

A core fragment of Arabidopsis thaliana COP9 signalosome (CSN) subunit 7 was expressed in Escherichia coli. The protein was purified to homogeneity and crystallized.
Keywords: COP9 signalosome, proteasome, PCI and MPN motifs
Abstract
A core fragment of Arabidopsis thaliana COP9 signalosome (CSN) subunit 7 was expressed in Escherichia coli. The protein was purified to homogeneity and screened for crystallization. Crystallization conditions were refined using the sitting-drop vapour-diffusion method. Crystals were obtained using polyethylene glycol 8000 as a precipitant and have a thick rod-like morphology. Their crystallographic symmetry is orthorhombic, space group C2221, with unit-cell parameters a = 57.2, b = 86.2, c = 72.6 Å and a diffraction limit of 2.06 Å.
1. Introduction
The COP9 signalosome or CSN is a multi-protein complex that is highly conserved among eukaryotes (reviewed in Chamovitz & Glickman, 2002 ▶). It was originally discovered in Arabidopsis as a repressor of photomorphogenetic growth patterns (Wei & Deng, 1992 ▶; Wei et al., 1994 ▶). More recently, it has been revealed as a major developmental regulator in a broad spectrum of eukaryotes (Schwechheimer & Deng, 2000 ▶). The CSN contains eight subunits numbered 1–8 in decreasing order of molecular weight. The important role of the CSN signalosome in eukaryote development is strongly related to its modulation of proteosome-dependent protein degradation (Schwechheimer, 2004 ▶).
In addition to the functional relationship between CSN and the proteosome, the CSN is similar structurally in size, subunit composition and sequence homology to a supramolecular component, the 19S proteasome regulatory lid, and, to a lesser extent, to a third multi-protein complex, the eukaryotic translation-initiation factor 3 (eIF3; Wei et al., 1998 ▶). Subunits of these three complexes share two unique sequence motifs known as PCI and MPN motifs (Hofmann & Bucher, 1998 ▶). Structural analysis has shown that certain variations of the MPN motif contain a metalloprotease-like (JAMM) domain (Tran et al., 2003 ▶) possessing protease activity (Yamoah et al., 2005 ▶). In contrast, the molecular function of the PCI motif, which is 100–150 residues in length, has not been clearly defined. In vitro and yeast two-hybrid studies suggest that it may play an important role in CSN assembly and stability (Freilich et al., 1999 ▶; Kapelari et al., 2000 ▶; Tsuge et al., 2001 ▶).
CSN7 is a 26 kDa PCI-containing subunit of the CSN (Karniol et al., 1999 ▶). The PCI motif in CSN7 has been delimited previously by bioinformatics methods (Hofmann & Bucher, 1998 ▶) and biochemical data obtained by limited proteolysis, mass-spectroscopy analysis and N-terminal sequencing have enabled an experimentally based definition of a CSN7 PCI domain (manuscript in preparation). To better understand the PCI domain in general and the specific role of CSN7 within the CSN complex, we pursued the crystallization of the experimentally defined CSN7 PCI domain from A. thaliana. Combined with biochemical and genetic data, the crystal structure of this engineered protein should help us to achieve this goal. Here, we present the expression, purification, crystallization and preliminary X-ray analysis of the AtCSN7 core, residues 1–169, which comprises the entire PCI domain.
2. Experimental procedures
2.1. Subcloning, expression and purification
AtCSN7 core cDNA (encoding residues 1–169) was amplified by PCR. Constructs were subcloned into pET-28a (Novagen) and expressed in Escherichia coli Tuner strain (Novagen) bearing the RIL Codon Plus plasmid (Stratagene). The protein was purified by sequential metal-chelate and gel-filtration chromatography.
PCR was used to engineer a TEV protease cleavage site between a His tag and the gene in the following manner: HisTag-linker-ENLYFQ↓G-CSN7 subunit, where the arrow indicates the cleavage site. The digested PCR product was ligated into doubly digested pET-28a vector (EcoRI, NotI). Subsequently, positive clones were identified by colony PCR and sequenced where appropriate.
Transformed bacteria were used to inoculate sequentially increasing volumes of LB growth media containing 50 µg ml−1 kanamycin and 34 µg ml−1 chloramphenicol at 310 K. The cells were grown for 2–3 h in a final volume of 4 l. Upon reaching A 600 = 0.3, the temperature was lowered to 289 K. At a culture density of A 600 = 0.5–0.6, protein expression was induced with 200 µM IPTG. Cells were harvested after 12–16 h by centrifugation (9700g) for 10 min at 277 K. The bacterial pellet was stored at 193 K.
Cells were suspended in a 1:10(w:v) ratio with lysis buffer (buffer L; 50 mM sodium phosphate pH 8.0, 300 mM NaCl, 0.1% Triton X-100, 10 mg ml−1 lysozyme, 1500 U DNase and 5% glycerol). After lysis of the bacterial cell suspension using a microfluidizer (Microfluidics), cell debris was removed by a 1 h centrifugation (20 000g) at 277 K. The soluble fraction was loaded onto a metal-chelate column (Ni-CAM, Sigma) pre-equilibrated with buffer A (50 mM sodium phosphate pH 8.0, 300 mM NaCl and 5% glycerol) at a flow rate of 1 ml min−1. The column was washed with buffer A containing 5 mM imidazole until a stable baseline was achieved. After a step elution with buffer A supplemented with 50 mM imidazole, the AtCSN7 core-containing fractions were pooled and subjected to TEV protease (Opatowsky et al., 2003 ▶). Proteolysis was carried out for 12–14 h at 292 K while dialyzing against 4 l buffer A containing no imidazole. Optimal digestions utilized a protein:protease ratio of 50:1(w:w). Subsequently, the sample was again loaded onto a Ni-CAM column and unbound fractions were collected and pooled. The protein was concentrated to volume of 5 ml using spin concentrators (Vivascience). Gel-filtration chromatography was then carried out using a pre-equilibrated Superdex200 HiLoad prep-grade column (Amersham Biosciences). The protein was eluted with buffer G (20 mM Tris pH 7.1, 100 mM NaCl and 5 mM β-mercaptoethanol). Pooled fractions were concentrated as above to 45 mg ml−1, divided into aliquots and flash-frozen in liquid N2. SDS–PAGE analysis (Coomassie-stained) of the various purification stages is shown in Fig. 1 ▶.
Figure 1.

SDS–PAGE of the AtCSN7 core purification steps. Lane 1, material loaded onto Ni2+-chelate column; lane 2, sample of unbound material; lane 3, elution from the Ni2+ -chelate column obtained with 50 mM imidazole; lane 4, protein after overnight TEV protease digestion; lane 5, sample of unbound material from second Ni2+-chelate column; lane 6, sample of material loaded onto Superdex 200 gel-filtration column; lane 7, relevant gel-filtration fractions. MW markers (extreme left) are from top to bottom: 97, 66, 45, 31, 21, 14 kDa. The gel was Coomassie-stained.
2.2. Crystallization
Initial crystallization screens were performed at 292 K in 96-well sitting-drop plates (Corning) using Hampton Crystal Screen and Crystal Screen 2 (Hampton Research). The drop size was 2 µl, with a 1:1 sample:reservoir ratio. The protein concentration was 20 mg ml−1, diluted with buffer G from the 45 mg ml−1 stock aliquots. Microcrystals and small crystals appeared using condition No. 18 of Crystal Screen [20%(w/v) PEG 8000, 0.1 M sodium cacodylate pH 6.5, 0.2 M magnesium acetate tetrahydrate]. Crystallization conditions were further refined using 24-well sitting-drop vapour-diffusion plates by varying the precipitant concentration against either varying pH values or magnesium acetate concentrations. Optimal crystal-growth conditions consisted of a protein concentration of 10 mg ml−1 (diluted from the frozen stock as above) with a reservoir content of 23%(w/v) PEG 8000, 0.1 M sodium cacodylate pH 7.2 and 0.06–0.08 M magnesium acetate tetrahydrate at 292 K. Thick rod-shaped crystals grew as clusters (Fig. 2 ▶). Crystals appeared after 2–3 h in 4 µl drops (protein:reservoir ratio of 2:2) and were harvested for diffraction experiments 72–96 h after setup with sizes ranging from 0.15 to 0.3 mm in the longest dimension. Only this single crystal form was pursued, which consistently gave low mosaicity (about 0.4°) and diffraction to about 2.0 Å.
Figure 2.

(a) A cluster of rod-shaped crystals of the AtCSN7 core as obtained in the initial screen. The cystals are approximately 0.05–0.1 mm in length. (b) Rod-shaped crystals of the AtCSN7 core obtained after lowering the magnesium acetate concentration. The crystals are approximately 0.15–0.3 mm in the longest dimension. The crystallization conditions for these crystals were a protein concentration of 10 mg ml−1 with a reservoir comprising 23%(w/v) PEG 8000, 0.1 M sodium cacodylate pH 7.2 and 0.08 M magnesium acetate.
2.3. Data collection
In preparation for freezing in cryoloops, crystals were gradually transferred to a cryoprotectant solution containing 15% glycerol and the mother-liquor components [23%(w/v) PEG 8000, 0.1 M sodium cacodylate pH 7.2 and 0.06–0.08 M magnesium acetate tetrahydrate]. Crystals were then frozen in a cryostream at 110 K for data collection. Using Cu Kα radiation from a rotating-anode generator operating at 50 kV and 90 mA with a Rigaku R-AXIS IV image-plate detector system, crystals were screened for diffraction quality. Diffraction data were measured and processed with HKL (Otwinowski & Minor, 1997 ▶). Statistics are shown in Table 1 ▶.
Table 1. Data-processing statistics for the AtCSN7 core crystals.
Values in parentheses are for the highest resolution shell. Data were collected on a rotating-anode source as described in §2.3.
| Wavelength (Å) | 1.5418 |
| Temperature (K) | 110 |
| Space group | C2221 |
| Unit-cell parameters (Å) | |
| a (Å) | 57.2 |
| b (Å) | 85.8 |
| c (Å) | 72.2 |
| α = β = γ (°) | 90 |
| Unit-cell volume (Å3) | 354340.3 |
| VM (Å3 Da−1) | 2.3 |
| Solvent content (%) | 48 |
| Resolution range (Å) | 50.0–2.1 (2.2–2.1) |
| Total reflections | 38640 (1942) |
| Unique reflections | 10424 (842) |
| Completeness (%) | 97.5 (80.6) |
| Rmerge† (%) | 4.7 (16.6) |
| I/σ(I) | 22.8 (3.7) |
R
merge = 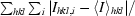
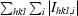 , where Ihkl is the intensity of a reflection and 〈I〉hkl is the average of all observations of this reflection and its symmetry equivalents.
, where Ihkl is the intensity of a reflection and 〈I〉hkl is the average of all observations of this reflection and its symmetry equivalents.
3. Results and discussion
The AtCSN7 PCI domain was overexpressed in E. coli, purified to homogeneity and crystallized using the sitting-drop vapour-diffusion method. One crystal form was pursued. This crystal form has a rod morphology and grew to 0.3 mm in the longest dimension. Moreover, it should provide us with a detailed atomic structure as we have obtained a complete data set with d min = 2.1 Å (see Fig. 3 ▶ and Table 1 ▶). Assuming the presence of one molecule per asymmetric unit gives a calculated V M value of 2.36 Å3 Da−1. To obtain experimental phase information, anomalous diffraction experiments will be performed on recombinant selenomethionine protein crystals. Substituted protein has been prepared and crystallized.
Figure 3.

Oscillation image (1°) of AtCSN7 core protein crystals. The crystal-to-detector distance was 180 mm. Diffraction data were observed to 2.06 Å. Data were collected and processed to a d min of 2.1 Å. Crystallization conditions were the same as for Fig. 2 ▶(b).
Acknowledgments
This work was supported by an Israel Science Foundation grant (783/05) to DC and JAH. MD was supported by a Tel Aviv University matching Rector fellowship.
References
- Chamovitz, D. A. & Glickman, M. (2002). Curr. Biol.12, R232. [DOI] [PubMed] [Google Scholar]
- Freilich, S., Oron, E., Kapp, Y., Nevo-Caspi, Y., Orgad, S., Segal, D. & Chamovitz, D. A. (1999). Curr. Biol.9, 1187–1190. [DOI] [PubMed] [Google Scholar]
- Hofmann, K. & Bucher, P. (1998). Trends Biochem. Sci.23, 204–205. [DOI] [PubMed] [Google Scholar]
- Kapelari, B., Bech-Otschir, D., Hegerl, R., Schade, R., Dumdey, R. & Dubiel, W. (2000). J. Mol. Biol.300, 1169–1178. [DOI] [PubMed] [Google Scholar]
- Karniol, B., Malec, P. & Chamovitz, D. A. (1999). Plant Cell, 11, 839–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opatowsky, Y., Chomsky-Hecht, O., Kang, M. G., Campbell, K. P. & Hirsch, J. A. (2003). J. Biol. Chem.278, 52323–52332. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Schwechheimer, C. (2004). Biochim. Biophys. Acta, 1695, 45–54. [DOI] [PubMed] [Google Scholar]
- Schwechheimer, C. & Deng, X. W. (2000). Semin. Cell Dev. Biol.11, 495–503. [DOI] [PubMed] [Google Scholar]
- Tran, H. J., Allen, M. D., Lowe, J. & Bycroft, M. (2003). Biochemistry, 42, 11460–11465. [DOI] [PubMed] [Google Scholar]
- Tsuge, T., Matsui, M. & Wei, N. (2001). J. Mol. Biol.305, 1–9. [DOI] [PubMed] [Google Scholar]
- Wei, N., Chamovitz, D. A. & Deng, X. W. (1994). Cell, 78, 117–124. [DOI] [PubMed] [Google Scholar]
- Wei, N. & Deng, X. W. (1992). Plant Cell, 4, 1507–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, N., Tsuge, T., Serino, G., Dohmae, N., Takio, K., Matsui, M. & Deng, X. W. (1998). Curr. Biol.8, 919–922. [DOI] [PubMed] [Google Scholar]
- Yamoah, K., Wu, K. & Pan, Z. Q. (2005). Methods Enzymol.398, 509–522. [DOI] [PubMed] [Google Scholar]