

E. coli bacterioferritin was crystallized in a novel crystal form from different conditions and the structure was solved. The crystals belonged to space group P213 and diffracted to a resolution of 2.5 Å.
Keywords: Escherichia coli bacterioferritin, iron storage and homeostasis, ferroxidase, merohedral twinning
Abstract
Escherichia coli bacterioferritin was serendipitously crystallized in a novel cubic crystal form and its structure could be determined to 2.5 Å resolution despite a high degree of merohedral twinning. This is the first report of crystallographic data on ‘as-isolated’ E. coli bacterioferritin. The ferroxidase active site contains positive difference density consistent with two metal ions that had co-purified with the protein. X-ray fluorescence studies suggest that the metal composition is different from that of previous structures and is a mix of zinc and native iron ions. The ferroxidase-centre configuration displays a similar flexibility as previously noted for other bacterioferritins.
1. Introduction
Bacterioferritins are bacterial iron-storage proteins that play an important role in the bacteria’s ability to utilize this element. They are hollow protein shells made up of 24 subunits and are members of the broad ferritin family. The iron is stored within the large cavity inside the protein shell as a mineral core of hydrated ferric oxide and phosphate. (Bacterio)ferritins generate the ferric form of iron by catalysing the oxidation of soluble FeII into its insoluble FeIII form by molecular oxygen. The so-called ferroxidase centre, a dinuclear iron site, plays an important role in this iron-oxidation and mineralization process, although the mechanistic details of its involvement may differ between ferritins and bacterioferritins (Lewin et al., 2005 ▶). Pores in the protein-shell subunit interfaces have been identified as potential routes of ion flow to and from the cavity, but the mechanisms of specific iron uptake and release in (bacterio)ferritins remain disputed. Even more obscure is the way the cell can regulate the accumulation and utilization of its iron (Andrews et al., 2003 ▶) and there may well be differences in regulatory control between different organisms.
Escherichia coli bacterioferritin was the first bacterioferritin to have its crystal structure determined (Frolow et al., 1994 ▶; Dautant et al., 1998 ▶). This structure showed that each four-helix bundle subunit has one catalytic ferroxidase centre consisting of a dinuclear metal site, which contains two Fe atoms in vivo (Lewin et al., 2005 ▶). In addition, bacterioferritins contain a haem which is sandwiched between two subunits related by twofold symmetry (Carrondo, 2003 ▶). Its function is not clear, but its reduction potential could allow it to play a mediating role in electron transfer during reduction and release of iron (Andrews et al., 2003 ▶). In vivo, bacterioferritin may therefore contain iron at three distinct locations: haem iron, iron from the ferroxidase centre and iron from the mineral core.
Mainly based on kinetic studies with E. coli bacterioferritin, a three-stage model was proposed for iron uptake in which the ferroxidase site plays a key role: (i) binding of two iron(II) ions per ferroxidase centre, (ii) rapid oxidation of these iron(II) ions to iron(III) by dioxygen and (iii) ferroxidase-dependent core formation (Lewin et al., 2005 ▶). However, the exact mechanism by which core formation depends on the ferroxidase site is not clear (Carrondo, 2003 ▶; Lewin et al., 2005 ▶). Two models have been proposed, one in which iron(III) ions transit from the ferroxidase centre into the inner cavity where they initiate or sustain core formation (Carrondo, 2003 ▶) and an alternative model in which iron(II) ions inside the cavity are oxidized and nucleated, coupled with the simultaneous reduction of iron(III) at the ferroxidase centre (Lewin et al., 2005 ▶). However, comparison of these models with the structure of E. coli bacterioferritin is confounded by the lack of crystallographic data on the iron-bound state of the ferroxidase site. All crystal forms of this bacterioferritin that have so far been obtained contain MnII from the crystallization solution (Frolow et al., 1994 ▶; Dautant et al., 1998 ▶). The different oxidation states of the ferroxidase centre have been characterized by X-ray crystallography in the bacterioferritins of the anaerobic sulfate-reducing bacterium Desulfovibrio desulfuricans (Macedo et al., 2003 ▶) and the nitrogen-fixing bacterium Azotobacter vinelandii (Swartz et al., 2006 ▶). These showed structural differences around the ferroxidase centre, which have generated some controversy about the mechanism by which ferroxidase activity is coupled to core formation.
Here, we report the serendipitous discovery of crystals of an at first unknown protein and the methods employed to simultaneously determine both its identity as E. coli bacterioferritin as well as its crystal structure. These crystals of ‘as-isolated’ E. coli bacterioferritin have a new crystal form. X-ray fluorescence studies have been performed to analyse the metal-ion content of this bacterioferritin.
2. Materials and methods
Crystal screens were performed with a recombinant Sulfolobus acidocaldarius 2-keto-3-deoxygluconate aldolase preparation purified from an E. coli BL21(DE3) culture grown in Luria–Bertani medium. The details of the expression, purification and structure determination of this aldolase will be published elsewhere (Wolterink-van Loo et al., manuscript in preparation). Briefly, crystals were obtained at 291 K using the hanging-drop vapour-diffusion technique, in which 1 µl of protein solution containing 18.9 mg ml−1 aldolase and an unknown but low amount of impurities was mixed with 1 µl reservoir solution containing 0.1 M HEPES pH 7.5, 30%(v/v) PEG 400 and 0.2 M MgCl2.
SDS–PAGE samples of crystals were prepared by washing the crystals with reservoir solution and then dissolving them in SDS–PAGE loading buffer. The samples were then applied onto a 20% Phast gel (Amersham Pharmacia Biotech). Silver staining was performed with a modification of the recommended protocol (Amersham Pharmacia Biotech) in which the silver nitrate concentration was doubled to 0.5% and the gel was stained twice.
X-ray diffraction data were collected at beamline ID14-2 (ESRF, France). Data (Table 1 ▶) were processed using MOSFLM (Leslie, 1992 ▶) and programs from the CCP4 package (Collaborative Computational Project, Number 4, 1994 ▶). Twinning was analysed with the program DETWIN (Collaborative Computational Project, Number 4, 1994 ▶). Molecular replacement was performed with Phaser (McCoy et al., 2005 ▶) using two subunits of E. coli bacterioferritin (PDB code 1bcf) as a search model. The data were subsequently transformed into ideally twinned data by averaging the twin-related intensities and an electron-density map was calculated with CNS (Brünger et al., 1998 ▶) using the scripts for perfectly twinned data. To allow cross-validation of the model despite the twinning and eightfold noncrystallographic symmetry (NCS), test-set reflections were selected in thin resolution shells. Model bias was removed using the simulated-annealing protocol in CNS (Brünger et al., 1998 ▶) and minor model rebuilding and placement of metal ions was performed in Coot (Emsley & Cowtan, 2004 ▶). The data were subsequently detwinned using the appropriate CNS scripts to allow final refinement with a maximum-likelihood target function, which was performed with REFMAC5 (Murshudov et al., 1999 ▶). Eightfold NCS restraints were applied on the bacterioferritin polypeptide chains throughout refinement; however, no NCS restraints were applied to the residues making up the ferroxidase sites.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Space group | P213 |
| Unit-cell parameter (Å) | 167.9 |
| Resolution (Å) | 60–2.5 (2.64–2.50) |
| Wavelength (Å) | 0.934 |
| Completeness (%) | 99.8 (98.7) |
| Redundancy | 7.5 (5.4) |
| Unique reflections | 54566 (7807) |
| I/σ(I) | 18.0 (4.4) |
| Rsym† (%) | 11 (34) |
| Refinement | |
| No. of atoms | |
| Protein | 10392 |
| Haem | 340 |
| Ferroxidase-site metals | 16 |
| Water | 98 |
| R/Rfree‡ (%) | 17.1/18.8 |
| Average B factor (Å2) | 23 |
| R.m.s. deviations | |
| Bonds (Å) | 0.009 |
| Angles (°) | 1.2 |
| Ramachandran plot§ (%) | |
| Favoured | 98.7 |
| Allowed | 0.7 |
| Outliers | 0.6 |
R
sym = 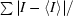
 , where I is the observed intensity and 〈I〉 is the average intensity.
, where I is the observed intensity and 〈I〉 is the average intensity.
R = 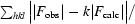
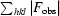 × 100, where F
obs is the observed structure factor and F
calc is the calculated structure factor. R
free is R calculated with 10% of the data in thin resolution shells that were omitted from the refinement.
× 100, where F
obs is the observed structure factor and F
calc is the calculated structure factor. R
free is R calculated with 10% of the data in thin resolution shells that were omitted from the refinement.
According to values from Lovell et al. (2003 ▶). The outliers are Ile79 of each chain.
The X-ray absorption edges of Mn, Fe, Cu and Zn were scanned at beamline BM16 (ESRF, France) and measured with a Roentec Xflash fluorescence detector using a narrow X-ray bandpass.
3. Results and discussion
3.1. Structure determination
During crystallization experiments on 2-keto-3-deoxygluconate aldolase from S. acidocaldarius (Wolterink-van Loo et al., manuscript in preparation), small cubic crystals with a bright red colour were observed with typical dimensions of 80 × 50 × 30 µm, appearing several weeks after the much larger aldolase crystals had formed (Fig. 1 ▶ a). One of them was suitable for X-ray diffraction analysis and diffracted to better than 2.5 Å using synchrotron radiation (Table 1 ▶). The data were initially scaled in the P432 point group; however, subsequent examination of the intensity statistics revealed the possibility that this crystal was merohedrally twinned with a high twinning fraction. The high symmetry of the P432 point group does not permit merohedral twinning. Therefore, the true space group would then have the lower P23 symmetry and the twinning operator (k, −h, l) would generate the additional apparent symmetry. Initial molecular-replacement attempts with the aldolase structure (33 kDa; Wolterink-van Loo et al., manuscript in preparation) failed. Moreover, SDS–PAGE analysis of dissolved crystals revealed that the crystals were made up of a ∼20 kDa protein (Fig. 1 ▶ b). This suggested that the crystals were not formed of the aldolase. Instead, an unknown red-coloured contaminating protein from the E. coli expression host appeared to have crystallized, even though the aldolase preparation had been highly purified (Wolterink-van Loo et al., manuscript in preparation).
Figure 1.

(a) A cubic crystal of E. coli bacterioferritin (red) growing between large hexagonal aldolase crystals; (b) silver-stained SDS–PAGE gel of washed crystals. Lane 1, contents of red cubic crystal; lane 2, contents of hexagonal aldolase crystal. Molecular-weight standards (in kDa) are indicated.
Before attempting to establish the identity of this protein by mass spectrometry, the Protein Data Bank was screened for E. coli protein structures of the approximate size of the unknown protein with a haem prosthetic group, the presence of which was suggested by the bright red colour of the crystals. E. coli bacterioferritin matched all of these criteria, since it is a 18.5 kDa protein, contains haem groups and in addition it is normally found as a 24-mer with 432 point symmetry (Frolow et al., 1994 ▶; Dautant et al., 1998 ▶). Therefore, it was selected for use in molecular replacement and the basic building block of the bacterioferritin sphere, consisting of a haem group sandwiched by two identical 18.5 kDa polypeptide chains, was used as a search model. Indeed, it was possible to obtain a high-scoring solution in space group P213. Despite the twinning, four of these dimers could be located in one of the two twin domains and inspection of the resulting electron-density maps revealed a very good fit of the model. The data were subsequently detwinned and the model was refined to acceptable R factors (Table 1 ▶). The good fit to the electron density over the entire sequence and the low R factors firmly establish that these crystals are formed of E. coli bacterioferritin.
3.2. The overall structure of bacterioferritin
E. coli bacterioferritin has previously been reported to form diffraction-quality crystals in four different crystal forms (Dautant et al., 1998 ▶). Here, bacterioferritin was identified in an additional cubic crystal form with different crystal packing. In this crystal form, eight of the polypeptide subunits of the 24-mer bacterioferritin shell (Fig. 2 ▶) were found in the asymmetric unit; they show a high overall similarity to the structures in space groups P21 and P42212 for which the coordinates are publicly available (Dautant et al., 1998 ▶; Frolow et al., 1994 ▶), as illustrated by the small r.m.s.d. of 0.4 Å between the 158 Cα atoms of a subunit. No clearly preferred orientation of the asymmetrical haem b in its symmetrical binding site is apparent, as there were no clear density differences between the two possible locations of the vinyl substituents. To represent this ambiguity, the two possible orientations were modelled with half occupancy, analogous to what has been found in the crystal structures of the Rhodobacter capsulatus, D. desulfuricans and A. vinelandii bacterioferritins (Cobessi et al., 2002 ▶; Macedo et al., 2003 ▶; Swartz et al., 2006 ▶), but in contrast to the high-resolution structures of the E. coli bacterioferritin in its tetragonal crystal form, where a certain preference for one orientation was observed (Frolow & Kalb, 2001 ▶). The bright red colour of the crystals (Fig. 1 ▶ a) had already indicated the presence of this haem and also suggests the absence of an elaborate mineral core of ferric oxide, which would have rendered the crystal darker.
Figure 2.

Overall structure of E. coli bacterioferritin. (a) Cartoon representation of the eight subunits found in the asymmetric unit. Additional subunits generating the entire protein shell are indicated as traces. (b) The bacterioferritin dimeric building block, consisting of two four-helix bundles. The short capping helices, which are involved in intersubunit contacts, are rendered in a different colour. The haem group is in stick representation and spheres indicate the metal positions of the ferroxidase centre. Figs. 2 ▶ and 3 ▶ were generated with PyMOL (DeLano Scientific; http://pymol.sourceforge.net/).
3.3. The ferroxidase centre
A more significant difference is present in the ferroxidase centre. Initial electron-density maps calculated with phases from the previously solved E. coli bacterioferritin model with the Mn ions removed showed the presence of positive difference density, characteristic of two transition metal ions in the previously identified FE1 and FE2 sites (Fig. 3 ▶ a). No Mn or other transition metals were added to the buffers used for purification or crystallization (Wolterink-van Loo et al., manuscript in preparation) and whereas divalent magnesium ions are present during crystallization, they could not account for the observed density levels. The coordination of the metal ions is quite similar to the MnII coordination in the previously determined structures of E. coli bacterioferritin (Frolow & Kalb, 2001 ▶; Dautant et al., 1998 ▶; Frolow et al., 1994 ▶). Glu18 and His54 act as ligands for site FE1, while Glu94 and His130 are ligands for site FE2 and the two glutamates Glu51 and Glu127 are bridging ligands (Figs. 3 ▶ a and 3 ▶ b). The largest difference in coordination involves Glu18, which donates both of its carboxylate O atoms as ligands to site FE1 in this structure, whereas in the Mn-occupied structures it acts as a monodentate ligand. As a result, the metal in site FE1 has five protein-derived ligands, whereas the metal occupying site FE2 has only four. Although the positions of atoms forming the ferroxidase sites were refined without applying NCS restraints, they do not show dramatic differences between the eight crystallographically independent molecules. Some residual density at ∼2.4 Å from the FE2 site is present in four out of eight of the independent ferroxidase sites in the asymmetric unit, which probably indicates an additional water ligand. A distance of 3.76 Å (range 3.53–3.92 Å) separates the two ions on average, which is clearly shorter than the average 4.0–4.3 Å Mn–Mn distances in previous structures (Dautant et al., 1998 ▶; Frolow et al., 1994 ▶), also supporting a different identity for these two ions.
Figure 3.

The ferroxidase centre. (a) Stereo figures of σA-weighted F o − F c density in the ferroxidase site contoured at 5σ, (b) superposition of the ferroxidase centre of the P213 structure (grey/orange) and the Mn-bound structure (yellow/purple; PDB code 1bcf; Frolow et al., 1994 ▶) of E. coli bacterioferritin and (c) superposition of the FE2 site environment in the P213 E. coli bacterioferritin structure (grey) with the corresponding region in the structure of reduced A. vinelandii bacterioferritin (pale green; PDB code 1fkz; Swartz et al., 2006 ▶). The location of the inner cavity (IC) is indicated. This view is rotated ∼90° with respect to the previous views.
To ascertain the identity of the metal ions in the structure, X-ray fluorescence spectra were recorded on a bacterioferritin crystal around the iron, manganese, copper and zinc K absorption edges (Fig. 4 ▶). These spectra indicated the presence of iron and also zinc, whereas manganese and copper were absent. Diffraction data collected around the Zn and Fe K edges would have unambiguously determined the specific location of the different metal ions at the centre, but none of the ∼25 crystals we tested were of sufficient quality to allow measurement of anomalous differences. Nevertheless, the X-ray fluorescence spectra do give some indication of the relative amount of metal ions present. The fluorescence signal, corrected for beam intensity, was weaker for zinc than for iron, even though zinc has a close to twofold larger fluorescence intensity than iron (Shi et al., 2005 ▶). Cautious estimation would then suggest the presence of more than twice as much iron than zinc in the bacterioferritin structure. Since the ferroxidase centre of E. coli bacterioferritin can bind zinc with higher affinity than iron (Baaghil et al., 2003 ▶; Le Brun et al., 1995 ▶), the zinc that was detected probably partly substituted iron at the ferroxidase sites. In agreement with this, the iron X-ray fluorescence spectrum has an irregular shape, indicating the presence of iron in multiple chemically different environments. This suggests that the signal of iron is not only derived from the haem b, but also from iron at the ferroxidase centre sites and perhaps from some ferric ions bound in the inner cavity that remained undetected in the crystallographic analysis.
Figure 4.

X-ray fluorescence scans around the K edges of Mn, Fe, Cu and Zn, as indicated. Plots are corrected for beam intensity and fluorescence is on an arbitrary scale.
The diffraction data were then re-examined with the knowledge that the ferroxidase FE1 and FE2 sites may be occupied by zinc and/or iron. The metal ions in the centre were modelled as either zinc or iron, but refinement did not lead to significant differences in coordination geometry, electron-density features or R factors. This is not entirely surprising since ZnII closely mimics FeII binding and has been used as a model for FeII binding to similar dinuclear sites in E. coli ferritin A (Stillman et al., 2001 ▶, 2003 ▶). The coordination arrangements of the FE1 and FE2 sites (Fig. 3 ▶ a) have been observed for both iron as well as zinc coordination in protein and small-molecule structures (Harding, 2000 ▶, 2006 ▶). The average distance between a zinc and a monodentate glutamate ligand is 1.99 Å, for example, which is quite comparable to the corresponding value for iron (2.03 Å). This structural similarity between zinc and iron therefore does not allow distinction between partial or complete substitution of iron for zinc, but it follows that the present configuration should resemble the native iron-bound state.
3.4. Comparison with ferroxidase centres from other bacterioferritins
Recent structural studies on D. desulfuricans and A. vinelandii bacterioferritins have suggested that redox-dependent conformational changes occur at the ferroxidase centre, consistent with transfer of iron from the ferroxidase centre to the interior (Macedo et al., 2003 ▶; Swartz et al., 2006 ▶). The occupancy of the metal sites and the flexibility at and around the ferroxidase centre in the current E. coli bacterioferritin structure were therefore analysed. All independent subunits display density for site FE1 of a level comparable to the density of the Fe atoms in the haem groups, indicating full occupancy, whereas the FE2 site is only partly occupied (estimated at 75%). This situation is very similar to the two independently determined homologous A. vinelandii bacterioferritin structures (66% sequence identity, average r.m.s.d. of 0.5 Å between 155 matching subunit Cα atoms), where occupancy levels of FE2 were estimated at 50 and 80% (reduced state), respectively (Liu et al., 2004 ▶; Swartz et al., 2006 ▶). Furthermore, the FE2 site of E. coli bacterioferritin was reported to have a lower affinity for MnII than the FE1 site (Frolow & Kalb, 2001 ▶). In all of these structures, the FE2 site therefore appears to have somewhat reduced occupancy levels.
The lower affinity for MnII of the FE2 site in E. coli bacterioferritin was attributed to the availability of an alternative conformation for the His130 side chain (Frolow & Kalb, 2001 ▶). Also, the present structure, obtained in the absence of Mn and from different conditions, indicates some flexibility of His130 and in addition of Glu94 and the nearby Glu47 and Tyr25, as indicated by their comparatively high B factors and electron-density features. Significantly, Glu47 and Tyr25 in the E. coli bacterioferritin structures have conformations that differ from the corresponding residues in the A. vinelandii bacterioferritin structures (Fig. 3 ▶ c). In E. coli bacterioferritin the Tyr25 ring is rotated by about 70°, while maintaining a hydrogen bond with Glu94 as in the A. vinelandii bacterioferritin structures, whereas the Glu47 carboxylate group points towards Tyr25 and makes an additional hydrogen bond. Moreover, the oxidized and reduced states of the A. vinelandii bacterioferritin display conformational variability for the His130, Glu94 and Glu47 side chains (Swartz et al., 2006 ▶). Given the high structural conservation between the ferroxidase centres and their environment between A. vinelandii and E. coli bacterioferritins, the conformations observed in the various A. vinelandii bacterioferritin structures for the side chains of His130, Glu47, Glu94 and Tyr25 may also be accessible for E. coli bacterioferritin (Fig. 3 ▶ c).
Such conformational changes could point to an iron-transfer mechanism from site FE2 towards the inner cavity (Swartz et al., 2006 ▶). However, the details of how such a mechanism would take place are not clear. The observed flexibility and conformational changes per se do not change the fact that the FE2 sites in E. coli and A. vinelandii bacterioferritins remain favourable binding sites for iron in either of its oxidation states. High-affinity binding of FeIII to the FE2 site is also consistent with spectroscopic data on reconstituted iron-loaded E. coli bacterioferritin. These suggested that the ferroxidase centre of bacterioferritins is stable and FeIII ions do not leave the ferroxidase centre upon oxidation (Yang et al., 2000 ▶). Indeed, cycling between the reduced and oxidized states of A. vinelandii bacterioferritin did not result in emptying of one or both sites of the ferroxidase centre (Swartz et al., 2006 ▶), which is inconsistent with a straightforward iron-transfer mechanism. Therefore, the structural data do not reveal how transfer of oxidized iron from the ferroxidase centre to the inner cavity could be the main determinant of the growth of the mineral core.
The available data also allow other interpretations for conformational changes at this location. Some flexibility may only be required to allow initial occupation of the ferroxidase centre. Also, the conformational changes may play a role in the alternative core-formation mechanism involving electron transfer from the cavity to the ferroxidase centre. As noted by Liu et al. (2004 ▶), some inherent flexibility around the FE2 site is necessary to accommodate iron in its different oxidation states. Different positions of key residues may also alter the electrostatic properties of the underlying surface of the inner cavity and promote initial nucleation of the core or favour electron transfer from the core to the ferroxidase centre.
Swartz et al. (2006 ▶) also observed conformational differences between oxidized and reduced forms of A. vinelandii bacterioferritin for some side chains of residues leading from the ferroxidase centre to a pore on the threefold subunit interface in some of their crystallographically independent molecules. On the basis of this, a general redox-dependent signalling mechanism for ferritins between the ferroxidase centre and the threefold pore was suggested. Regarding this, we note that many of the residues proposed to be involved in this signalling mechanism are not conserved in the highly homologous E. coli bacterioferritin; A. vinelandii bacterioferritin residues Ser126, Glu129, Glu121 and Glu118 are Asp, Gly, Ile and Glu, respectively, in E. coli bacterioferritin, making such a general signalling mechanism highly unlikely.
4. Conclusion
A structure of the E. coli bacterioferritin has been serendipitously obtained in a novel crystal form. Of the five bacterioferritin crystal structures known to date, three have been determined after crystals were unintentionally obtained (this study; Cobessi et al., 2002 ▶; Zhao et al., 2004 ▶). This highlights the ease of crystallization of proteins with this architecture. In vivo, crystallization of human ferritin has even been implicated in disease (Brooks et al., 2002 ▶). The structure of the ‘as isolated’ E. coli bacterioferritin that we have obtained in this fortuitous way has a different metal content to previous E. coli bacterioferritin structures, with an undefined mix of zinc and native iron ions occupying the ferroxidase centre. Flexibility of residues around the FE2 iron site appears to be a general feature of this type of bacterioferritins, whose function remains ill-defined.
Supplementary Material
PDB reference: bacterioferritin, 2htn, r2htnsf
Acknowledgments
We would like to thank Dr G. Fox for help with fluorescence scans and group members for fruitful discussions. This research was supported in part by The Netherlands Foundation of Chemical Research (CW), with financial aid from The Netherlands Foundation for Scientific Research (NWO).
References
- Andrews, S. C., Robinson, A. K. & Rodríguez-Quiñones, F. (2003). FEMS Microbiol. Rev.27, 215–237. [DOI] [PubMed] [Google Scholar]
- Baaghil, S., Lewin, A., Moore, G. R. & Le Brun, N. E. (2003). Biochemistry, 42, 14047–14056. [DOI] [PubMed] [Google Scholar]
- Brooks, D. G., Manova-Todorova, K., Farmer, J., Lobmayr, L., Wilson, R. B., Eagle, R. C. Jr, St Pierre, T. G. & Stambolian, D. (2002). Invest. Ophthalmol. Vis. Sci.43, 1121–1126. [PubMed] [Google Scholar]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed] [Google Scholar]
- Carrondo, M. A. (2003). EMBO J.22, 1959–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobessi, D., Huang, L. S., Ban, M., Pon, N. G., Daldal, F. & Berry, E. A. (2002). Acta Cryst. D58, 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Dautant, A., Meyer, J. B., Yariv, J., Precigoux, G., Sweet, R. M., Kalb, A. J. & Frolow, F. (1998). Acta Cryst. D54, 16–24. [DOI] [PubMed] [Google Scholar]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Frolow, F. & Kalb, A. J. (2001). Handbook of Metalloproteins, edited by A. Messerschmidt, Vol. 2, pp. 782–790. New York: Wiley.
- Frolow, F., Kalb, A. J. & Yariv, J. (1994). Nature Struct. Biol.1, 453–460. [DOI] [PubMed] [Google Scholar]
- Harding, M. M. (2000). Acta Cryst. D56, 857–867. [DOI] [PubMed] [Google Scholar]
- Harding, M. M. (2006). Acta Cryst. D62, 678–682. [DOI] [PubMed] [Google Scholar]
- Le Brun, N. E., Andrews, S. C., Guest, J. R., Harrison, P. M., Moore, G. R. & Thomson, A. J. (1995). Biochem. J.312, 385–392. [PMC free article] [PubMed] [Google Scholar]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl.Protein Crystallogr.26
- Lewin, A., Moore, G. R. & Le Brun, N. E. (2005). Dalton Trans., pp. 3597–3610. [DOI] [PubMed] [Google Scholar]
- Liu, H.-L., Zhou, H.-N., Xing, W.-M., Zhao, J.-F., Li, S.-X., Huang, J.-F. & Bi, R.-C. (2004). FEBS Lett.573, 93–98. [DOI] [PubMed] [Google Scholar]
- Lovell, S. C., Davis, I. W., Arendall, W. B. III, de Bakker, P. I., Word, J. M., Prisant, M. G., Richardson, J. S. & Richardson, D. C. (2003). Proteins, 50, 437–450. [DOI] [PubMed] [Google Scholar]
- McCoy, A. J., Grosse-Kunstleve, R. W., Storoni, L. C. & Read, R. J. (2005). Acta Cryst. D61, 458–464. [DOI] [PubMed] [Google Scholar]
- Macedo, S., Romão, C. V., Mitchell, E., Matias, P. M., Liu, M. Y., Xavier, A. V., LeGall, J., Teixeira, M., Lindley, P. & Carrondo, M. A. (2003). Nature Struct. Biol.10, 285–290. [DOI] [PubMed] [Google Scholar]
- Murshudov, G. N., Vagin, A. A., Lebedev, A., Wilson, K. S. & Dodson, E. J. (1999). Acta Cryst. D55, 247–255. [DOI] [PubMed] [Google Scholar]
- Shi, W., Zhan, C., Ignatov, A., Manjasetty, B. A., Marinković, N., Sullivan, M., Huang, R. & Chance, M. R. (2005). Structure, 13, 1473–1486. [DOI] [PubMed] [Google Scholar]
- Stillman, T. J., Connolly, P. P., Latimer, C. L., Morland, A. F., Quail, M. A., Andrews, S. C., Treffry, A., Guest, J. R., Artymiuk, P. J. & Harrison, P. M. (2003). J. Biol. Chem.278, 26275–26286. [DOI] [PubMed] [Google Scholar]
- Stillman, T. J., Hempstead, P. D., Artymiuk, P. J., Andrews, S. C., Hudson, A. J., Treffry, A., Guest, J. R. & Harrison, P. M. (2001). J. Mol. Biol.307, 587–603. [DOI] [PubMed] [Google Scholar]
- Swartz, L., Kuchinskas, M., Li, H., Poulos, T. L. & Lanzilotta, W. N. (2006). Biochemistry, 45, 4421–4428. [DOI] [PubMed] [Google Scholar]
- Yang, X., Le Brun, N. E., Thomson, A. J., Moore, G. R. & Chasteen, N. D. (2000). Biochemistry, 39, 4915–4923. [DOI] [PubMed] [Google Scholar]
- Zhao, J.-F., Liu, H.-L., Zhou, H.-N., Wang, Z.-P., Zhao, Y., Bian, S. M., Li, S.-X., Bi, R.-C. & Huang, J.-F. (2004). Acta Bot. Sin.46, 1331–1337.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: bacterioferritin, 2htn, r2htnsf