

Proteolysis in situ by a protease secreted by a contaminating fungus is essential for the crystallization of yeast CPSF-100.
Keywords: cleavage and polyadenylation specificity factor complex, in situ proteolysis, pre-mRNA processing, in-drop proteolysis, ribonucleases, metallo-β-lactamase, artemis
Abstract
The cleavage and polyadenylation specificity factor (CPSF) complex is required for the cleavage and polyadenylation of the 3′-end of messenger RNA precursors in eukaryotes. During structural studies of the 100 kDa subunit (CPSF-100, Ydh1p) of the yeast CPSF complex, it was serendipitously discovered that a solution that is infected by a fungus (subsequently identified as Penicillium) is crucial for the crystallization of this protein. Further analyses suggest that the protein has undergone partial proteolysis during crystallization, resulting in the deletion of an internal segment of about 200 highly charged and hydrophilic residues, very likely catalyzed by a protease secreted by the fungus. With the removal of this segment, yeast CPSF-100 (Ydh1p) has greatly reduced solubility and can be crystallized in the presence of a minute amount of precipitant.
1. Introduction
Most eukaryotic messenger RNA precursors (pre-mRNAs) must undergo co-transcriptional processing in the nucleus before they can be exported to the cytoplasm and function as mRNAs (Zhao et al., 1999 ▶; Calvo & Manley, 2003 ▶; Proudfoot, 2004 ▶; Zorio & Bentley, 2004 ▶). Cleavage and polyadenylation specificity factor (CPSF) is required for both the cleavage and the polyadenylation reactions of pre-mRNA 3′-ends. CPSF contains four subunits, CPSF-30, CPSF-73, CPSF-100 and CPSF-160, which form a stable complex and can be copurified. Recent evidence suggests that CPSF-73 is the nuclease for the cleavage reaction (Daiyasu et al., 2001 ▶; Callebaut et al., 2002 ▶; Ryan et al., 2004 ▶; Mandel et al., 2006 ▶). CPSF-100 shares weak amino-acid sequence homology with CPSF-73 in the N-terminal segment, but is catalytically inactive.
Currently, there is no structural information on any of the CPSF subunits. To help understand the molecular basis of the functional roles of CPSF-73 and CPSF-100, we have begun to study the three-dimensional structures of these proteins (Mandel et al., 2006 ▶). Several problems were encountered during the crystallization and structure determination of yeast CPSF-100 (also known as Ydh1p), including a requirement for in situ partial proteolysis for crystallization, a lack of induction of selenomethionyl protein in bacteria and a lack of non-crystallographic symmetry (NCS) within individual crystal forms for NCS averaging and phase improvement. Our solutions to these problems are described in more detail in this paper, some of which may be useful for the crystallization and structure determination of other proteins. In fact, the protocol of in situ partial proteolysis with the fungal protease has led to the crystallization of two other proteins in our laboratory (data not shown). The in situ proteolysis removed a highly charged and hydrophilic segment of about 200 internal residues from yeast CPSF-100 (Ydh1p).
2. Materials and methods
2.1. Protein expression and purification
Yeast CPSF-100 (Ydh1p) was subcloned into the pET28a vector (Novagen) and overexpressed in Escherichia coli. After reaching an OD of 0.6, the cells were induced with 0.4 mM IPTG and grown for 17 h at 293 K. The soluble protein was purified by nickel-agarose affinity chromatography, TMAE anion-exchange chromatography and S-300 (GE Healthcare) gel-filtration chromatography. The protein was concentrated to 10 mg ml−1 in a buffer containing 20 mM Tris pH 8.5, 250 mM NaCl, 5% glycerol and 10 mM DTT. The recombinant protein contained an N-terminal hexa-His tag with the sequence MGSSHHHHHHSSGLVPRGSH, which was not removed for crystallization.
2.2. Protein crystallization
Crystals of yeast CPSF-100 (Ydh1p) were obtained by the hanging-drop vapor-diffusion method. Details of the crystallization procedures are described in §3.
The crystals were cryoprotected with Paratone-N or in mother liquor supplemented with 25% ethylene glycol and were flash-frozen in liquid propane for diffraction analysis and data collection at 100 K.
2.3. Data collection and processing
X-ray diffraction data were collected on an ADSC CCD at the X4A beamline of Brookhaven National Laboratory (BNL) and the diffraction images were processed and scaled with the HKL package (Otwinowski & Minor, 1997 ▶). Native data sets for three different crystal forms (I, II and III) were collected at up to 2.5 Å resolution. A single-wavelength anomalous diffraction data set for a gold derivative of crystal form II was collected at the gold absorption peak. The data-processing statistics are summarized in Table 1 ▶.
Table 1. Summary of crystallographic information.
Values in parentheses are for the highest resolution shell.
| Crystal form | I | II | II | III |
|---|---|---|---|---|
| Heavy-atom compound | — | — | KAu(CN)2 | — |
| Space group | P212121 | P212121 | P212121 | I222 |
| Unit-cell parameters | ||||
| a (Å) | 63.5 | 70.2 | 70.4 | 75.6 |
| b (Å) | 73.1 | 75.9 | 75.8 | 122.8 |
| c (Å) | 107.8 | 109.8 | 110.0 | 126.9 |
| Maximum resolution (Å) | 2.6 (2.7–2.6) | 2.9 (3.0–2.9) | 2.9 (3.0–2.9) | 2.5 (2.6–2.5) |
| No. of observations | 61446 | † | 103668 | 234576 |
| No. of reflections | 16034 | † | 13408 | 20806 |
| Completeness (%) | 100 (100) | † | 99 (97) | 100 (100) |
| Rmerge‡ (%) | 7.4 (26.2) | † | 7.2 (23.7) | 8.7 (40.8) |
| I/σ(I) | 14.7 (4.6) | † | 16.4 (4.7) | 17.3 (6.3) |
The raw data for this crystal form were unfortunately destroyed.
R
merge = 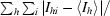
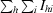 .
.
2.4. Phase improvement through NCS averaging among three crystal forms
The native and the SAD data for the gold derivative of crystal form II were loaded into the program SOLVE/RESOLVE for phase determination to 3.0 Å resolution and phase improvement (Terwilliger, 2003 ▶). The figure of merit from the SIRAS phasing was 0.28 and the Z score was 6.8. The automatic chain-tracing protocol in RESOLVE was able to place several segments of the protein into the electron-density map and also built many segments as polyalanine models. Clear secondary-structure elements (β-strands and α-helices) could be recognized for many of these segments, but the phase information was not of sufficient quality to allow complete tracing of the protein structure.
To carry out noncrystallographic symmetry (NCS) averaging, the electron density from SIRAS phasing for the molecule in crystal form II was selected using a spherical envelope and placed in a large P1 unit cell and structure factors were generated for this molecule using reciprocal-space methods (Main & Rossmann, 1966 ▶) with the locally written program Ghkl (L. Tong, unpublished work). These were then used for molecular-replacement (MR) calculations to solve the structures of crystal forms I and III with the REPLACE program package (Tong, 1993 ▶). Based on the MR solution, reflections in crystal forms I and III were phased to 3.0 Å resolution, again with the program Ghkl. The MR solution also defined the NCS operators among the molecules in the three crystal forms.
The NCS averaging among the three crystal forms was carried out using a locally written program (L. Tong, unpublished work). The mask for the protein was generated based on the model built by RESOLVE for crystal form II. The electron-density values of the three crystal forms were brought onto the same scale before they were averaged and the scaling was based on the r.m.s. values of the electron density within a sphere of 25 Å radius near the center of the molecule (Tong et al., 1992 ▶).
The phase information to 2.5 Å resolution for crystal form III after NCS averaging was input to the RESOLVE program for automated chain tracing and model building (Terwilliger, 2003 ▶). A total of 338 residues were located, 110 of which had complete side chains. The complete model was built manually with the program O (Jones et al., 1991 ▶). The refined atomic model for this crystal form was placed into crystal forms I and II with the molecular-replacement method using the program COMO (Jogl et al., 2001 ▶). The structure shows that the gold compound is coordinated by the side chain of Cys373 in crystal form II.
3. Results and discussion
3.1. Crystallization requires in situ partial proteolysis
We were able to quickly obtain a soluble bacterial expression construct for yeast CPSF-100 (Ydh1p), covering residues 1–733, and the purified protein behaved as monomers based on gel-filtration and solution light-scattering studies (data not shown). Crystallization conditions were then screened at 277 and 294 K using commercially available kits (Hampton Research, Emerald Biosystems and others; Jancarik & Kim, 1991 ▶; Cudney et al., 1994 ▶). Several conditions from the PEG/Ion screen (Hampton Research) produced small needle-shaped crystals.
However, when we tried to reproduce these initial hits using homemade solutions, we invariably failed. We examined the solutions in the commercial kit that were used to produce the original hits and found that they all appeared to have some particulate matter in them. These solutions had been used in the laboratory for more than 18 months. Noting that the precipitation may have changed the composition of the solutions, we redesigned the crystallization grid using homemade solutions but still failed to reproduce the crystals.
Next, we only used the solution from the commercial kit to optimize the crystallization conditions. The original solution from the kit contained 20% PEG 3350 and 0.2 M ammonium citrate. We mixed the protein solution with this solution and equilibrated the mixture over a reservoir of homemade solution. To vary the composition of the drop, we screened various dilutions of the solution from the commercial kit (from no dilution up to 1:10 dilution with water). We also varied the amount of PEG and ammonium citrate in the reservoir in the optimization. Using this protocol, we were able to produce crystals of sufficient size for X-ray diffraction analysis and data collection (Fig. 1 ▶).
Figure 1.

Crystals of yeast CPSF-100 (Ydh1p).
Once the unit-cell parameters had been characterized for these crystals (crystal form I, Table 1 ▶), it was clear that the unit cell was too small to accommodate even one molecule of the CPSF-100 molecule. The calculated V M would be 1.5 Å3 Da−1 and the solvent content would be 18%. This was inconsistent with the diffraction quality of the crystal, which extended only to about 3 Å resolution, and suggested that some proteolysis was very likely to have happened during the crystallization process.
With this knowledge, we examined the crystallization solution from the commercial kit under a microscope and realised that it may be contaminated with fungal growth. This immediately suggested a source for the protease. The fungus may have secreted the enzyme into the solution and we could only produce crystals with this solution as in situ partial proteolysis is required for the crystallization of yeast CPSF-100 (Ydh1p). In an attempt to obtain experimental evidence for the protease, we concentrated the fungal solution up to 100-fold and examined the resulting solution by SDS–PAGE. Unfortunately, we were not able to observe any protein after staining, suggesting that the fungal protease must be present at very low concentrations. By sequencing a small segment of the genome of this fungus, we have identified it as Penicillium. This is a very common fungus/mold; remarkably, the same one that led to the discovery of penicillin by Sir Alexander Fleming in 1928.
Further experiments showed that the protein had very low solubility after the partial proteolysis. Crystals could be obtained by mixing the protein solution and the fungal solution diluted fourfold in water and equilibrating the mixture over a reservoir of pure water. The resulting drop contained only about 2% PEG 3350 as the precipitant, which was apparently sufficient to produce crystals of the protein. This suggests that the fungal protease may be removing segments of yeast CPSF-100 (Ydh1p) that greatly enhance its solubility (see below).
3.2. The partial proteolysis produced three fragments
To obtain direct evidence for the partial proteolysis by the fungal solution, we incubated the protein with this solution and took samples at various time points. Because of the presence of PEG 3350 in the fungal solution, there was a significant amount of precipitation during this reaction. Nonetheless, examination of the soluble material by SDS–PAGE clearly showed that CPSF-100 has been proteolyzed (Fig. 2 ▶). Three distinct bands were observed, with molecular weights of about 45, 25 and 10 kDa. The total molecular weight of these three bands (80 kDa) is about the same as that of the original protein (82 kDa). These three bands can be explained based on the structure of this protein (see below), which suggests that the 25 kDa segment is missing in the structure.
Figure 2.

Evidence for proteolysis of yeast CPSF-100 (Ydh1p) by the fungal solution. The protein was incubated with the fungal solution and samples were taken at various time points. The resulting protein species was separated by SDS–PAGE and stained with Coomassie Blue. The three major species are indicated by the arrowheads and their molecular weights (and possible residue range for the fragment) are labeled.
Once we understood the mechanism for the crystallization of yeast CPSF-100 (Ydh1p), we tried to replace the fungal protease with known proteases such as trypsin, subtilisin and chymotrypsin in the crystallization solution. We were able to produce crystals of the protein by including a minute amount (1:1000 weight-to-weight ratio) of trypsin or subtilisin in the crystallization solution. Unfortunately, the crystals were highly mosaic and not suitable for structural studies and these experiments were not pursued further. This protocol may be useful with other protein samples.
With the successful crystallization of the initial expression construct containing residues 1–733, we made additional constructs that sampled the N- and C-terminal positions near this construct. This led to a new construct, covering residues 1–720, that produced more soluble protein in E. coli and could also be crystallized more readily. The crystals were grown by the hanging-drop vapor-diffusion method at 294 K. The fungal solution was diluted 500-fold into the protein solution and 1 µl of this mixture was combined with 1 µl of reservoir solution containing 18%(w/v) PEG 3350 and 0.3 M ammonium citrate. All the data reported here for crystal forms II and III were obtained using this new construct.
3.3. Identification of one heavy-atom derivative with a single site
We would have liked to solve the structure by the selenomethionyl anomalous diffraction method (Hendrickson, 1991 ▶). This segment of CPSF-100 has a normal abundance of methionines (ten residues or about 1.4%). However, the selenomethionyl protein is not induced in bacteria using either the methionine-auxotroph strains in defined LeMaster media (Hendrickson et al., 1990 ▶) or using conventional E. coli strains and blocking endogenous methionine biosynthesis (Doublié et al., 1996 ▶).
We then turned to the isomorphous replacement method to solve the structure. The search for heavy-atom derivatives was complicated by the fact that the native crystals displayed a significant degree of non-isomorphism, even though they were crystallized under similar conditions and had the same morphology. In fact, more than five different crystal forms have been observed, three of which (crystal forms I, II and III) were of sufficient quality for data collection (Table 1 ▶).
From the X-ray diffraction data, we were able to locate the position of a heavy atom for the KAu(CN)2 compound (soaked for 14 h at 0.5 mM concentration) based on the isomorphous difference Patterson map, but the occupancy of the compound was low. To increase the occupancy, we soaked another crystal at 5 mM KAu(CN)2 for 14 h and collected a diffraction data set at the gold absorption peak at the X4A beamline of BNL. Both the isomorphous and the anomalous difference Patterson maps based on this new data set showed a single strong Au site in the derivative.
We also tried to solve the structure using anomalous diffraction from soaked or cocrystallized halide ions (Dauter et al., 2001 ▶; Nagem et al., 2003 ▶). The highest concentration of NaBr that could be used for soaking without destroying the crystal was only 0.2 M. We then tried to crystallize the protein in the presence of NaBr and found that the highest concentration we could use was 0.25 M. A new crystal form (form III) of the protein was obtained in the presence of NaBr, grown at 277 K, which diffracted to 2.5 Å resolution (Table 1 ▶). Unfortunately, we could not locate any Br sites based on the SAD data.
3.4. Phase improvement through NCS averaging among three crystal forms
Noncrystallographic symmetry (NCS) averaging is a powerful method for improving phase information (Rossmann, 1990 ▶). Unfortunately, all our crystals contain only one molecule in the crystallographic asymmetric unit. However, the presence of three different crystal forms of the protein (Table 1 ▶) allowed us to carry out NCS averaging among them. This produced about 40° average phase shift (60° r.m.s.) for reflections of all three crystal forms and moderate improvement (from 0.8 to 0.9) in the correlation coefficient between the observed structure-factor amplitudes and those calculated from back-transformation of the electron-density map. NCS averaging also allowed the phase information to be extended to the limits of diffraction data for the three crystal forms. Most importantly, the quality of the electron-density map, especially for side chains, was improved by this NCS averaging process (Figs. 3 ▶ a and 3 ▶ b).
Figure 3.

Representative electron-density map for yeast CPSF-100 (Ydh1p). (a) Electron-density map for residues 180–183 and 668–676 after SIRAS phasing at 3.0 Å resolution. (b) Electron-density map for the same residues after NCS averaging among the three crystal forms. The maps were weighted by the figure of merit from phasing or NCS averaging and contoured at 1σ. This figure was produced with the program SETOR (Evans, 1993 ▶).
3.5. A highly hydrophilic segment of 200 residues was removed by the in situ proteolysis
The structures for the three crystal forms show that residues 423–625 of the protein are missing. These residues could be disordered in structure, but it is more likely that these residues were removed by the fungal protease during the crystallization process.
Examination of the amino-acid sequence of this segment of yeast CPSF-100 (Ydh1p) shows that it is highly hydrophilic, with 75 charged residues (Asp, Glu, Lys or Arg) and 45 hydrophilic residues (Asn, Gln, His, Ser or Thr) (Fig. 4 ▶ a). Therefore, more than 60% of the residues in this segment are hydrophilic and it should be highly flexible in structure and highly susceptible to proteolysis.
Figure 4.

A highly hydrophilic segment in yeast CPSF-100 (Ydh1p). (a) Residues 401–650 of yeast CPSF-100 (Ydh1p) are highly hydrophilic. Negatively charged residues (Asp and Glu) are shown in red, positively charged residues (Lys and Arg) in blue and hydrophilic residues (Ser, Thr, Asn, Gln, and His) in magenta. (b). Schematic drawing of the domain architecture of yeast CPSF-100 (Ydh1p). The metallo-β-lactamase domain is shown in cyan, the β-CASP domain in yellow (Callebaut et al., 2002 ▶; Mandel et al., 2006 ▶) and the hydrophilic segment in gray.
Our structural observations are consistent with the studies on the proteolysis of yeast CPSF-100 (Ydh1p) by the fungal solution (Fig. 2 ▶). The bands at 45 and 10 kDa may correspond to residues 1–420 and 631–720, respectively (Fig. 4 ▶ b). Interestingly, we also observed a band at 25 kDa (Fig. 2 ▶), which may correspond to residues 421–630. Several additional bands of lower molecular weight in this region suggest that the fungal protease may cut at several places in this segment. The V M value for the three crystal forms excluding this and other disordered segments in the protein is about 2.5 Å3 Da−1, which is well within the expected range (Matthews, 1968 ▶).
4. Conclusions
Partial proteolysis to remove flexible segments is an important component of the crystallization of many protein samples. In most instances, the proteolysis is carried out separately from the crystallization experiments. The domain of the protein that is stable to proteolysis is identified first; for example, by N-terminal sequencing or mass spectrometry (MS). Recently, hydrogen/deuterium exchange followed by MS analysis has been found to be a useful method for identifying stable protein cores (Hamuro et al., 2003 ▶). New expression constructs are then engineered based on this information. Alternatively, preparative-scale proteolysis can be carried out and the modified protein sample is purified before crystallization experiments.
In our structural studies of yeast CPSF-100 (Ydh1p), we discovered serendipitously that a fungus-infected solution is required for the crystallization of this protein. Our additional experiments showed that the fungal solution may contain a protease that removed a highly hydrophilic segment of the protein (Fig. 4 ▶ b). This in situ proteolysis has rarely been used in protein crystallization. Our studies suggest that this protocol may be crucial for the crystallization of selected proteins. In fact, there are recent reports of successful crystallization of other proteins by in situ proteolysis with subtilisin as the enzyme (Johnson et al., 2006 ▶; Gaur et al., 2004 ▶; Taneja et al., 2006 ▶). In our laboratory, in situ proteolysis with subtilisin or the fungal protease was crucial for the crystallization of a domain of the 77 kDa subunit of the cleavage stimulation factor (CstF-77), another factor that is important for pre-mRNA 3′-end processing (Y. Bai, T. Auperin & L. Tong, manuscript in preparation).
This internal highly hydrophilic segment may function as a ‘solubility tag’ by greatly enhancing the solubility of the protein. Once it has been removed by proteolysis, the solubility of the protein decreases dramatically and the modified protein can be crystallized in the presence of minute amounts of precipitant (2% PEG 3350). This also suggests that if an expression construct for CPSF-100 is engineered that is missing this segment, the resulting protein is likely to have low solubility in bacteria and we probably would not be able to produce a sufficient amount of protein sample for structural studies. Removal of internal flexible segments by protein engineering is crucial for the crystallization of many other proteins and some recent examples include the SR protein kinase Sky1p (Nolen et al., 2001 ▶), the nuclear cap binding complex (Mazza et al., 2002 ▶) and the pre-mRNA splicing factor U2AF (Sickmier et al., 2006 ▶).
Acknowledgments
We thank Randy Abramowitz, John Schwanof and Xiaochun Yang for setting up the X4A beamline at the NSLS, Javed Khan and Yang Shen for help with data collection at the synchrotron and Benjamin Tweel for identifying the fungus.
References
- Callebaut, I., Moshous, D., Mornon, J.-P. & de Villartay, J.-P. (2002). Nucleic Acids Res.30, 3592–3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo, O. & Manley, J. L. (2003). Genes Dev.17, 1321–1327. [DOI] [PubMed] [Google Scholar]
- Cudney, R., Patel, S., Weisgraber, K., Newhouse, Y. & McPherson, A. (1994). Acta Cryst. D50, 414–423. [DOI] [PubMed] [Google Scholar]
- Daiyasu, H., Osaka, K., Ishino, Y. & Toh, H. (2001). FEBS Lett.503, 1–6. [DOI] [PubMed] [Google Scholar]
- Dauter, Z., Li, M. & Wlodawer, A. (2001). Acta Cryst. D57, 239–249. [DOI] [PubMed] [Google Scholar]
- Doublié, S., Kapp, U., Aberg, A., Brown, K., Strub, K. & Cusack, S. (1996). FEBS Lett.384, 219–221. [DOI] [PubMed] [Google Scholar]
- Evans, S. V. (1993). J. Mol. Graph.11, 134–138. [DOI] [PubMed] [Google Scholar]
- Gaur, R. K., Kupper, M. B., Fischer, R. & Hoffmann, K. M. V. (2004). Acta Cryst. D60, 965–967. [DOI] [PubMed] [Google Scholar]
- Hamuro, Y., Coales, S. J., Southern, M. R., Nemeth-Cawley, J. F., Stranz, D. D. & Griffin, P. R. (2003). J. Biomol. Tech.14, 171–182. [PMC free article] [PubMed] [Google Scholar]
- Hendrickson, W. A. (1991). Science, 254, 51–58. [DOI] [PubMed] [Google Scholar]
- Hendrickson, W. A., Horton, J. R. & LeMaster, D. M. (1990). EMBO J.9, 1665–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst.24, 409–411. [Google Scholar]
- Jogl, G., Tao, X., Xu, Y. & Tong, L. (2001). Acta Cryst. D57, 1127–1134. [DOI] [PubMed] [Google Scholar]
- Johnson, S., Roversi, P., Espina, M., Deane, J. E., Birket, S., Picking, W. D., Blocker, A., Picking, W. L. & Lea, S. M. (2006). Acta Cryst. F62, 865–868. [DOI] [PMC free article] [PubMed]
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed] [Google Scholar]
- Main, P. & Rossmann, M. G. (1966). Acta Cryst.21, 67–72. [Google Scholar]
- Mandel, C. R., Kaneko, S., Zhang, H., Gebauer, D., Vethantham, V., Manley, J. L. & Tong, L. (2006). Submitted. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Mazza, C., Segref, A., Mattaj, I. W. & Cusack, S. (2002). Acta Cryst. D58, 2194–2197. [DOI] [PubMed] [Google Scholar]
- Nagem, R. A. P., Polikarpov, I. & Dauter, Z. (2003). Methods Enzymol.374, 120–137. [DOI] [PubMed] [Google Scholar]
- Nolen, B., Yun, C. Y., Wong, C. F., McCammon, J. A., Fu, X.-D. & Ghosh, G. (2001). Nature Struct. Biol.8, 176–183. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Method Enzymol.276, 307–326. [DOI] [PubMed]
- Proudfoot, N. J. (2004). Curr. Opin. Cell Biol.16, 272–278. [DOI] [PubMed] [Google Scholar]
- Rossmann, M. G. (1990). Acta Cryst. A46, 73–82. [DOI] [PubMed] [Google Scholar]
- Ryan, K., Calvo, O. & Manley, J. L. (2004). RNA, 10, 565–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickmier, E. A., Frato, K. E. & Kielkopf, C. L. (2006). Acta Cryst. F62, 457–459. [DOI] [PMC free article] [PubMed]
- Taneja, B., Patel, A., Slesarev, A. & Mondragon, A. (2006). EMBO J.25, 398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger, T. C. (2003). Methods Enzymol.374, 22–37. [DOI] [PubMed] [Google Scholar]
- Tong, L. (1993). J. Appl. Cryst.26, 748–751. [Google Scholar]
- Tong, L., Choi, H.-K., Minor, W. & Rossmann, M. G. (1992). Acta Cryst. A48, 430–442. [DOI] [PubMed] [Google Scholar]
- Zhao, J., Hyman, L. & Moore, C. L. (1999). Microbiol. Mol. Biol. Rev.63, 405–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorio, D. A. R. & Bentley, D. (2004). Exp. Cell Res.296, 91–97. [DOI] [PubMed] [Google Scholar]