

l-Methionine γ-lyase 2 from E. histolytica, a key enzyme in sulfur-containing amino-acid degradation in this protozoan parasite, has been crystallized in a form suitable for X-ray structure analysis.
Keywords: l-methionine γ-lyase, Entamoeba histolytica
Abstract
l-Methionine γ-lyase (MGL) is considered to be an attractive target for rational drug development because the enzyme is absent in mammalian hosts. To enable structure-based design of drugs targeting MGL, one of the two MGL isoenzymes (EhMGL2) was crystallized in the orthorhombic space group P212121, with unit-cell parameters a = 88.89, b = 102.68, c = 169.87 Å. The crystal diffracted to a resolution of 2.0 Å. The presence of a tetramer in the asymmetric unit (4 × 43.1 kDa) gives a Matthews coefficient of 2.2 Å3 Da−1. The structure was solved by the molecular-replacement method and structure refinement is now in progress.
1. Introduction
Entamoeba histolytica is the causative agent of amoebiasis, which affects over 50 million people and results in 70 000 deaths annually (World Health Organization, 1998 ▶). The major clinical manifestations of amoebiasis are amoebic dysentery and extraintestinal abscesses (i.e. hepatic, pulmonary and cerebral; Stanley, 2003 ▶). E. histolytica has several unique features in its sulfur-containing amino-acid metabolism (Nozaki et al., 2005 ▶). In particular, all the major pathways responsible for sulfur-containing amino-acid degradation are missing in this pathogen and instead it possesses a unique enzyme l-methionine γ-lyase (MGL; EC 4.4.1.11), which is concerned with the degradation of all major sulfur-containing amino acids such as methionine, homocysteine and cysteine (Tokoro et al., 2003 ▶). MGL contains pyridoxal 5′-phosphate (PLP) as a prosthetic group essential for its enzymatic activity and is categorized into the γ-family of PLP-dependent enzymes (Alexander et al., 1994 ▶). MGL catalyzes α,γ- or α,β-elimination of sulfur-containing amino acids and produces ammonia, α-keto acids and a volatile thiol such as hydrogen sulfide or methanethiol (Tanaka et al., 1985 ▶). MGL is known to be present only in a limited range of bacteria and parasitic protozoa and is absent in fungi, plants and mammals (Coombs & Mottram, 2001 ▶; Tokoro et al., 2003 ▶; Nozaki et al., 2005 ▶). MGLs from Pseudomonas putida, Clostridium sporogenes, Porphyromonas gingivalis, Brevibacterium linens, Citrobacter freundii, Trichomonas vaginalis and E. histolytica have been biochemically characterized so far (Nakayama et al., 1984 ▶; Kreis & Hession, 1973 ▶; Yoshimura et al., 2000 ▶; Dias & Weimer, 1998 ▶; Manukhov et al., 2005 ▶; Lockwood & Coombs, 1991 ▶; Tokoro et al., 2003 ▶) and in addition crystal structures have been reported for the MGLs from P. putida (Motoshima et al., 2000 ▶; Sridhar et al., 2000 ▶), T. vaginalis (PDB code 1e5f; unpublished) and C. freundii (Mamaeva et al., 2005 ▶).
Unlike bacterial MGLs, MGL from E. histolytica is unique in that it exists as two isoenzymes (EhMGL1 and EhMGL2), which differ significantly in substrate specificity (Tokoro et al., 2003 ▶, Nozaki et al., 2005 ▶). However, neither the structural factors causing the marked differences in their substrate specificity nor the biological significance of this apparent redundancy are well understood at this point. MGL is a target for the design of a prodrug and trifluoromethionine, which is converted into a toxic compound by MGL in P. gingivalis, T. vaginalis and E. histolytica (Yoshimura et al., 2002 ▶; Coombs & Mottram, 2001 ▶; Tokoro et al., 2003 ▶), has been proven to be effective both in in vitro culture and by peritoneal administration to cure animal infections caused by these pathogens (Coombs & Mottram, 2001 ▶; Tokoro et al., 2003 ▶; Nozaki et al., unpublished work).
In order to enable structure-based design of other prodrugs targeting MGL, we attempted to crystallize EhMGL1 and EhMGL2. In the present study, we describe the overexpression, purification, crystallization and preliminary X-ray diffraction studies of EhMGL2.
2. Materials and methods
2.1. Overexpression and purification of recombinant MGL2
A protein-coding region for EhMGL2 (accession No. AB094500) was inserted into pGEX-6P-1 vector (GE Healthcare Biosciences) as described in Tokoro et al. (2003 ▶). The plasmid was introduced into Escherichia coli BL21 Star (DE3) strain (Invitrogen). The transformant was grown in 600 ml Luria–Bertani medium containing 50 µg ml−1 ampicillin and 20 µM PLP at 310 K. When the culture reached a mid-log growth phase (optical density at 660 nm = 0.5), expression of the recombinant protein was induced with 1 mM isopropyl β-d-thiogalactopyranoside at 291 K for 24 h. The cells were harvested by centrifugation at 5000g for 20 min and suspended in 40 ml lysis buffer (50 mM sodium phosphate buffer pH 7.3, 150 mM NaCl and 20 µM PLP) supplemented with 0.5 mM PMSF, 28 µM E-64 protease inhibitor (Sigma–Aldrich) and 1 mM EDTA. After sonication, the lysate was centrifuged at 43 000g at 277 K for 30 min. The supernatant containing EhMGL2 expressed as a fusion protein with glutathione S-transferase (GST) was filtrated with a 0.45 µm filter and applied onto a GSTrap HP column (1.6 × 2.5 cm; GE Healthcare Biosciences) pre-equilibrated with lysis buffer. The column was then washed with 50 ml PBS (137 mM NaCl, 8.1 mM Na2HPO4, 2.68 mM KCl and 1.47 mM KH2PO4 pH 7.4) containing 20 µM PLP and further with 10 ml of 50 mM Tris–HCl pH 8.0, 150 mM NaCl, 20 µM PLP, 1 mM EDTA and 1 mM DTT. The column was incubated with PreScission Protease (GE Healthcare Biosciences) for 12 h at 277 K to digest the GST-EhMGL2 fusion protein. The EhMGL2 portion was eluted from the column with 50 mM Tris–HCl pH 8.0, 150 mM NaCl and 20 µM PLP. The pooled fractions were diluted tenfold with buffer A (20 mM Tris–HCl pH 8.0 and 20 µM PLP) and applied onto a MonoQ HR 5/5 column pre-equilibrated with buffer A. The recombinant EhMGL2 was eluted with a linear gradient of NaCl (0.0–1.0 M in 30 ml) with a flow rate of 1.0 ml min−1. EhMGL2 was eluted with ∼300 mM NaCl. The final recombinant EhMGL2 contained an additional ten amino-acid residues (GPLGSPEFPG) at the amino-terminus, which were derived from the expression vector. The fractions containing EhMGL2 were subjected to buffer-exchange and concentration using a Vivaspin centrifugal concentrator (Vivaspin 20, 10 kDa moelcular-weight cutoff) to yield 10 mg ml−1 EhMGL2 in 10 mM Tris–HCl pH 7.4. The purified EhMGL2 was estimated to be >95% pure by densitometric quantitation of the corresponding band on SDS–PAGE (Fig. 1 ▶, lane 4) and retained activity comparable to the previous study (Tokoro et al., 2003 ▶).
Figure 1.
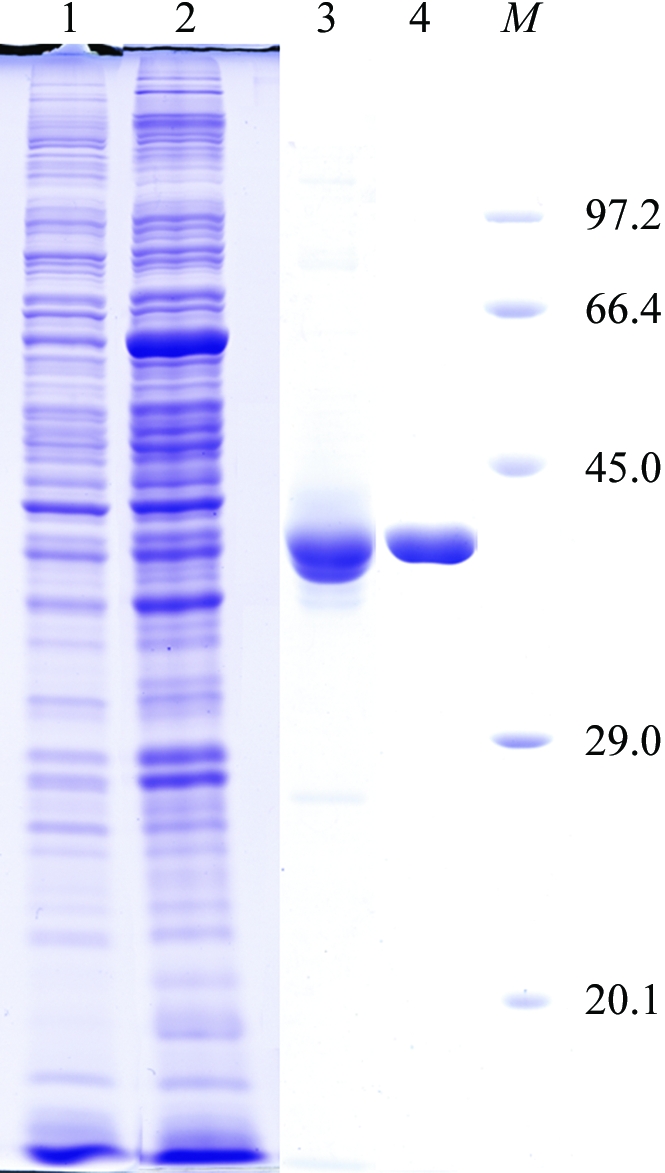
A 12% SDS–PAGE gel stained with Coomassie Brilliant Blue showing the apparent homogeneity of the purified EhMGL2. Lane 1, E. coli soluble extract before IPTG induction; lane 2, E. coli soluble extract after IPTG induction; lane 3, eluate from the GSTrap column; lane 4, purified EhMGL2 after HPLC chromatography using a MonoQ column; lane M, molecular-weight markers (kDa).
2.2. Crystallization and X-ray diffraction data collection
Crystallization conditions were screened at 293 and 277 K by the sitting-drop vapour-diffusion method using 96-well CrystalClear Strips (Hampton Research). A 0.5 µl droplet containing about 10 mg ml−1 EhMGL2 dissolved in 10 mM Tris–HCl pH 7.4 was mixed with an equal volume of reservoir solution and the droplet was allowed to equilibrate against 150 µl reservoir solution. In the initial screening experiment, Crystal Screen (Jancarik & Kim, 1991 ▶), Crystal Screen II (Hampton Research) and Wizard Screens I and II (Emerald BioStructures) were used as the reservoir solution. Out of 194 conditions, reagents containing polyethylene glycol 400 (PEG 400) as a precipitant gave single crystals at 277 K. Conditions were further optimized at 277 K by varying the PEG 400 concentration and the buffer pH. The effects of additives were also examined using Additive Screen kits from Hampton Research. The best crystals grew at 277 K from the reservoir solution containing 26%(w/v) PEG 400, 100 mM HEPES–NaOH pH 7.4 and 0.2 M CaCl2.
X-ray diffraction experiments were performed under liquid-nitrogen-cooled conditions at 100 K. A crystal mounted in a nylon loop was transferred and soaked briefly in a solution containing 30%(w/v) PEG 400, 100 mM HEPES–NaOH pH 7.4, 0.2 M CaCl2 and 5%(w/v) glycerol and then frozen by rapidly submerging it in liquid nitrogen. X-ray diffraction data were collected by the rotation method at the BL44XU beamline of SPring-8 using a DIP6040 detector at a wavelength of 0.900 Å. A total of 180 frames were recorded with an oscillation angle of 1°, an exposure time of 5 s per frame and a crystal-to-detector distance of 250 mm. The intensities were integrated with MOSFLM (Leslie, 1992 ▶) and scaled with SCALA (Evans, 1993 ▶) from the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶).
3. Results and discussion
Of the 194 crystallization conditions screened using commercially available screening kits, reagents containing PEG 400 gave rectangular single crystals. After optimization of the pH, PEG 400 concentration and additives, rectangular crystals grew to typical dimensions of 0.03 × 0.03 × 0.2 mm (Fig. 2 ▶) in one week using 26%(w/v) PEG 400, 0.1 M HEPES NaOH pH 7.4 and 0.2 M CaCl2 at 277 K. Diffraction patterns were recorded on 180 imaging plates at 100 K using one crystal. Analysis of the symmetry and systematic absences in the recorded diffraction pattern indicates that the crystals belong to the orthorhombic space group P212121, with unit-cell parameters a = 88.89, b = 102.68, c = 169.87 Å. Assuming the presence of four EhMGL2 molecules (4 × 43.1 kDa) in the asymmetric unit, the V M value is calculated to be 2.2 Å3 Da−1, with an estimated solvent content of 45%; these are within the range commonly observed for protein crystals (Matthews, 1968 ▶). A total of 690 565 observed reflections were merged to 105 693 unique reflections in the 50.0–2.0 Å resolution range. The data-collection and processing statistics are summarized in Table 1 ▶.
Figure 2.

Crystals of EhMGL2 obtained by the sitting-drop vapour-diffusion method using 26% PEG 400 as a precipitant.
Table 1. Data-collection and processing statistics.
Values for the highest resolution shell are given in parentheses.
| Wavelength (Å) | 0.900 |
| Space group | P212121 |
| Unit-cell parameters (Å) | a = 88.89, b = 102.68, c = 169.87 |
| Solvent content† (%) | 45 |
| Resolution range (Å) | 50.0–2.0 (2.07–2.00) |
| No. of reflections | 690565 |
| Unique reflections | 105693 |
| Multiplicity | 6.5 |
| Completeness (%) | 96.3 (87.6) |
| Rmerge‡ (%) | 9.2 (34.5) |
| I/σ(I) | 6.5 (2.2) |
Assuming the presence of four molecules in the asymmetric unit.
R
merge(I) = 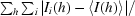
 .
.
An attempt to solve the structure using the molecular-replacement method with the MOLREP program (Navaza, 1994 ▶) as implemented within the CCP4 package (Collaborative Computational Project, Number 4, 1994 ▶) was carried out using the refined coordinates of MGL from P. putida (PDB code 1gc2; 41% sequence identity with EhMGL2) as a model. X-ray diffraction data in the resolution range 15.0–2.0 Å were used. A promising solution with a homotetrameric structure was obtained (correlation coefficient and R factor of 0.65 and 51.8%, respectively) and the model was subsequently subjected to rigid-body refinement to give an R factor of 0.45. Amino-acid residues of the initial homotetramer model that differed from those of the search model were replaced with Ala. Using the molecular-replacement solution, the structure is being subjected to iterative cycles of manual model correction and crystallographic refinement. In parallel with the refinement, we are also attempting to obtain crystals of EhMGL2 complexed with substrate analogues and inhibitors.
References
- Alexander, F. W., Sandmeier, E., Mehta, P. K. & Christen, P. (1994). Eur. J. Biochem.219, 953–960. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Coombs, G. H. & Mottram, J. C. (2001). Antimicrob. Agents Chemother.45, 1743–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias, B. & Weimer, B. (1998). Appl. Environ. Microbiol.64, 3327–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, P. R. (1993). Proceedings of the CCP4 Study Weekend. Data Collection and Processing, edited by L. Sawyer, N. Isaacs & S. Bailey, pp. 114–122. Warrington: Daresbury Laboratory.
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst.24, 409–411. [Google Scholar]
- Kreis, W. & Hession, C. (1973). Cancer Res.33, 1862–1865. [PubMed] [Google Scholar]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Lockwood, B. C. & Coombs, G. H. (1991). Biochem. J.279, 675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamaeva, D. V., Morozova, E. A., Nikulin, A. D., Revtovich, S. V., Nikonov, S. V., Garber, M. B. & Demidkina, T. V. (2005). Acta Cryst. F61, 546–549. [DOI] [PMC free article] [PubMed]
- Manukhov, I. V., Mamaeva, D. V., Rastorguev, S. M., Faleev, N. G., Morozova, E. A., Demidkina, T. V. & Zavilgelsky, G. B. (2005). J. Bacteriol.187, 3889–3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Motoshima, H., Inagaki, K., Kumasaka, T., Furuichi, M., Inoue, H., Tamura, T., Esaki, N., Soda, K., Tanaka, N., Yamamoto, M. & Tanaka, H. (2000). J. Biochem.128, 349–354. [DOI] [PubMed] [Google Scholar]
- Nakayama, T., Esaki, N., Sugie, K., Beresov, T. T., Tanaka, H. & Soda, K. (1984). Anal. Biochem.138, 421–424. [DOI] [PubMed] [Google Scholar]
- Navaza, J. (1994). Acta Cryst. A50, 157–163. [Google Scholar]
- Nozaki, T., Ali, V. & Tokoro, M. (2005). Adv. Parasitol.60, 1–99. [DOI] [PubMed] [Google Scholar]
- Sridhar, V., Xu, M., Han, Q., Sun, X., Tan, Y., Hoffman, R. M. & Prasad, G. S. (2000). Acta Cryst. D56, 1665–1667. [DOI] [PubMed] [Google Scholar]
- Stanley, S. Jr (2003). Lancet, 361, 1025–1034. [DOI] [PubMed] [Google Scholar]
- Tanaka, H., Esaki, N. & Soda, K. (1985). Enzyme Microb. Technol.7, 530–537.
- Tokoro, M., Asai, T., Kobayashi, S., Takeuchi, T. & Nozaki, T. (2003). J. Biol. Chem.43, 42717–42727. [DOI] [PubMed]
- World Health Organization (1998). The World Health Report. Geneva: World Health Organization.
- Yoshimura, M., Nakano, Y. & Koga, T. (2002). Biochem. Biophys. Res. Commun.292, 964–968. [DOI] [PubMed] [Google Scholar]
- Yoshimura, M., Nakano, Y., Yamashita, Y., Oho, T., Saito, T. & Koga, T. (2000). Infect. Immun. 68, 6912–6916. [DOI] [PMC free article] [PubMed] [Google Scholar]