

LmACR2 from L. major is the first rhodanese-like enzyme directly involved in the reduction of arsenate and antimonate to be crystallized. Diffraction data have been collected to 1.99 Å resolution using synchrotron X-rays.
Keywords: arsenate/antimonate reductase, arsenic detoxification, rhodanese, Leishmania sp
Abstract
Arsenic is present in the biosphere owing either to the presence of pesticides and herbicides used in agricultural and industrial activities or to leaching from geological formations. The health effects of prolonged exposure to arsenic can be devastating and may lead to various forms of cancer. Antimony(V), which is chemically very similar to arsenic, is used instead in the treatment of leishmaniasis, an infection caused by the protozoan parasite Leishmania sp.; the reduction of pentavalent antimony contained in the drug Pentostam to the active trivalent form arises from the presence in the Leishmania genome of a gene, LmACR2, coding for the protein LmACR2 (14.5 kDa, 127 amino acids) that displays weak but significant sequence similarity to the catalytic domain of Cdc25 phosphatase and to rhodanese enzymes. For structural characterization, LmACR2 was overexpressed, purified to homogeneity and crystallized in a trigonal space group (P321 or P3121/P3221). The protein crystallized in two distinct trigonal crystal forms, with unit-cell parameters a = b = 111.0, c = 86.1 Å and a = b = 111.0, c = 175.6 Å, respectively. At a synchrotron beamline, the diffraction pattern extended to a resolution limit of 1.99 Å.
1. Introduction
Rhodanese proteins (thiosulfate:cyanide sulfurtransferases; TSTs; EC 2.8.1.1) are widespread enzymes found in the three major evolutionary phyla that catalyze the in vitro transfer of a sulfane S atom from thiosulfate to cyanide (Westley et al., 1983 ▶). A second group of enzymes that are closely related to rhodanese are the 3-mercaptopyruvate:cyanide sulfutransferases (MSTs; EC 2.8.1.2). Rhodanese structural domains appeared very early during natural evolution, prior to eukaryota/prokaryota diversification (Bordo & Bork, 2002 ▶). In present-day proteins, they are found in distinct structural contexts, either as tandem repeats with the C-terminal domain hosting the properly structured active-site Cys residue, as single-domain proteins or in combination with distinct protein domains. A third class of eukaryotic enzymes evolutionarily related to rhodanese are the Cdc25 phosphatases (EC 3.1.3.48; Galaktionov & Beach, 1991 ▶). In both rhodanese and Cdc25 phosphatase, the active site is formed by a loop connecting the central strand of the β-sheet that forms the hydrophobic core of the protein structure to the contiguous α-helix. The active-site loop hosts a Cys residue at the first position and assumes a cradle-like structure with the Cys Sγ atom at its geometrical centre (Bordo et al., 2000 ▶). The relevant structural difference between sulfurtransferases and Cdc25 phosphatases is the length of the active-site loop, which is six and seven amino acids long, respectively. The distinct active-site loop length and amino-acid composition determine the affinity for the known substrates containing S or P atoms.
As/Sb reductases are closely related to Cdc25 phosphatase. These cytoplasmic enzymes are involved in the reduction of arsenate (AsV) to arsenite (AsIII) or antimonate (SbV) to antimonite (SbIII), which is then removed from the cytosol by specific carrier proteins and extruded by selective pumps. One of these enzymes is encoded by the gene LmACR2 identified in the genome of Leishmania major (Zhou et al., 2004 ▶). The corresponding 14.5 kDa protein (LmACR2, 127 amino acids) displays weak but significant similarity (23% identity) to the catalytic domain of human Cdc25 phosphatases and shares the same active-site loop motif, Cys-X 5-Arg, with these enzymes (see Fig. 1 ▶). Biochemical characterization has shown that LmACR2 is able to reduce both AsV and SbV to AsIII and SbIII, respectively (Zhou et al., 2004 ▶). Furthermore, evidence has been provided that eukaryotic metalloid reductases evolved from phosphatases (Mukhopadhyay & Rosen, 2002 ▶) and that LmACR2 is a bifunctional enzyme which dephosphorylates tyrosine residues (Zhou et al., 2006 ▶). The presence of the gene LmACR2 in the Leishmania genome has medical relevance in the clinical treatment of leishmaniasis, a parasitic disease caused by Leishmania and transmitted by the bite of some species of sand fly, which is endemic in numerous tropical and subtropical countries. LmACR2 is in fact involved in the reduction of the pentavalent antimonial drug Pentostam (sodium stibogluconate) used in treatment of leishmaniasis to the active trivalent form of the drug (Zhou et al., 2004 ▶). Thus, determination of the three-dimensional structure of LmACR2 will help to understand the two distinct in vitro catalytic mechanisms and to identify the in vivo biological substrates. We describe here the purification and crystallization of LmACR2, together with a preliminary analysis of the X-ray diffraction data.
Figure 1.

Sequence alignment of LmACR2 and the catalytic domain of the human Cdc25A phosphatase. In Cdc25A phosphatase secondary-structure elements have been identified in the crystallographic structure (PDB code 1c25; Fauman et al., 1998 ▶) with the program DSSP (Kabsch & Sander, 1983 ▶). Residues assuming α-helical and β-strand conformation are shown on a black and grey background, respectively.
2. Materials and methods
2.1. Protein expression and purification
LmACR2 was cloned in the plasmid pBAD-MYC-HISA from Invitrogen. The resulting chimeric protein also contains a 25-amino-acid C-terminal domain composed of the c-Myc epitope and a six-His tail (sequence KLGPEQKLISEEDLNSAVDHHHHHH). The protein was overexpressed in Escherichia coli strain TOP10 and purified as previously described (Zhou et al., 2004 ▶). Briefly, cells from a 4 l culture were harvested by centrifugation, washed once with buffer A (10 mM Tris–HCl, 0.1 M KCl pH 7.5), suspended in buffer B [50 mM MOPS–KOH pH 7.5 containing 20 mM imidazole, 0.5 M NaCl, 10 mM β-mercaptoethanol and 20%(v/v) glycerol] and then lysed by a single passage through a French pressure cell at 138 MPa. The lysate was centrifuged at 100 000g for 60 min at 277 K and the supernatant solution was loaded at a flow rate of 0.5 ml min−1 onto an Ni–NTA column pre-equilibrated with buffer B. The column was then washed with 250 ml buffer B followed by elution with 125 ml buffer B with the concentration of imidazole increased to 0.2 M. Concentrated fractions from the Ni–NTA column were applied onto a Sephacryl S-100 (Amersham Biosciences) column pre-equilibrated with buffer C (50 mM MOPS–KOH pH 6.5 containing 0.5 M NaCl, 10 mM β-mercaptoethanol, 20% glycerol and 0.5 mM EDTA), eluted with buffer C, pooled and concentrated.
2.2. Crystallization
For crystallization purposes, the protein was concentrated to 44 mg ml−1 in a solution containing 50 mM MOPS–KOH pH 6.5, 250 mM NaCl and 10%(v/v) glycerol. Initial crystallization conditions were determined by the sparse-matrix method (Jancarik & Kim, 1991 ▶) as implemented in Hampton Crystal Screens I and II (Hampton Research, CA, USA). Crystals were grown by the hanging-drop vapour-diffusion method by mixing an equal amount (1 µl) of protein and reservoir solutions. The optimization of the preliminary crystallization conditions resulted in crystals of optimal size, having a hexagonal shape and typical dimensions of 0.1 × 0.1 × 0.2 mm, which grew after 5–7 d equilibration against a reservoir solution containing 2.0 M (NH4)2SO4, 100 mM HEPES pH 8.0 and 15 mM Na2S2O3 at 277 K (Fig. 2 ▶). The presence of thiosulfate in the protein solution was essential to promote crystal growth and to avoid the formation of unstable and weakly diffracting crystals; this is likely to be a consequence of the intrinsic capability of the reduced thiol group of the active-site cysteine to form disulfide bridges, leading to the destabilization of the protein native structure. Sulfane sulfur-donor compounds such as Na2S2O3 are likely to either keep the protein in the persulfurated form or to prevent intermolecular disulfide bridges leading to unfolding and aggregation (Bordo et al., 2001 ▶).
Figure 2.

Hanging drop with crystal of LmACR2 grown in a precipitating solution of 2.0 M ammonium sulfate, 100 mM HEPES pH 8.0 and 15 mM sodium thiosulfate. The crystal, displaying an approximate hexagonal shape, has dimensions of about 0.2 × 0.2 × 0.2 mm.
3. Results and discussion
LmACR2 crystals are thermolabile and rather sensitive to changes in mother-liquor composition. Small temperature changes arising from manipulation or heavy-atom soaking easily resulted in substantial decay of the diffraction power. To maintain good diffraction quality, crystal manipulation was carried out at 277 K and crystals were immersed as soon as possible in a cryoprotectant solution identical to the reservoir solution but containing an additional 20%(v/v) glycerol, mounted on a cryoloop and immersed in liquid nitrogen. The skin present on the surface of the crystallization drop (Fig. 2 ▶) could easily be removed with microtools before further crystal manipulation. Data collections were carried out at 100 K on an MSC R-AXIS II image-plate system coupled to a Rigaku RU-200 rotating-anode generator at the Cu Kα wavelength. Diffraction data were also collected at the ID23-1 and ID14-2 beamlines at ESRF, Grenoble (Fig. 3 ▶) and at the BW7B beamline at EMBL-DESY, Hamburg. Images were integrated with MOSFLM (Powell, 1999 ▶) and scaled using SCALA. Further data manipulations were performed using programs from the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶).
Figure 3.

Diffraction pattern of LmACR2 crystals collected at the ID14-2 beamline, ESRF, Grenoble on a ADSC Q4 CCD area detector. The outer ring is at 2.50 Å resolution.
Data reduction showed that LmACR2 crystals belong to space group P321 or P3121/P3221. Notably, two different unit cells were found, with a = b = 111.02, c = 86.08 Å (resolution, 2.50 Å) and a = b = 110.98, c = 175.59 Å (resolution, 1.99 Å). Data-collection statistics are summarized in Table 1 ▶. In spite of the length of the c axis of the greater unit cell (175.59 Å) being approximately twice that of the smaller unit cell (2 × 86.08 = 172.16 Å), the resulting difference (3.43 Å) is too large to be accommodated within measurement errors or to be a data-reduction artefact. In fact, attempts to reduce the measured data using the smaller unit cell proved unsuccessful. Furthermore, no preference for one of the two crytal forms was observed within the range of conditions leading to crystal formation and in a few cases both crystal forms were observed in the same crystallization drop.
Table 1. Data-collection statistics.
Values in parentheses are for the outer resolution shell.
| Resolution (Å) | 1.99 (2.10–1.99) | 2.50 (2.64–2.50) |
| Source | X-13 beamline, DESY, Hamburg | ID14-2 beamline, ESRF, Grenoble |
| Unit-cell parameters (Å) | a = b = 110.98, c = 175.59 | a = b = 111.02, c = 86.08 |
| Wavelength (Å) | 0.801 | 0.933 |
| Temperature (K) | 100 | 100 |
| No. of observations | 345080 | 105392 |
| Unique reflections | 82490 | 21306 |
| Data completeness (%) | 96.0 (74.1) | 99.0 (98.3) |
| 〈I〉/σ(I) | 14.3 (2.9) | 19.0 (5.6) |
| Rmerge† (%) | 6.9 (39.8) | 6.6 (20.9) |
| Multiplicity | 4.2 (3.6) | 4.9 (4.7) |
R
merge = 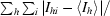
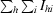 .
.
In order to determine the number of LmACR2 molecules in the asymmetric unit, estimation of the Matthews coefficient V M took place for both unit cells (Matthews, 1968 ▶). For the small unit cell, the calculated V M suggests the presence of 3–6 molecules per asymmetric unit (V M from 3.5 to 1.7 Å3 Da−1; solvent content in the range 65–29%, respectively), with twice as many in the larger unit cell (6–12 molecules). Notably, calculation of the self-rotation function indicates the presence of a noncrystallographic twofold axis in both crystal forms (see Fig. 4 ▶), supporting the hypothesis of a similar protein dimeric assembly in both crystal lattices. Structure solution was attempted by using the molecular-replacement method as implemented in the programs AMoRe, Phaser and MOLREP (Navaza, 1994 ▶; McCoy et al., 2005 ▶; Vagin & Teplyakov, 2000 ▶) on both crystal forms. For this purpose, several search models were derived from the three-dimensional structures of the catalytic domain of Cdc25A and Cdc25B phosphatases (Fauman et al., 1998 ▶; Reynolds et al., 1999 ▶). Unfortunately, no solution could be found to correctly place the template molecules in the crystal unit cells; also, the calculation of potential pseudo-translation vectors did not produce useful hints about the number and location of the molecular chains in the unit cell. The elucidation of the three-dimensional structure of LmACR2 is presently being pursued through multiple isomorphous replacement methods by way of heavy-atom screening.
Figure 4.

The χ = 180° sectin of the self-rotation function, as calculated with the program MOLREP (Collaborative Computational Project, Number 4, 1994 ▶), indicates the presence of a noncrystallographic twofold axis (θ = 46.5, ϕ = 90.0°).
Acknowledgments
We are grateful to Martino Bolognesi for helpful discussions. Data collection at both ESRF, Grenoble and DESY, Hamburg beamlines was supported by the EU ‘Access to Research Infrastructure Action FP6 Programme’. This research was partly supported by National Institutes of Health Grants AI58170 to RM and GM52216 to BPR.
References
- Bordo, D. & Bork, P. (2002). EMBO Rep.3, 741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordo, D., Deriu, D., Colnaghi, R., Carpen, A., Pagani, S. & Bolognesi, M. (2000). J. Mol. Biol.298, 691–704. [DOI] [PubMed] [Google Scholar]
- Bordo, D., Forlani, F., Spallarossa, A., Colnaghi, R., Carpen, A., Bolognesi, M. & Pagani, S. (2001). Biol. Chem.382, 1245–1252. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Fauman, E. B., Cogswell, J. P., Lovejoy, B., Rocque, W. J., Holmes, W., Montana, V. G., Piwnica-Worms, H., Rink, M. J. & Saper, M. A. (1998). Cell, 93, 617–25. [DOI] [PubMed] [Google Scholar]
- Galaktionov, K. & Beach, D. (1991). Cell, 67, 1181–1194. [DOI] [PubMed] [Google Scholar]
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst.24, 409–411. [Google Scholar]
- Kabsch, W. & Sander, C. (1983). Biopolymers, 12, 2577–2637. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Storoni, L. C. & Read, R. J. (2005). Acta Cryst. D61, 458–464. [DOI] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay, R. & Rosen, B. P. (2002). Environ. Health Perspect.110, 745–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navaza, J. (1994). Acta Cryst. A50, 157–163. [Google Scholar]
- Powell, H. R. (1999). Acta Cryst. D55, 1690–1695. [DOI] [PubMed] [Google Scholar]
- Reynolds, R. A., Yem, A. W., Wolfe, C. L., Deibel, M. R. Jr, Chidester, C. G. & Watenpaugh, K. D. (1999). J. Mol. Biol.293, 559–568. [DOI] [PubMed] [Google Scholar]
- Vagin, A. & Teplyakov, A. (2000). Acta Cryst. D56, 1622–1624. [DOI] [PubMed] [Google Scholar]
- Westley, J., Adler, H., Westley, L. & Nishida, C. (1983). Fundam. Appl. Toxicol.3, 377–382. [DOI] [PubMed] [Google Scholar]
- Zhou, Y., Bhattacharjee, H. & Mukhopadhyay, R. (2006). Mol. Biochem. Parasitol.148, 161–168. [DOI] [PubMed] [Google Scholar]
- Zhou, Y., Messier, N., Ouellette, M., Rosen, B. P. & Mukhopadhyay, R. (2004). J. Biol. Chem.279, 37445–37451. [DOI] [PubMed] [Google Scholar]