

Yeast Sfh1p, a close homolog of the Sec14p phosphatidylinositol transfer protein, was crystallized in the absence of detergent. X-ray data have been collected to 2.5 Å.
Keywords: Sfh1p, phosphatidylinositol transfer proteins, Saccharomyces cerevisiae
Abstract
Sec14p is the major phosphatidylinositol (PtdIns)/phosphatidylcholine (PtdCho) transfer protein in the budding yeast Saccharomyces cerevisiae and is the founding member of a large eukaryotic protein superfamily. This protein catalyzes the exchange of either PtdIns or PtdCho between membrane bilayers in vitro and this exchange reaction requires no external input of energy or of other protein cofactors. Despite the previous elucidation of the crystal structure of a detergent-bound form of Sec14p, the conformational changes that accompany the phospholipid-exchange reaction remain undefined. Moreover, a structural appreciation of how Sec14p or its homologs bind their various phospholipid substrates remains elusive. Here, the purification and crystallization of yeast Sfh1p, the protein most closely related to Sec14p, are reported. A combination of electrospray ionization mass-spectrometry and collision-induced decomposition mass-spectrometry methods indicate that recombinant Sfh1p loads predominantly with phosphatidylethanolamine. Unlike phospholipid-bound forms of Sec14p, this form of Sfh1p crystallizes readily in the absence of detergent. Sfh1p crystals diffract to 2.5 Å and belong to the orthorhombic primitive space group P212121, with unit-cell parameters a = 49.40, b = 71.55, c = 98.21 Å, α = β = γ = 90°. One Sfh1p molecule is present in the asymmetric unit (V M = 2.5 Å3 Da−1; V s = 50%). Crystallization of a phospholipid-bound Sec14p-like protein is a critical first step in obtaining the first high-resolution picture of how proteins of the Sec14p superfamily bind their phospholipid ligands. This information will significantly extend our current understanding of how Sec14p-like proteins catalyze phospholipid exchange.
1. Introduction
The Saccharomyces cerevisiae SEC14 gene product (Sec14p) is the major phosphatidylinositol (PtdIns)/phosphatidylcholine (PtdCho) transfer protein of this organism and plays an essential role in stimulating protein transport from the yeast trans-Golgi network (TGN; Bankaitis et al., 1990 ▶; Cleves, McGee & Bankaitis, 1991 ▶). The mechanism by which it does so involves Sec14p coordinating critical aspects of the so-called housekeeping pathways of lipid metabolism with the action of proteins that catalyze the biogenesis of transport vesicles on TGN membranes (Cleves, McGee, Whitters et al., 1991 ▶; McGee et al., 1994 ▶; Xie et al., 1998 ▶; Li et al., 2002 ▶; Yanagisawa et al., 2002 ▶; Routt & Bankaitis, 2004 ▶). Sec14p is a founding member of a large eukaryotic protein superfamily that is represented from yeast to mammals (Phillips et al., 2006 ▶). This Sec14p superfamily is relevant to human disease as mutations in specific members of this superfamily are associated with inherited human disorders (for examples, see Aravind et al., 1999 ▶; Bomar et al., 2003 ▶; Meier et al., 2003 ▶).
The available data indicate that Sec14p-like proteins generally regulate cellular functions associated with membrane trafficking and/or membrane biogenesis. These include pathways for polarized membrane growth in yeast (Carmen-Lopez et al., 1994 ▶; Nakase et al., 2001 ▶; Rudge et al., 2004 ▶) and in higher plants (Vincent et al., 2005 ▶). A role in specific stress responses is also indicated (Kearns et al., 1998 ▶). The ability of Sec14p-like proteins to bind PtdIns is a characteristic property of many, but not all, members of the Sec14p superfamily (Li et al., 2000 ▶; Peterman et al., 2004 ▶; Routt et al., 2005 ▶; Phillips et al., 2006 ▶).
The first detailed picture of the Sec14p fold was provided by solution of the crystal structure of a detergent-bound form (Sha et al., 1998 ▶). The Sec14p fold was novel at the time that structure was solved and exhibits a single large hydrophobic pocket that is sufficient in size to accommodate one phospholipid monomer. The crystallized form is considered to represent a conformer that exists only transiently on the membrane surface during the course of the phospholipid-exchange reaction. Since all presently known conditions conducive for the formation of Sec14p crystals require β-octylglucoside at concentrations above its critical micellar concentration (Sha et al., 1998 ▶; Phillips et al., 1999 ▶), we suggest that the phospholipid ligand that occupied Sec14p is replaced by two detergent molecules during an abortive exchange cycle on the surface of the detergent micelles (Phillips et al., 2006 ▶). The Sec14p fold is distinct from that of the recognized metazoan PtdIns-transfer proteins that share similar biochemical properties (Yoder et al., 2001 ▶; Phillips et al., 2006 ▶).
While the β-octylglucoside-bound Sec14p structure provided important new information, it fails to address a number of critical issues regarding how Sec14p binds its phospholipid ligands. An understanding of the mechanism by which Sec14p distinguishes and binds its phospholipid substrates is essential for any progress in elucidating what conformational changes within Sec14p accompany the phospholipid-exchange reaction and what the individual biological functions are of specific phospholipid-bound forms of Sec14p. While the application of sophisticated electron spin resonance spectroscopy techniques provides some general clues to the chemistry associated with phospholipid binding by Sec14p (Smirnova et al., 2006 ▶), those data do not provide great insight and more precise structural information is required. To this end, we have undertaken efforts to crystallize a phospholipid-bound form of Sec14p or any other Sec14p-like protein that binds phospholipids. Here, we report the successful crystallization of a phospholipid-bound form of yeast Sfh1p (a close homolog of Sec14p; Li et al., 2000 ▶) under detergent-free conditions and describe the preliminary characterization of those crystals.
2. Materials, methods and results
2.1. Expression of recombinant octahistidine-tagged Sfh1p
DNA manipulations were carried out using standard protocols. The SFH1 open-reading frame was amplified from yeast genomic DNA by the polymerase chain reaction using 5′-ATGACAACCAGCATACTCGATACTTATCCTC-3′ and 5′-TTAGCTGGTAACAGTAAATTTACCAAAAATG-3′ as the forward and reverse oligonucleotide primers, respectively. The amplified product was adenine-tailed and cloned into the pGEM-T Easy Vector (Promega, Madison, WI, USA) following the manufacturer’s instructions to generate plasmid pGEM-SFH1. To create an expression system for the octahistidine-tagged version of Sfh1p, the SFH1-coding region was amplified again using pGEM-SFH1 as template and oligonucleotides 5′-CCATGGGTCATCATCATCATCATCATCATCATATGACAACCAGCATACTCGATAC-3′ and 5′-GAGCTCTTAGCTGGTAACAGTAAATTTACCAAAAATG-3′ as the forward and reverse primers, respectively. The resulting amplified product was subcloned into the bacterial expression vector PET28b (Novagen, Madison, WI, USA) by virtue of the NcoI and SacI sites that clamped the respective primers used to amplify the product (shown in bold in the primer sequences given above). The identity and fidelity of the constructs was confirmed by DNA-sequence analysis using the chain-termination method and differentially labeled fluorescent dideoxynucleotides.
2.2. Purification of recombinant Sfh1p
Escherichia coli BL21-CodonPlus (DE3)-RIL cells (Stratagene, La Jolla, CA, USA) harboring PET28-SFH1 were induced for Sfh1p production by incorporation of IPTG (20 µM) into the growth medium (Luria–Bertani broth; Silhavy et al., 1984 ▶) and cultures were incubated for an additional 20 h with shaking at 289 K prior to harvest. Cells were pelleted, resuspended in ice-cold lysis buffer (300 mM NaCl, 50 mM sodium phosphate pH 7.5 and 2 mM β-mercaptoethanol) and disrupted as follows. Cell integrity was compromised first by the addition of lysozyme (1 mg ml−1 final concentration) to the resuspended cells and incubation at 298 K for 20 min. Following sonication, the resultant lysate was serially clarified by centrifugation at 5000g and 26 000g followed by filtration of the 26 000g supernatant through a 0.45 µm syringe filter (Fisherbrand).
Soluble His8-tagged Sfh1p was purified by incubation of clarified extract with Talon cobalt-affinity resin (BD Biosciences Clontech). The affinity resin was packed into a column, washed exhaustively with lysis buffer containing 5 mM imidazole (>50 bed volumes) and bound protein was eluted with a linear 5–200 mM imidazole gradient. Peak fractions enriched in His8-Sfh1p were identified by SDS–PAGE of individual fractions and Coomassie staining. Fractions where His8-Sfh1p comprised at least 95% of the total protein were pooled and buffer-exchanged against lysis buffer using a Centricon Plus-30 centrifugal filter (Millipore, MA, USA) and the fractions were subsequently concentrated to 18.75 mg ml−1 His8-Sfh1p. Final preparations were essentially pure as judged by SDS–PAGE and staining with Coomassie Brilliant Blue. Typical yields of soluble His8-Sfh1p ranged from 22 to 38 mg per litre of culture.
2.3. Mass spectrometry
Lipids were extracted from purified His8-Sfh1p (2.25 mg) with a single-phase chloroform/methanol/H2O system (1:2:0.8; Bligh & Dyer, 1959 ▶). A tube containing only the buffer (300 mM NaCl, 50 mM sodium phosphate pH 7.5, 2 mM β-mercapthoethanol) was extracted alongside the protein sample as a control. After sonication in a bath sonicator for 2 min, the denatured protein was sedimented by centrifugation and the supernatant transferred to a new tube. This single-phase extract was converted into a two-phase system by the addition of chloroform and water to a final ratio of 2:2:1.8 chloroform:methanol:H2O. Subsequently, the aqueous and organic phases were resolved by centrifugation and the organic (lower) phase was collected and dried under a stream of anhydrous N2 gas. Dried lipid films were dissolved in 200 µl chloroform/methanol (2:1), diluted 1:10 in the same and infused into a quadrupole time-of-flight tandem mass spectrometer (QStar XL, Applied Biosystems) at 6 µl min−1. The mass spectra were obtained scanning from 40–2000 Da in the negative-ion multi-channel acquisition mode with the electrospray ionization source operating at the following settings: nebulizer gas, 138 kPa; curtain gas, 138 kPa; ion-spray voltage, −4200 V; declustering potential, −55 V; focusing potential, −265 V; declustering potential 2, −15 V. Spectra were acquired for 1 min at one scan per second and analyzed using Analyst QS 1.1 software (Applied Biosystems). Collision-induced decomposition mass spectrometry (CID–MS) was performed using a collision energy of −40.0 V (laboratory frame of reference) and N2 as the collision gas. Spectra were acquired for 2 min at one scan per second.
The electrospray ionization mass-spectrometry (ESI–MS) profiles for material extracted from recombinant His8-Sfh1p are shown in Fig. 1 ▶. While very little organic material was recovered from mock buffer control (Fig. 1 ▶ a), considerable organic matter was extracted from His8-Sfh1p. Analyses of the 650–800 Da region of the spectrum identify phosphatidylethanolamine (PtdEtn) as the predominant molecular species in the His8-Sfh1p organic extract (Fig. 1 ▶ b). Expansion of the PtdEtn-derived component of the spectrum revealed nine distinct molecular species (Fig. 1 ▶ c and Table 1 ▶). CID–MS using N2 as the collision gas and a collision energy of −40.0 V indicates these mass species correspond to PtdEtn species with 30:0, 32:1, 33:0 cp, 34:2, 34:1, 35:1 cp, 35:0 cp, 36:2 and 37:1 cp acyl chains (see Table 1 ▶ for notation). Fig. 1 ▶(e) shows the CID–MS spectrum of the most abundant PtdEtn species.
Figure 1.

(a) Negative-ion mode ESI–MS of the organic extract from buffer control. The ions present in this spectrum are similar in composition and intensity to the ions in a negative-ion mode ESI–MS spectrum of 2:1 chloroform:methanol alone. (b) Negative-ion mode ESI–MS of the organic extract from the His8-Sfh1p sample. Note the difference in intensity scale between (a) and (b). (c) Expansion of the 650–800 Da region of the spectrum in (b). The predominant ions are derived from PtdEtn species (marked by an asterisk). (d) Expansion of the 720–800 Da region of the spectrum in (b). This region highlights ions derived from PtdGro species (marked with a diamond). (e) CID–MS of the major PtdEtn species, showing the structures of select product ions consistent with the structure of the parent ion at 702.49 Da (Pulfer & Murphy, 2003 ▶). CID–MS of the other PtdEtn species revealed similar product ions, with differences consistent with differences in acyl-chain length and saturation.
Table 1. List of the identified PtdEtn and PtdGro molecular species.
The mass of each molecular species is indicated. Molecular species are identified by the total number of acyl-chain C atoms:double bonds and by the presence or absence of a cyclopropane group (cp) in the acyl chains.
| PtdEtn species | Mass (Da) |
|---|---|
| 30:0 | 662.55 |
| 32:1 | 688.50 |
| 33:0 cp | 702.51 |
| 34:2 | 714.52 |
| 34:1 | 716.53 |
| 35:1 cp | 728.53 |
| 35:0 cp | 730.55 |
| 36:2 | 742.55 |
| 37:1 cp | 756.53 |
| PtdGro species | |
| 33:0 cp | 733.52 |
| 34:1 | 747.53 |
| 35:0 cp | 761.54 |
| 36:2 | 773.55 |
| 37:1 cp | 787.56 |
Analyses of the 720–800 Da region identified five predominant phosphatidylglycerol (PtdGro) molecular species (Fig. 1 ▶ d and Table 1 ▶). CID–MS using N2 as the collision gas and a collision energy of −40.0 V identifies these as corresponding to PtdGro molecular species with 33:0 cp, 34:1, 35:0 cp, 36:2 and 37:1 cp acyl chains, respectively (Table 1). Other E. coli-derived acidic phospholipids such as phosphatidic acid or cardiolipin were not evident. As PtdGro ionizes much more efficiently than PtdEtn in negative-ion mode ESI–MS (unpublished data), the raw data suggest that Sfh1p is predominantly loaded with PtdEtn. Because PtdEtn represents the most abundant phospholipid in the E. coli inner membrane (∼75% of bulk lipid) and E. coli does not synthesize PtdIns or PtdCho, it is not surprising that His8-Sfh1p is purified in a form loaded predominantly with PtdEtn. As a point of comparison, we note that recombinant Sec14p expressed in E. coli loads almost exclusively with PtdGro (unpublished data).
2.4. Optimization of crystallization conditions
Initial crystallization screens were carried out using an in-house robotic platform that sampled an array of 288 different conditions at 293 and 278 K (a total of 576 conditions) that included, but were not limited to, the Classics and Classics Lite screens from Nextal and the PEG/Ion screen from Hampton Research. In these initial screens, His8-Sfh1p was reconstituted to 18.75 mg ml−1 in lysis buffer and a hanging-drop geometry was employed. The hanging drop consisted of 0.5 µl 18.75 mg ml−1 protein solution and 0.5 µl well solution. The conditions of the primary screen generally did not include cryoprotectant. Crystals appeared in approximately 100 conditions at both 293 and 278 K and conditions where medium-molecular-weight PEG was present as a precipitant were particularly effective. To further optimize crystallization, we used the Crystal Screen Cryo system from Hampton Research (Aliso Viejo, CA, USA) to inspect 50 conditions at 293 K. Of the 50 conditions tested, 18 yielded crystals and the most promising of these corresponded to cryo solutions Nos. 22, 37, 39 and 43 in the Crystal Screen array.
Based on these conditions, we generated an expanded screen in which three parameters were systematically varied. The first parameter was pH, which was varied from 5.5 to 8.5 in increments of 0.5 pH units. The second was His8-Sfh1p concentration, which was varied from 5 to 12.5 mg ml−1. Finally, the buffer in which His8-Sfh1p was reconstituted included either lysis buffer (300 mM NaCl, 50 mM sodium phosphate pH 7.5) or phosphate-free buffer (300 mM NaCl, 20 mM HEPES pH 7.2). Condition No. 22 of the Crystal Screen array at pH 8.5 with an initial His8-Sfh1p concentration of 8.75 mg ml−1 in lysis buffer (final protein concentration in the hanging drop of 4.38 mg ml−1) yielded the best results of the 192 optimization conditions tested. We also successfully obtained His8-Sfh1p crystals in phosphate-free buffer systems. However, those crystals were not as large nor as well formed as those obtained when His8-Sfh1p was crystallized in the presence of phosphate.
2.5. Optimized crystallization and data collection
His8-Sfh1p crystals were grown by the sitting-drop vapour-diffusion method at 293 K under optimized conditions using Cryschem Plates (sitting-drop geometry; Hampton Research, Aliso Viejo, CA, USA). The well solution consisted of 170 mM sodium acetate, 85 mM Tris–HCl pH 8.5, 25.5%(w/v) PEG 4000 and 11.9%(v/v) glycerol. The sitting drop consisted of a 1:1 ratio of protein (1.8 µl, 8.75 mg ml−1 in lysis buffer) to well solution (1.8 µl) in a total of 3.6 µl. Crystals grew as thin two-dimensional plates ranging from approximately 100 to 500 µm in length and 10 to 15 µm in width (Fig. 2 ▶). Because His8-Sfh1p crystals formed in the absence of detergent, we anticipate that the crystallizing form of Sfh1p is bound to phospholipid, likely PtdEtn. This expectation is confirmed by ESI–MS analysis of the chloroform:methanol-extractable material from washed crystals. PtdEtn is identified as the only detectable species in the organic extract of crystallized Sfh1p and similar analyses indicate bound PtdEtN can be quantitatively exchanged for PtdCho with a resulting PtdCho-loaded Sfh1p that is also amenable to crystallization (data not shown).
Figure 2.

Typical crystals of Sfh1p formed under the established conditions.
His8-Sfh1p crystals were transferred directly from the mother liquor and placed under a stream of cold nitrogen vapor (100 K). Diffraction data were collected at the University of North Carolina Facility for Biomolecular X-Ray Crystallography on a Rigaku RU-H3R class rotating copper-anode generator and an R-AXIS IV++ image-plate detector with Osmic confocal blue optics. Frames using 0.5° oscillation and 10 min exposure time were collected from two independent crystals (a total of 461 frames) and data were processed using the HKL package (Otwinowski & Minor, 1997 ▶). The processing statistics are summarized in Table 2 ▶. We calculated a Matthews coefficient of 2.5 Å3 Da−1 for the unit cell (V M; Matthews, 1968 ▶). This value is consistent with one 36 kDa Sfh1p monomer per asymmetric unit and a unit-cell solvent content (V s) of 50%.
Table 2. Processing statistics for His8-Sfh1p crystals.
Values in parentheses are for the highest resolution bin (2.59–2.5 Å).
| X-ray source | Rigaku RU-H3R rotating copper-anode generator |
| Wavelength (Å) | 1.5418 |
| Space group | P212121 |
| Unit-cell parameters (Å, °) | a = 49.40, b = 71.55, c = 98.21, α = β = γ = 90° |
| Resolution range (Å) | 50–2.5 |
| No. of observed/unique reflections | 512862/12389 |
| Redundancy | 5.1 |
| Completeness (%) | 98.0 (94.1) |
| Rmerge† (linear, SCALEPACK) (%) | 11.2 (35.5) |
| I/σ(I) | 15 (3.2) |
| Matthews coefficient (VM) (Å3 Da−1) | 2.5 |
R
merge = 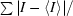
 ; data from two crystals are included.
; data from two crystals are included.
3. Concluding remarks
S. cerevisiae Sfh1p is the closest homolog to the major yeast PtdIns/PtdCho-transfer protein (PITP) Sec14p. These two proteins share 64% primary sequence identity, yet remarkably do not execute redundant physiological functions (Li et al., 2000 ▶). This tight physiological coupling of PITPs to specific biological functions is a hallmark feature of PITPs (Ile et al., 2006 ▶). The physiological function of Sfh1p is unknown as it is nonessential for yeast viability and genetic analyses fail to support a functional redundancy between Sfh1p and Sec14p (Li et al., 2000 ▶). We purified recombinant Sfh1p from E. coli, determined that purified Sfh1p is bound predominantly to PtdEtn and successfully crystallized the protein under detergent-free conditions. The crystals diffract to a resolution of 2.5 Å and the data strongly indicate that the crystallizing form of Sfh1p contains bound phospholipid. Solution of a phospholipid-bound Sfh1p structure will be greatly facilitated by molecular-replacement approaches using the available detergent-bound Sec14p structure as the search model. We expect the phospholipid-bound Sfh1p structure will prove informative regarding how members of the Sec14p superfamily of proteins accommodate a phospholipid monomer into the single hydrophobic cavity of the protein fold. We also find that Sfh1p is readily amenable to loading with other phospholipids such as PtdIns and PtdCho in vitro (unpublished data). Given the ease with which Sfh1p crystallizes, we now face the exciting prospect of being in the position to elucidate the structures of a family of phospholipid-bound forms of Sfh1p. These structures will collectively reveal high-resolution insights into how Sec14p-like proteins accommodate the lipid phosphate group, how these proteins accommodate a wide diversity of acyl-chain species within the phospholipid-binding pocket and how Sec14p-like proteins generate the phospholipid head-group specificity that provides the discriminating factor which distinguishes between potential phospholipid-binding ligands.
Although Sec14p and Sfh1p are functionally distinct in the context of the yeast cell, we further anticipate that the novel information gained from the Sfh1p model will translate directly to Sec14p. In this regard, we have isolated Sfh1p derivatives that endow these mutant proteins with Sec14p-like function in cells (unpublished data). An understanding at the structural level of the mechanism underlying the switch in molecular identity between these similar (yet functionally distinct) proteins promises to be highly informative from the viewpoint of understanding how proteins of the large Sec14p protein superfamily function.
Acknowledgments
This work was supported by grant GM44530 to VAB from the National Institutes of Health. GS is a postdoctoral fellow of the Deutsche Forschungsgemeinshaft (SCHA 1274/1-1). TAG and CRHR are supported by the LIPID MAPS Large Scale Collaborative Grant No. GM069338 from the National Institutes of Health. The services of the University of North Carolina Lineberger Comprehensive Cancer Center Genome Analysis and Nucleic Acids Core facilities are also acknowledged.
References
- Aravind, L., Neuwald, A. F. & Ponting, C. P. (1999). Curr. Biol.9, R195–R197. [DOI] [PubMed] [Google Scholar]
- Bankaitis, V. A., Aitken, J. R., Cleves, A. E. & Dowhan, W. (1990). Nature (London), 347, 561–562. [DOI] [PubMed] [Google Scholar]
- Bligh, E. G. & Dyer, W. J. (1959). Can. J. Biochem. Physiol.37, 911–917. [DOI] [PubMed] [Google Scholar]
- Bomar, J. M., Benke, P. J., Slattery, E. L., Puttagunta, R., Taylor, L. P., Seong, E., Nystuen, A., Chen, W., Albin, R. L., Patel, P. D., Kittles, R. A., Sheffield, V. C. & Burmeister, M. (2003). Nature Genet.35, 264–269. [DOI] [PubMed] [Google Scholar]
- Carmen-Lopez, M., Nicaud, J.-M., Skinner, H. B., Vergnolle, C., Kader, J. C., Bankaitis, V. A. & Gaillardin, C. (1994). J. Cell Biol.124, 113–127. [DOI] [PMC free article] [PubMed]
- Cleves, A. E., McGee, T. & Bankaitis, V. A. (1991). Trends Cell. Biol.1, 31–34. [DOI] [PubMed]
- Cleves, A. E., McGee, T. P., Whitters, E. A., Champion, K. M., Aitken, J. R., Dowhan, W., Goebl, M. & Bankaitis, V. A. (1991). Cell, 64, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ile, K. E., Schaaf, G. & Bankaitis, V. A. (2006). In the press. [DOI] [PubMed]
- Kearns, M. A., Monks, D. E., Fang, M., Rivas, M. P., Courtney, P. D., Chen, J., Prestwich, G. D., Theibert, A. B., Dewey, R. E. & Bankaitis, V. A. (1998). EMBO J.17, 4004–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X., Rivas, M. P., Fang, M., Marchena, J., Mehrotra, B., Chaudhary, A., Feng, L., Prestwich, G. D. & Bankaitis, V. A. (2002). J. Cell Biol.157, 63–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X., Routt, S. M., Xie, Z., Cui, X., Fang, M., Kearns, M. A., Bard, M., Kirsch, D. & Bankaitis, V. A. (2000). Mol. Biol. Cell, 11, 1989–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee, T. P., Skinner, H. B., Whitters, E. A., Henry, S. A. & Bankaitis, V. A. (1994). J. Cell Biol.124, 273–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Meier, R., Tomizaki, T., Schulze-Briese, C., Baumann, U. & Stocker, A. (2003). J. Mol. Biol.331, 725–734. [DOI] [PubMed] [Google Scholar]
- Nakase, Y., Nakamura, T., Hirata, A., Routt, S. M., Skinner, H. B., Bankaitis, V. A. & Shimoda, C. (2001). Mol. Biol. Cell, 4, 901–917. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Peterman, T. K., Ohol, Y. M., McReynolds, L. J. & Luna, E. J. (2004). Plant Physiol.136, 3080–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips, S. E., Sha, B.-D., Topalof, L., Xie, Z., Alb, J. G. Jr, Clenchin, V., Swigart, P., Cockcroft, S., Martin, T. F. J., Luo, M. & Bankaitis, V. A. (1999). Mol. Cell, 4, 187–197. [DOI] [PubMed] [Google Scholar]
- Phillips, S. E., Vincent, P., Rizzieri, K., Schaaf, G., Gaucher, E. A. & Bankaitis, V. A. (2006). Crit. Rev. Biochem. Mol. Biol.41, 1–28. [DOI] [PubMed]
- Pulfer, M. & Murphy, R. C. (2003). Mass Spectrom. Rev.22, 332–364. [DOI] [PubMed] [Google Scholar]
- Routt, S. M. & Bankaitis, V. A. (2004). Biochem. Cell Biol.82, 254–262. [DOI] [PubMed] [Google Scholar]
- Routt, S. M., Ryan, M. M., Tyeryar, K., Rizzieri, K., Roumanie, O., Brennwald, P. J. & Bankaitis, V. A. (2005). Traffic, 6, 1157–1172. [DOI] [PubMed] [Google Scholar]
- Rudge, S. A, Sciorra, V. A., Iwamoto, M., Zhou, C., Strahl, T., Morris, A. J., Thorner, J. & Engebrecht, J. (2004). Mol. Biol. Cell, 1, 207–218. [DOI] [PMC free article] [PubMed]
- Sha, B.-D., Phillips, S. E., Bankaitis, V. A. & Luo, M. (1998). Nature (London), 391, 506–510. [DOI] [PubMed] [Google Scholar]
- Silhavy, T. J., Berman, M. L. & Enquist, L. W. (1984). Experiments with Gene Fusions, p. 217. Cold Spring Harbor Laboratory, NY, USA: Cold Spring Harbor Laboratory Press.
- Smirnova, T. I., Chadwick, T. G., MacArthur, R., Poluektov, O., Song, L., Ryan, M. M., Schaaf, G. & Bankaitis, V. A. (2006). In the press. [DOI] [PubMed]
- Vincent, P., Chua, M., Nogue, F., Fairbrother, A., Mekheel, H., Xu, Y., Allen, N., Bibikova, T. N., Gilroy, S. & Bankaitis, V. A. (2005). J. Cell Biol.168, 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, Z., Fang, M., Rivas, M. P., Faulkner, A., Sternweis, P. C., Engebrecht, J. & Bankaitis, V. A. (1998). Proc. Natl Acad. Sci. USA, 95, 12346–12351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa, L., Marchena, J., Xie, Z., Li, X., Poon, P. P., Singer, R. A., Johnston, G. C., Randazzo, P. A. & Bankaitis, V. A. (2002). Mol. Biol. Cell, 13, 2193–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder, M. D., Thomas, L. M., Tremblay, J. M., Oliver, R. L., Yarbrough, L. R. & Helmkamp, G. M. Jr (2001). J. Biol. Chem.276, 9246–9252. [DOI] [PubMed] [Google Scholar]