to mjAK). Four protein molecules were present in the asymmetric unit as calculated from the Matthews coefficient (2.6 Å3 Da−1; Matthews, 1968 ▶). An MR search with a single subunit of the homology model failed to yield a solution; however, on performing a separate search with a truncated N-terminal kinase domain (residues 25–323), a clear solution was obtained with a Z score of 13. The kinase domain was then held in a fixed position while a second search was conducted with a truncated portion of the C-terminal ACT regulatory domain (residues 336–481). Again, a clear solution was found with a final Z score of 17.
Following rigid-body and restrained refinement of mjAK with REFMAC5, the initial R factor was 37% with an R free of 45%. However, the electron density for much of this structure was clearly biased by the molecular-replacement model used for phasing. The electron density calculated from this data was significantly improved by noncrystallographic symmetry averaging and density modification as implemented in the program NCSref from CCP4. Manual model building into the NCS-averaged electron-density map using the Coot software package (Emsley & Cowtan, 2004 ▶) allowed a complete model of mjAK to be built with the exception of the N-terminal methionine, residues 384–388 and the C-terminal residues 471–473 owing to lack of electron density in these regions. Refinement statistics are given in Table 2 ▶.
Table 2. Refinement statistics for the aspartokinase complex.
| Refinement | |
| Resolution range (Å) | 30–2.7 |
| Rcryst (%) | 0.241 |
| Rfree (%) | 0.276 |
| No. of protein atoms | 14160 |
| No. of nonprotein atoms | 148 |
| No. of water molecules | 123 |
| Stereochemistry | |
| R.m.s.d. for bond lengths (Å) | 0.009 |
| R.m.s.d. for bond angles (°) | 1.22 |
| Average Wilson B (Å2) | 53 |
| Residues in the Ramachandran plot | |
| Most favored region (%) | 91.2 |
| Additional allowed regions (%) | 8.3 |
| Disallowed region (glycine) (%) | 0.5 |
2.5. Gel filtration and dynamic light scattering
Gel-filtration studies were conducted on a Superdex 200 size-exclusion column (26 mm diameter, 250 ml bed volume). The AK enzymes (1.6 mg ecAKIII and 1.0 mg mjAK) were mixed in 25 mM Tris buffer pH 8.0 with 25 mM KCl and 1 mM DTT, loaded onto the column and eluted with the same buffer. DLS studies of mjAK and ecAKIII were carried out on a DynaPro Titan DLS instrument (Wyatt Technologies) at 1 mg ml−1 of each protein in 50 mM Tris pH 8.0 with 50 mM KCl and were also conducted using the crystallization conditions (50 mM Tris pH 8.0 with 800 mM ammonium formate, 2 mM MgCl2, 2 mM ADP and 5 mM l-aspartate). Subunit dissociation studies on mjAK were conducted in the same buffer in the presence of increasing levels of guanidine–HCl.
3. Results and discussion
3.1. Subunit structure and oligomeric state of mjAK
The structure of mjAK contains four subunits in the asymmetric unit organized into a dimer of dimers (Fig. 1 ▶ a). The interface within each dimer is quite extensive amd is comprised of about 2800 Å2 of buried surface area, 14% of the overall subunit surface. In contrast, the interface between the dimers is much smaller (Fig. 1 ▶ a), only consisting of about 500 Å2 or 3% of the overall surface area. These differences in subunit contacts suggest that dissociation into a dimer may yield the functional catalytic unit of aspartokinase in solution.
Figure 1.

Overall structure of M. jannaschii aspartokinase (mjAK) determined in the presence of the amino-acid substrate l-aspartic acid and the nucleotide product MgADP. (a) Subunit packing in the dimer of dimers with each subunit separately colored. (b) Monomer structure showing the relationship between the two C-terminal ACT regulatory domains (dark blue) and the N-terminal catalytic domain (light blue).
The domain structure of the aspartokinase from M. jannaschii (mjAK) is similar to that of atAK (Mas-Droux et al., 2006 ▶), with an N-terminal catalytic domain (residues 2–300) and a C-terminal regulatory domain (residues 310–470) joined through a hinge region (residues 301–309) (Fig. 1 ▶ b).
The catalytic domain of mjAK belongs to the amino-acid kinase family (Pfam 00696) and shares significant homology to the kinase domain of atAK (r.m.s.d. = 1.22 Å for 1428/1857 aligned atoms). The catalytic domain is built around an eight-stranded parallel β-sheet (β1, β2, β3, β6, β9, β10, β11 and β12) with five α-helices (α1, α2, α3, α4 and α5) on one side and four α-helices (α6, α7, α8 and α9) on the other side joined by a cluster of four β-strands (β4, β5, β7 and β8). This domain is further divided into two lobes, one making up the l-aspartate-binding site, while the other provides a nucleotide-binding pocket for ATP (Fig. 1 ▶ b).
The regulatory domain of mjAK is composed of two ACT subdomains (Aravind & Koonin, 1999 ▶; Chipman & Shaanan, 2001 ▶), ACT1 and ACT2, each constructed from four-stranded antiparallel β-strands and two α-helices. The two ACT domains each have the same overall topology and relative antiparallel orientation to each other as observed in the atAK structure (Mas-Droux et al., 2006 ▶). However, the ACT1 subdomain of mjAK gives a poorer structural alignment to the ACT1 subdomain from atAK (r.m.s.d. = 1.40 Å with 248/357 aligned atoms) compared with the alignment of the ACT2 subdomain (r.m.s.d. = 0.88 Å with 278/406 aligned atoms). Superimposition of the catalytic domains of mjAK and atAK reveals that the orientation of the ACT1 subdomain differs between the plant and archaeal enzyme forms. The atAK structure was solved as a complex with its allosteric effectors, lysine and AdoMet, both of which bind to ACT1, while our mjAK structure does not have its allosteric site occupied. The missing electron density for residues 384–388 in the mjAK structure is at the end of a long loop in the ACT1 subdomain of mjAK which has been proposed to be involved in the allosteric inhibitor-binding site (Mas-Droux et al., 2006 ▶). These differences between mjAK and atAK suggest that the ACT1 subdomain becomes more ordered in the presence of the feedback inhibitors and undergoes a domain movement that is part of the communication between the regulatory site and the active site of AK.
Both atAK and mjAK crystallize as a dimer of dimers, with the dimer interface mediated through both ACT subdomains, with formation of the tetramer (dimer of dimers) stabilized by the interaction of complementary residues from α4 of the catalytic domain. However, native gel electrophoresis studies suggested that the plant enzyme exists as a functional dimer in solution (Mas-Droux et al., 2006 ▶) and earlier analytical ultracentrifugation and gel-filtration studies clearly established the dimer as the catalytically active unit of the monofunctional AK in Escherichia coli (ecAKIII; Richaud et al., 1973 ▶. Gel-filtration and dynamic light-scattering studies were conducted on mjAK to address the discrepancy in the quaternary structure of AK between that observed in the crystal and that reported in solution. Co-injection of mjAK and ecAKIII onto a size-exclusion column results in a clear separation between the two enzymes (Fig. 2 ▶). The mjAK species elutes first, followed by the well characterized ecAKIII dimer, showing that mjAK remains associated into a dimer of dimers in solution under these conditions. These results are confirmed by dynamic light-scattering (DLS) studies of mjAK and ecAKIII. A fit to the DLS data gives a hydrodynamic radius for ecAKIII consistent with an 89 kDa spherical particle, while under the same conditions the mjAK data fit a 154 kDa particle molecular weight. There is no change in the mjAK particle size when examined under the high-salt crystallization conditions. However, the addition of increasing levels of guanidine–HCl up to 2 M causes a decrease in the mjAK particle size to a molecular weight of 83 kDa, consistent with dissociation into dimers. At a lower guanidine–HCl concentration (1 M) high polydispersity is observed by DLS, consistent with an equilibrium in solution between the associated tetramer and the dissociated dimer. A further increase to 4 M guanidine–HCl leads to dissociation of the mjAK dimers into monomers with a molecular weight of 58 kDa predicted by DLS. While the contact surface observed between the dimers of mjAK in the crystal structure is quite small, there must be sufficient binding affinity for the dimers to remain associated in solution.
Figure 2.

Native gel-filtration chromatography showing the separation of the mjAK tetramer (subunit MW = 51 392 Da) from the ecAKIII dimer (subunit MW = 48 532 Da).
3.2. Catalytic activity, feedback regulation and active-site organization
The M. jannaschii genome contains only one gene encoding a monofunctional aspartokinase, in contrast to bacteria and plants, which typically have multiple AK isozymes. mjAK catalyzes phosphoryl transfer to l-aspartate at 310 K with a turnover of 12 s−1. This rate is about eightfold lower than the monofunctional AK isozyme (k cat = 98 s−1) from E. coli; however, when measured closer to its physiological temperature (343 K), mjAK exceeds the activity of ecAKIII, with a catalytic turnover of 245 s−1. This gene was annotated as a lysine-sensitive monofunctional AK (lysC) in the M. jannaschii genome (Bult et al., 1996 ▶). However, lysine has been found to have no effect on the activity of mjAK at concentrations up to 10 mM. Each of the end-product amino acids were then tested as feedback inhibitors of mjAK, with only threonine inhibiting the enzyme, with a K i of 0.3 mM. Therefore, this gene should be correctly annotated as a threonine-sensitive AK (thrA). The structure of this threonine-sensitive monofunctional mjAK was determined as a substrate–product complex to identify the enzyme functional groups involved in catalysis, most of which are conserved in the bacterial and plant AKs. The substrate and product were fitted into well defined electron density as seen in an F o − F c OMIT map (Fig. 3 ▶).
Figure 3.

An F o − F c OMIT map showing the fit of the substrate (l-aspartate) and product (MgADP) to the electron density.
The active site of mjAK is found in a deep cleft formed between the regulatory and catalytic domains within each subunit of the enzyme, with all of the contacts identified between the substrates and the enzyme provided by residues from the catalytic domain. The amino-acid substrate l-aspartic acid, which accepts the phosphoryl group from MgATP, is oriented at the active site through binding interactions with each of its functional groups. The α-carboxyl group forms a bidentate interaction with the bridging and terminal guanido N atoms of Arg207 and also makes hydrogen bonds with the side-chain hydroxyl of Thr46 and the backbone amide of Gly206 (Fig. 4 ▶ a). The α-amino group of l-aspartate makes only a single electrostatic interaction with the Glu130 carboxyl group, while the substrate β-carboxyl group, which is the phosphoryl acceptor, is positioned by hydrogen bonds to the side-chain hydroxyl of Ser40 and the backbone amide of Ser210. The atAK structure was not determined in the presence of substrates, but does have a second molecule of the feedback-inhibitor lysine bound at the active site (Mas-Droux et al., 2006 ▶). The α-amino group of lysine correctly interacts with the active-site glutamate in the atAK structure; however, the active-site arginine side chain that is responsible for a bidentate interaction with the substrate α-carboxyl group (Fig. 4 ▶ a) is not properly oriented in their structure and this change in position of this side chain is consistent with their proposed feedback-inhibition mechanism (Mas-Droux et al., 2006 ▶).
Figure 4.

Expansion of the mjAK structure showing the substrate-binding sites. (a) The amino-acid binding site, with each interaction <3.4 Å annotated. The α-carboxyl group of l-aspartate (L-ASP) forms an ion pair with Arg207 and a hydrogen bond with the backbone amide of Gly206 and the side chain of Thr46. The α-amino group of l-aspartate interacts electrostatically with the carboxyl group of Glu130. The β-carboxyl group, the phosphoryl acceptor, forms hydrogen bonds with the backbone amide of Ser210 and the side chain of Ser40. (b) The nucleotide-binding site. The divalent cation, Mg2+, forms a bridge between the α- and β-phosphate groups of ADP. The ribose moiety is bound by interactions with the side chains of Asp231, Asp239 and Arg241, while the adenine ring is positioned through hydrophobic interactions with Val267 as well as a hydrogen bond to the hydroxyl group of Tyr236.
The nucleotide product, MgADP, is bound in an extended conformation through interactions with each of its structural components. The divalent cation Mg2+ bridges between the α- and β-phosphate groups of ADP (Fig. 4 ▶ b). The β-phosphate is further positioned through a hydrogen bond to the side-chain hydroxyl of Thr230. The ribose hydroxyl groups hydrogen bond to the side-chain carboxyls of Asp231 and Asp239, with an additional interaction between the 3′-hydroxyl group and one of the terminal N atoms from the guanido group of Arg241. The adenine ring is positioned through a hydrophobic interaction with the side chain of Val267 and a hydrogen bond from the backbone carbonyl of Tyr236 to the exocyclic amino group on carbon 6.
3.3. Proposed catalytic mechanism
This structure of mjAK was determined in the presence of both the amino acid and nucleotide reactants, albeit lacking the phosphoryl group to be transferred during the catalytic cycle. The position between these reactants that would contain the γ-phosphate of ATP in the catalytically active complex is occupied in this structure by a salt bridge between the side chains of Lys6 and Asp211. Binding of ATP would disrupt this salt bridge and also cause a shift of the Mg2+ ion to include coordination to the γ-phosphate group. Metal-ion coordination will stabilize the negative charge density on this terminal phosphoryl group and lower the barrier towards transfer to the β-carboxyl group of l-aspartate. This transfer can be further catalyzed through an interaction with the side chain of Lys6 that would be properly positioned to assist in the phosphoryl transfer.
Supplementary Material
PDB reference: aspartokinase, 2hmf, r2hmfsf
Acknowledgments
Use of the Argonne National Laboratory at the Advanced Photon Source was supported by the US Department of Energy, Office of Energy Research under Contract No. W-31-109-ENG-38. We thank the staff members at BioCARS for their assistance with data collection. The authors wish to thank Dr Jean-Luc Ferrer (Université Joseph Fourier) for providing the coordinates of their A. thaliana aspartokinase structure prior to its release.
References
- Aravind, L. & Koonin, E. V. (1999). J. Mol. Biol.287, 1023–1040. [DOI] [PubMed] [Google Scholar]
- Brzozowski, A. M. & Walton, J. (2001). J. Appl. Cryst.34, 97–101. [Google Scholar]
- Bult, C. J. et al. (1996). Science, 273, 1058–1073. [DOI] [PubMed] [Google Scholar]
- Chipman, D. M. & Shaanan, B. (2001). Curr. Opin. Struct. Biol.11, 694–700. [DOI] [PubMed] [Google Scholar]
- Cohen, G. N. (1983). Amino Acids: Biosynthesis and Genetic Regulation, edited by K. M. Herrmann & R. L. Somerville, pp. 147–171. Reading, MA, USA: Addison–Wesley.
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Dautry-Varsat, A., Sibilli-Weill, L. & Cohen, G. N. (1977). Eur. J. Biochem.76, 1–6. [DOI] [PubMed] [Google Scholar]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Faehnle, C. R., Ohren, J. F. & Viola, R. E. (2005). J. Mol. Biol.353, 1055–1068. [DOI] [PubMed] [Google Scholar]
- McCoy, A., Grosse-Kunstleve, R. W., Storoni, L. C. & Read, R. J. (2005). Acta Cryst. D61, 458–464. [DOI] [PubMed] [Google Scholar]
- Mas-Droux, C., Curien, G., Robert-Genthon, M., Laurencin, M., Ferrer, J.-L. & Dumas, R. (2006). Plant Cell, 18, 1681–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Paidhungat, M., Setlow, B., Driks, A. & Setlow, P. (2000). J. Bacteriol.182, 5505–5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richaud, C., Mazat, J. P., Gros, C. & Patte, J. C. (1973). Eur. J. Biochem.40, 619–629. [DOI] [PubMed] [Google Scholar]
- Roberts, C. J. & Selker, E. U. (1995). Nucleic Acids Res.23, 4818–4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtman, E. R., Cohen, G. N., LeBras, G. & DeRobichon-Szulmajster, H. (1961). J. Biol. Chem.236, 2033–2038.
- Van Heijenoort, J. (2001). Nat. Prod. Rep.18, 503–519. [DOI] [PubMed] [Google Scholar]
- Veron, M., Guillou, Y., Fazel, A. & Cohen, G. N. (1985). Eur. J. Biochem.151, 521–524. [DOI] [PubMed] [Google Scholar]
- Viola, R. E. (2001). Acc. Chem. Res.34, 339–349. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: aspartokinase, 2hmf, r2hmfsf