

Mouse carnosinase was crystallized in complex with Zn2+ or Mn2+ and the complexes are undergoing structure determination by the MAD method.
Keywords: carnosine, mouse, metalloenzymes
Abstract
Mammalian tissues contain several histidine-containing dipeptides, of which l-carnosine is the best characterized and is found in various tissues including the brain and skeletal muscles. However, the mechanism for its biosynthesis and degradation have not yet been fully elucidated. Crystallographic study of carnosinase CN2 from mouse has been undertaken in order to understand its enzymatic mechanism from a structural viewpoint. CN2 was crystallized by the hanging-drop vapour-diffusion technique using PEG 3350 as a precipitant. Crystals were obtained in complex with either Mn2+ or Zn2+. Both crystals of CN2 belong to the monoclinic space group P21 and have almost identical unit-cell parameters (a = 54.41, b = 199.77, c = 55.49 Å, β = 118.52° for the Zn2+ complex crystals). Diffraction data were collected to 1.7 and 2.3 Å for Zn2+ and Mn2+ complex crystals, respectively, using synchrotron radiation. Structure determination is ongoing using the multiple-wavelength anomalous diffraction (MAD) method.
1. Introduction
l-Carnosine (β-alanyl-l-histidine) and structurally related dipeptides such as homocarnosine (γ-aminobutyryl-l-histidine) and anserine (β-alanyl-N-methyl-l-methylhistidine) are distributed in a wide variety of vertebrate tissues (Bonfanti et al., 1999 ▶). l-Carnosine is present at particularly high concentrations in mammalian skeletal muscles and brain, but its physiological functions remain to be determined. We have recently found that intracranial injection of l-carnosine suppresses 2-deoxy-d-glucose-induced hyperglycaemia and that this effect is suppressed by thioperamide, a histamine H3 antagonist (Yamano et al., 2001 ▶). We have also found that l-carnosine decreases the blood-pressure levels of DOCA-salt hypertension rats (Niijima et al., 2002 ▶). In addition, l-carnosine facilitates the activity of parasympathetic nerves innervating the pancreas and kidney, while it inhibits the activity of sympathetic nerves innervating the pancreas (Yamano et al., 2001 ▶). Moreover, l-carnosine is thought to be released from muscles by physical activity (Nagai et al., 2003 ▶). These findings suggest that l-carnosine regulates the autonomic nervous system by modulating the activities of histaminergic neurons in the brain and thus functions as a beneficial factor of physical activity in diabetes and hypertension.
l-Carnosine is synthesized from β-alanine and l-histidine by carnosine synthetase and is degraded by carnosinase. However, the molecular characteristics of these enzymes are as yet largely unknown. Recently, cDNAs encoding dipeptidases that can degrade l-carnosine (CN1 and CN2) have been reported (Teufel et al., 2003 ▶). We have independently found a carnosine-degrading enzyme that matched the nucleotide sequence of CN2 in mouse brain (Otani et al., 2005 ▶). These enzymes belong to the metalloproteinase M20 family and digest a wide range of peptides, including l-carnosine. Immunohistochemical analysis showed that CN2 is highly concentrated in the tuberomammillary nucleus of the hypothalamus, where the neuronal cell bodies of histaminergic neurons are located (Otani et al., 2005 ▶). This raised the possibility that l-carnosine is degraded by CN2 to supply the substrate of the histamine-synthesizing enzyme histidine decarboxylase. CN2 requires Mn2+ for full activity, whereas CN2 in complex with Zn2+ has around 10% of the activity of CN2 in complex with Mn2+.
We initiated crystallographic study of CN2 from mice in order to understand its enzymatic mechanism from a structural viewpoint. CN2 was crystallized by the hanging-drop vapour-diffusion technique using PEG 3350 as a precipitant. The most highly homologous protein to CN2 of known structure is the aminopeptidase PepV from Lactobacillus delbrueckii (Jozic et al., 2002 ▶). Its sequence identity is only 17% and hence the molecular-replacement method is not promising. Two kinds of crystals were obtained, in complex with Mn2+ or Zn2+; the presence of the metal ions was confirmed by XAFS (X-ray absorption fine structure) spectra. We collected diffraction data sets from both types of crystal for independent MAD phasing in order to determine the two crystal structures and to compare them.
2. Expression and purification
A cDNA encoding full-length CN2 was obtained by reverse transcriptase-polymerase chain reaction from mouse brain mRNA as previously described (Otani et al., 2005 ▶) and subcloned into the EcoRI–SmaI sites of the pGEX 4T3 vector (Amersham Biosciences). CN2 protein fused with glutathione-S-transferase at the N-terminus was then induced in Escherichia coli strain BL21(DE3) pLysS in the presence of 1 mM IPTG and 2 mM MnCl2 for 12–16 h at 298 K. The E. coli cells were collected and sonicated in buffer containing 25 mM Tris–HCl pH 7.4, 50 mM NaCl, 0.2 mM MnCl2, 1 mM DTT. After centrifugation, the supernatant was mixed with glutathione-Sepharose beads (Amersham Biosciences), washed and incubated with thrombin (Amersham Biosciences) for 12–16 h. The soluble fraction was then applied onto a gel-filtration column (HiLoad 26/60 Superdex 200pg, Amersham Biosciences) and an anion-exchange column (HiTrap Q, Amersham Biosciences) and protein was eluted with a linear gradient of 50–750 mM NaCl in the same buffer. The buffer of the protein solution was changed to that for crystallization by ultrafiltration with Centriprep YM-30 (Millipore). The final product, after thrombin cleavage, represents the full-length CN2 sequence (SWISS-PROT code CPGL1_MOUSE) preceded by five residues (GSPNS) of the expression vector. The number of amino-acid residues including N-terminal artificial five residues is 480.
3. Crystallization
Preparation and crystallization of the CN2 Zn2+ and Mn2+ complexes were carried out in the same way. CN2 was crystallized using the hanging-drop vapour-diffusion method. Initial screening for crystallization conditions was performed using Crystal Screens 1 and 2 (Hampton Research). However, we were not able to obtain crystals.
Thermostable proteins have a tendency to crystallize more easily (Yamagata et al., 2001 ▶). Because the denaturation temperature of a protein depends on the pH and the additives present in the protein solution, finding conditions that maximize a protein’s denaturation temperature may be assumed to increase its crystallization potential. To search for conditions leading to good crystals, we measured the thermal denaturation temperatures of the protein using a differential scanning calorimeter (MicroCal VP-DSC) under various conditions by changing the pH and adding inhibitors and additives, the details of which will be described elsewhere. We describe the crystallization conditions obtained by the differential scanning calorimeter below.
2 µl protein solution (20 mg ml−1) in buffer containing 25 mM Tris–HCl pH 7.4, 50 mM NaCl, 0.2 mM MnCl2, 1 mM DTT and 30 mM bestatin (an inhibitor) was mixed with an equal volume of reservoir solution and was allowed to equilibrate against 0.5 ml of the reservoir solution at 293 K. Wing-shaped crystals were obtained using a reservoir solution containing 20%(w/v) PEG 3350 and 0.2 M KF. Crystal size and quality were further improved with a combination of macroseeding and microseeding techniques (Stura, 1999 ▶). As a result, single crystals of dimensions 0.4 × 0.2 × 0.1 mm were obtained in hanging drops using 20%(w/v) PEG 3350, 20%(w/v) glycerol, 0.2 M KF as the reservoir solution (Fig. 1 ▶). In order to remove an excess of Mn2+, crystals were transferred to harvesting solution consisting of 20%(w/v) PEG 3350, 20%(w/v) glycerol, 0.2 M KF for 1 d prior to X-ray data collection.
Figure 1.

Crystals of mouse carnosinase.
4. Data collection and processing
Mn2+ and Zn2+ complex crystals of CN2 were mounted in nylon CryoLoops (Hampton Research) and placed directly into a nitrogen stream at 100 K. For identification of the metal type in the CN2 complex crystals, XAFS spectra were measured and used to discriminate the metal type. We selected crystals containing only Mn2+ or Zn2+ ions and used them for the Zn and Mn MAD method. Synchrotron-radiation oscillation data were collected using a Quantum 210 CCD detector (ADSC, USA) at beamline NW-12 of Photon Factory (KEK, Tsukuba, Japan) for a Mn2+ complex crystal at two wavelengths at the Mn K absorption edge of 1.8941 Å (edge) and 1.8926 Å (peak) and at one remote wavelength of 1.7926 Å; data were collected from a Zn2+ complex crystal at two wavelengths at the Zn K absorption edge of 1.2834 Å (edge), 1.2827 Å (peak) and two remote wavelengths of 1.000 and 1.2573 Å. Complete data sets were collected through contiguous rotation ranges at a given wavelength before proceeding to the next wavelength. The total oscillation ranges were 360° for all data collections except for the Zn high-resolution data set. Oscillation data were recorded in frames of 1° oscillation with 1 and 2 s exposures per image for Mn MAD and Zn MAD, respectively. One crystal was used for the Mn MAD data collection and one for the Zn MAD data collection. The data collected at the various wavelengths were processed with the program HKL-2000 (Otwinowski & Minor, 1997 ▶). Statistics of the MAD data sets are summarized in Table 1 ▶. The Mn complex crystal suffer appreciable radiation damage and its effects together with our data-collection conditions were reflected in the statistics. The space groups of all CN2 crystals were determined to be monoclinic P21. Assuming the presence of two molecules of CN2 in the asymmetric unit, the value of the Matthews coefficient V M (Matthews, 1968 ▶) is 2.50 Å3 Da−1, corresponding to a solvent content of 49.9%, both of which are within the normal range of values for protein crystals (Matthews, 1968 ▶). The self-rotation function calculation showed strong peaks indicating twofold rotation symmetry. Structure analysis of the Zn2+ and Mn2+ complex crystals by MAD are currently in progress.
Table 1. Statistics of X-ray diffraction data collection.
Values in parentheses are for the highest resolution shell.
| Mn2+ complex | Zn2+ complex | ||||||
|---|---|---|---|---|---|---|---|
| Unit-cell parameters (Å, °) | a = 54.49, b = 199.18, c = 55.21, β = 118.92 | a = 54.41, b = 199.77, c = 55.49, β = 118.52 | |||||
| Space group | P21 | P21 | |||||
| Data set | Edge | Peak | Remote | High resolution | Edge | Peak | Remote |
| Wavelength (Å) | 1.8941 | 1.8926 | 1.7926 | 1.000 | 1.2834 | 1.2827 | 1.2573 |
| Resolution (Å) | 50.0–2.8 (2.91–2.81) | 50.0–2.8 (2.90–2.80) | 50.0–2.3 (2.38–2.30) | 50.0–1.7 (1.76–1.70) | 50.0–1.8 (1.86–1.80) | 50.0–1.8 (1.86–1.80) | 50.0–1.8 (1.86–1.80) |
| Redundancy | 7.3 (6.3) | 7.4 (6.9) | 6.6 (3.1) | 5.0 (3.4) | 7.0 (4.4) | 6.9 (4.4) | 7.0 (4.6) |
| No. of observed reflections | 183799 | 186077 | 264160 | 545738 | 617481 | 618712 | 638696 |
| No. of unique reflections | 25204 (2534) | 25248 (2390) | 39799 (1412) | 109231 (8405) | 88807 (5360) | 89035 (5495) | 90757 (6112) |
| Completeness (%) | 99.9 (99.1) | 99.3 (94.4) | 86.8 (30.9) | 96.1 (74.4) | 92.8 (56.1) | 93.2 (57.5) | 94.7 (63.8) |
| I/σ(I) | 31.8 (6.4) | 45.2 (15.6) | 35.3 (3.4) | 38.4 (2.8) | 46.2 (5.4) | 47.1 (6.3) | 45.5 (5.0) |
| Rsym† (%) | 6.9 (27.8) | 5.9 (14.2) | 5.8 (23.5) | 3.8 (34.2) | 4.0 (19.9) | 4.1 (18.0) | 4.2 (22.1) |
R
sym = 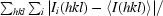
 , where I
i(hkl) is the ith intensity measurement of reflection hkl, including symmetry-related reflections, and 〈I(hkl)〉 is its average.
, where I
i(hkl) is the ith intensity measurement of reflection hkl, including symmetry-related reflections, and 〈I(hkl)〉 is its average.
Acknowledgments
We are grateful to the beamline staff of Photon Factory BL12NW for their assistance during data collection. This research is supported by the National Project on Protein Structural and Functional Analyses from the Ministry of Education, Culture, Sports, Science and Technology of Japan. We are thankful to Dr Kyoko Ogasahara at the Institute for Protein Research, Osaka University and Dr Katsuhide Yutani at RIKEN SPring-8 Center, Harima Institute for the differential scanning calorimetry measurements and analysis.
References
- Bonfanti, L., Peretto, P., De Marchis, S. & Fasolo, A. (1999). Prog. Neurobiol.59, 333–353. [DOI] [PubMed] [Google Scholar]
- Jozic, D., Bourenkow, G., Bartunik, H., Scholze, H., Dive, V., Henrich, B., Huber, R., Bode, W. & Maskos, K. (2002). Structure, 10, 1097–1106. [DOI] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Nagai, K., Niijima, A., Yamano, T., Otani, H., Okumura, N., Tsuruoka, N., Nakai, M. & Kiso, Y. (2003). Exp. Biol. Med.228, 1138–1145. [DOI] [PubMed]
- Niijima, A., Okui, T., Matsumura, Y., Yamano, T., Tsuruoka, N., Kiso, Y. & Nagai, K. (2002). Auton Neurosci.97, 99–102. [DOI] [PubMed] [Google Scholar]
- Otani, H., Okumura, N., Okumura, A. & Nagai, K. (2005). J. Biochem.137, 167–175. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Stura, E. A. (1999). Crystallization of Nucleic Acid and Proteins: A Practical Approach, 2nd ed., edited by A. Ducruix & R. Giegé, pp. 177–208. Oxford University Press.
- Teufel, M., Saudek, V., Ledig, J. P., Bernhardt, A., Boularand, S., Carreau, A., Cairns, N. J., Carter, C., Cowley, D. J., Duverger, D., Ganzhorn, A. J., Guenet, C., Heintzelmann, B., Laucher, V., Sauvage, C. & Smirnova, T. (2003). J. Biol. Chem.278, 6521–6531. [DOI] [PubMed] [Google Scholar]
- Yamagata, Y., Ogasahara, K., Hioki, Y., Lee, S. J., Nakagawa, A., Nakamura, H., Ishida, M., Kuramitsu, S. & Yutani, K. (2001). J. Biol. Chem.276, 11062–11071. [DOI] [PubMed] [Google Scholar]
- Yamano, T., Niijima, A., Iimori, S., Tsuruoka, N., Kiso, Y. & Nagai, K. (2001). Neurosci. Lett.313, 78–82. [DOI] [PubMed] [Google Scholar]