

PmSOD1 and PmSOD2, two superoxide dismutases from the protozoan parasite P. marinus, have been crystallized and their structures have been solved to 1.8 and 2.3 Å resolution, respectively.
Keywords: superoxide dismutases, Perkinsus marinus
Abstract
Perkinsus marinus, a facultative intracellular parasite of the eastern oyster Crassostrea virginica, is responsible for mass mortalities of oyster populations. P. marinus trophozoites survive and proliferate within oyster hemocytes, invading most tissues and fluids, thus causing a systemic infection that eventually kills the host. The phagocytosis of P. marinus trophozoites lacks a respiratory burst, suggesting that the parasite has mechanisms that actively abrogate the host’s oxidative defense responses. One mechanism and the first line of defense against oxidative damage is the dismutation of superoxide radical to molecular oxygen and hydrogen peroxide by superoxide dismutases (SODs). P. marinus possesses two iron-cofactored SODs, PmSOD1 and PmSOD2. Here, the crystallization and X-ray structures of both PmSOD1 and PmSOD2 are presented.
1. Introduction
Perkinsus marinus, a facultative intracellular parasite of the eastern oyster Crassostrea virginica, is responsible for mass mortalities of both natural and farmed oyster populations from the Gulf of Mexico to the Eastern seaboard (Ford, 1996 ▶). Efforts to control the epizootic through parasite diagnosis and oyster stock management have been largely unsuccessful and new intervention approaches utilizing molecular and biochemical information are being explored. Taxonomically, P. marinus (phylum Perkinsozoa) is within the protistan group Alveolata, which also contains the phylum Apicomplexa, represented by the human pathogens Toxoplasma gondii and Plasmodium falciparum (Saldarriaga et al., 2003 ▶).
The infection cycle of P. marinus begins with the engulfment of trophozoites or zoospores by phagocytic oyster hemocytes. Unlike most phagocytosed microorganisms, P. marinus survives, proliferates in the intracellular environment and is disseminated throughout the mollusk host via migrating hemocytes. A major antimicrobial activity of oyster hemocytes is the generation of reactive oxygen species (ROS) during phagocytosis. However, the phagocytosis of live P. marinus trophozoites by oyster hemocytes fails to yield the intense ROS burst that takes place upon the uptake of other biotic particles (Volety & Chu, 1995 ▶; Anderson et al., 1992 ▶, 1999 ▶). The absence of this ROS burst suggests that the parasite has an efficient antioxidation or ROS-detoxification system. The detoxification of ROS is a widespread defense strategy of intracellular parasites and is typically carried out by catalyzing the conversion of ROS to less harmful species by enzymes such as superoxide dismutase (De Groote et al., 1997 ▶). Our previous findings that (i) P. marinus is resistant to in vitro-produced superoxide and hydrogen peroxide (Schott, Pecher et al., 2003 ▶), (ii) P. marinus extracts display Fe-type SOD activity (Wright et al., 2002 ▶) and (iii) P. marinus expresses two Fe-SOD genes, PmSOD1 and PmSOD2 (Wright et al., 2002 ▶), strongly suggest that this parasite has the capacity to neutralize ROS within the hemocyte phagosome.
The amino-acid sequences and biochemical characterization of the PmSOD1 and PmSOD2 products indicate that they encode iron-containing enzymes with similar active sites, although they have less than 40% amino-acid identity (Wright et al., 2002 ▶). We present here the crystallization and X-ray structures of recombinant PmSOD1 and PmSOD2.
2. Materials and methods
2.1. Crystallization
The expression and purification of PmSOD2 and PmSOD1 took place as described previously (Ahmed et al., 2003 ▶). Both samples of protein used for crystallization studies were the full-length mature protein and lacked the signal peptide (Ahmed et al., 2003 ▶). The PmSOD2 sample also has a short vector-derived N-terminal peptide with the sequence AMADIGS. The protein solutions were concentrated to 18 mg ml−1 in 500 mM Tris–HCl pH 8.5 prior to crystallization experiments. Crystals were grown at 293 K by vapor diffusion in hanging drops. Drops were prepared by mixing 2 µl protein solution with an equal volume of reservoir solution. The reservoir solution contained 0.17 M sodium acetate trihydrate, 0.085 M Tris–HCl pH 8.5, 25.5%(w/v) PEG 4000 and 15% glycerol. Small light-brown rod-like crystals of less than 0.1 mm on the smallest face were obtained within a few days. The crystals were enlarged to their final size through several cycles of washing in protein solution and macroseeding by placing washed crystals into fresh crystallization drops. The seeded crystals maintained their rod shape and resulted in thicker, longer rods for both PmSOD1 and PmSOD2. Fragments of the PmSOD1 and PmSOD2 crystals were cut and flash-cooled directly in a stream of N2 prior to data collection at 100 K. The fragments were approximately 0.2 × 0.4 × 0.3 mm in size.
2.2. Data collection and structure determination
X-ray diffraction data extending to 1.86 and 2.5 Å resolution were collected for PmSOD1 and PmSOD2, respectively. Both data sets were collected using a MAR 345 detector (MAR Research). The X-ray source was a Rigaku RU-200 rotating-anode generator operating at 50 kV and 100 mA with a double focusing-mirror system. A complete data set was collected from a single crystal using a crystal-to-detector distance of 150 mm and exposure times of 20 min for 1.0° oscillations. X-ray data sets were processed using the programs DENZO and SCALEPACK (Otwinowski & Minor, 1997 ▶). The PmSOD1 crystal belonged to space group P21, with unit-cell parameters a = 53.8, b = 62.5, c = 57.3 Å, β = 95.50°, while the PmSOD2 crystal belonged to space group P212121, with unit-cell parameters a = 61.4, b = 73.9, c = 94.5 Å. The structures were solved by molecular replacement with AMoRe (Navaza, 1994 ▶) using a monomer of PDB entry 3sdp (Stoddard et al., 1990 ▶) as the search model. Both molecular-replacement solution models had R factors in the low 40% and free R factors in the mid 40% range. Most of the amino-acid residues were clearly visible in 2F o − F c OMIT electron-density maps calculated from molecular-replacement phases. The final models were obtained through iterative cycles of model building in O (Jones et al., 1991 ▶) followed by structure refinement with CNS (Brünger et al., 1998 ▶) using a maximum-likelihood refinement procedure (Brünger et al., 1990 ▶, 1997 ▶; Adams et al., 1997 ▶; Brünger, 1992a ▶,b ▶). The structure of PmSOD1 refined to an R factor of 17.1% and a free R factor of 21.6%, while that of PmSOD2 refined to an R factor 19.7% and a free R factor of 27.6%. Data and model statistics are shown in Table 1 ▶.
Table 1. Crystallographic data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data | PmSOD1 | PmSOD2 |
|---|---|---|
| Resolution (Å) | 56.8–1.86 (1.93–1.86) | 30–2.5 (2.57–2.5) |
| Rmerge† (%) | 4.6 (29.5) | 8.6 (33.0) |
| Completeness (%) | 95 (91) | 96.7 (92.9) |
| Redundancy | 5.5 (4.0) | 8.0 (3.0) |
| I/σ(I) | 21.6 (4.1) | 20.0 (2.6) |
| Refinement statistics | ||
| R factor‡ (%) | 17.1 (24.8) | 19.7 (18.5) |
| Rfree§ | 21.6 (26.2) | 27.6 (33.9) |
| No. of atoms | ||
| Protein | 3147 | 3211 |
| Water | 408 | 203 |
| Fe | 2 | 2 |
| Mean B factor (Å2) | 16.8 | 15.1 |
| R.m.s. deviations from ideal | ||
| Bond lengths (Å) | 0.014 | 0.015 |
| Bond angles (°) | 1.3 | 1.7 |
| Chiral (Å3) | 0.108 | 0.099 |
R
merge = 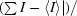
 , where I is the observed intensity and 〈I〉 is the average intensity obtained from multiple observations of symmetry-related reflections after rejections.
, where I is the observed intensity and 〈I〉 is the average intensity obtained from multiple observations of symmetry-related reflections after rejections.
R factor = 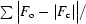
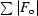 , where F
o are observed and F
c are calculated structure factors.
, where F
o are observed and F
c are calculated structure factors.
The R free set uses 5% of randomly chosen reflections.
The PmSOD1 protein sample used for crystallization has 201 amino-acid residues and in the refined model of PmSOD1 one C-terminal residue and four C-terminal residues are disordered in monomer A and monomer B, respectively. Of the 203 amino-acid residues of PmSOD2, the refined model of PmSOD2 monomer A includes residues 2–201, while monomer B includes residues 9–203. The vector-derived sequence is not visible in any of the electron-density maps.
3. Results and discussion
3.1. Structure
Both PmSOD1 and PmSOD2 have typical overall SOD topology, with monomers consisting of two domains. The C-terminal domain is made up of two long antiparallel α-helices and the N-terminal domain displays a central three-stranded antiparallel β-sheet surrounded by five helices (Fig. 1 ▶). In both structures the locations of the Fe-ion sites were unambiguously identified by strong signals in the corresponding electron-density maps. In addition, the dimers of both PmSOD1 and PmSOD2 have the classic topology of Fe/Mn-SOD (Fig. 1 ▶). The refined model of both enzymes unambiguously define their typical metal active sites with three histidine residues, an aspartic acid residue and a solvent molecule as metal ligands, which is the case in all reported Fe/Mn-SOD structures.
Figure 1.

(a) Ribbon diagram of the PmSOD1 dimer showing Fe ions (purple spheres) at the active sites with their ligands. At the center of the figure, Glu165 interacts across the dimer interface with the metal ligand His166. (b) Close-up of the active site of PmSOD1 shows the Fe ion (represented as a large yellow sphere) coordinated by three histidine residues, an aspartic acid residue and a water molecule. The water molecule, whose O-atom location is represented as a small red sphere, is coordinated by Gln76.
The main-chain conformation for most residues of PmSOD1 and PmSOD2 fall within allowed regions of the Ramachandran plots; the only exceptions are Asp146 and Arg173 in both subunits of PmSOD1 and the equivalent residues Asp152 and Gln180 in both subunits of PmSOD2. These residues lie in two different tight turns on the dimer interface. Similar main-chain conformations have been observed in other SODs and they correspond to Asn140 and Arg167 in Escherichia coli Fe-SOD (Lah et al., 1995 ▶).
3.2. Active site and dimerization
The biochemical characterization indicated that both enzymes were Fe-dependent SODs (Wright et al., 2002 ▶). PmSOD1 and PmSOD2 were confirmed to be carrying Fe instead of Mn based on their susceptibility to inhibition by cyanide or hydrogen peroxide. Native PmSOD1 and PmSOD2 have activities that are resistant to cyanide and susceptible to hydrogen peroxide. Furthermore, recombinant PmSOD1 and PmSOD2 were produced in the presence of iron. However, the amino-acid sequences of both enzymes showed some anomalies for such classification. PmSOD1, while showing high overall sequence homology to Fe-dependent SODs, has the unusual sequence Phe-Phe-Phe at residue numbers 82–84, instead of the highly conserved consensus Fe-SOD sequence of Phe-Tyr-Trp, in the region that builds up the hydrophobic cavity containing the metal sites and its ligands. A Phe residue at position 83 is considered to be one of the markers for Mn-dependent SODs (Yamano & Maruyama, 1999 ▶). However, biochemical data, particularly inhibition by cyanide or hydrogen peroxide, reveals that the enzymes are indeed Fe-SOD. Other consensus sequence deviations include the triad Phe-Tyr-Trp becoming Met-Tyr-Trp in PmSOD2 (Fig. 2 ▶). This deviation from the consensus sequence does not appear to be important from a structural perspective since Met is relatively far from the metal site and compensatory changes are not evident.
Figure 2.

An overlay of the PmSOD1 (green) and PmSOD2 (blue) structures reveals key mutations in a highly conserved triad (Phe-Try-Trp) of Fe-SOD. PmSOD1 has the unusual sequence Phe-Phe-Phe. The Tyr84 to Phe83 change is structurally compensated by the Ile23 to Met23 change owing to the larger volume of Met relative to Ile. Overall, PmSOD1 displays a more hydrophobic second shell of amino-acid residues than PmSOD2 around the metal site, i.e. Phe83, Phe84 and Tyr183.
The metal sites of both structures are in close proximity to the active dimer interface. Consequently, there are interactions with metal ligands across the dimer interface. For PmSOD1 these interactions include Glu165 with the metal ligand His166 (Fig. 1 ▶), while Phe125 and Tyr169 interact across the dimer interface with the second layer of residues of the metal cavity. Equivalent interactions are observed in PmSOD2. Furthermore, PmSOD1 shows typical dimer-interface interactions as E. coli Fe-SOD, whereas PmSOD2, which is more hydrophobic, lacks some conserved interactions (Fig. 3 ▶). The dimer interface for both structures also forms solvent-filled funnels which are assumed to drive the substrate to the active site for Fe-SODs.
Figure 3.

(a) The PmSOD1 dimer interface reveals several polar interactions, notably the Glu26–Arg173 salt bridges, that are conserved in most Fe/Mn-SODs. (b) The PmSOD2 dimer interface is more hydrophobic and lacks the Glu26–Arg173 salt bridge; the equivalent amino-acid residues are Gln180 and Ala26. The absence of such salt bridges is very unusual in both Mn-SODs and Fe-SODs.
4. Final remarks
It is widely accepted that the extant protozoa are derived from one or more ancestral endosymbiotic events. The evolutionary origins of the two Fe-SODs of P. marinus are of interest to researchers attempting to decipher the origins of protozoa, in particular the Alveolata. The apicomplexan parasites T. gondii and P. falciparum each contain multiple Fe-SODs that are highly similar to each other and are presumably the result of relatively recent gene duplication. P. marinus diverges from this pattern in that its two Fe-SODs share less than 40% amino-acid identity and thus gene duplication may not be the most parsimonious explanation for their origins. Two alternative interpretations could be that (i) the fact that their active sites appear to function in much the same way may be evidence of convergent selective pressure acting on ancestrally different proteins or (ii) the structural differences may reflect selective pressures for related/duplicated SODs to fulfill distinct roles in specific compartments within the cell or in the extracellular environment. PmSOD1 is targeted to the mitochondrion, whereas PmSOD2 is localized within a different cellular compartment below the plasma membrane (Schott, Robledo et al., 2003 ▶). The structural differences between PmSOD1 and PmSOD2 are most striking at the dimer interface and in residues on the second shell around the active site. Both are likely to have effects on enzymatic activity since the active site is very close to the dimer interface. The possibility that this is a reflection of their different cellular location and function is an intriguing hypothesis.
Supplementary Material
PDB reference: PmSOD1, 2cw2, r2cw2sf
PDB reference: PmSOD2, 2cw3, r2cw3sf
Acknowledgments
This study was supported by Grant IOB 06 18409 from the National Science Foundation to GRV.
References
- Adams, P. D., Pannu, N. S., Read, R. J. & Brünger, A. T. (1997). Proc. Natl Acad. Sci. USA, 94, 5018–5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed, H., Schott, E. J., Gauthier, J. D. & Vasta, G. R. (2003). Anal. Biochem.318, 132–141. [DOI] [PubMed] [Google Scholar]
- Anderson, R. S., Oliver, L. M. & Brubacher, L. L. (1992). J. Invert. Pathol.59, 303–307.
- Anderson, R. S., Patel, K. M. & Roesijadi, G. (1999). Dev. Comput. Immunol.23, 443–449. [DOI] [PubMed]
- Brünger, A. T. (1992a). X-PLOR. A System for X-ray Crystallography & NMR, v.3.1. New Haven, CT, USA: Yale University Press.
- Brünger, A. T. (1992b). Nature (London), 355, 472–474. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst D54, 905–921. [DOI] [PubMed] [Google Scholar]
- Brünger, A. T., Adams, P. D. & Rice, L. M. (1997). Structure, 5, 325–336. [DOI] [PubMed] [Google Scholar]
- Brünger, A. T., Krukowski, A. & Erickson, J. (1990). Acta Cryst. A46, 585–593. [DOI] [PubMed] [Google Scholar]
- De Groote, M. A., Ochsner, U. A., Shiloh, M. U., Nathan, C., McCord, J. M., Dinauer, M. C., Libby, S. J., Vazquez-Torres, A., Xu, Y. & Fang, F. C. (1997). Proc. Natl Acad. Sci. USA, 94, 13997–14001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford, S. E. (1996). J. Shellfish Res.15, 45–56.
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed] [Google Scholar]
- Lah, M. S., Dixon, M. M., Pattridge, K. A., Stallings, W. C., Fee, J. A. & Ludwig, M. L. (1995). Biochemistry, 34, 1646–1660. [DOI] [PubMed] [Google Scholar]
- Navaza, J. (1994). Acta Cryst. A50, 157–163. [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Saldarriaga, J. F., McEwan, M. L., Fast, N. M., Taylor, F. J. & Keeling, P. J. (2003). Int. J. Syst. Evol. Microbiol.53, 355–365. [DOI] [PubMed] [Google Scholar]
- Schott, E. J., Pecher, W. T., Okafor, F. & Vasta, G. R. (2003). Exp. Parasitol.105, 232–240. [DOI] [PubMed] [Google Scholar]
- Schott, E. J., Robledo, J. A., Wright, A. C., Silva, A. M. & Vasta, G. R. (2003). Gene, 309, 1–9. [DOI] [PubMed] [Google Scholar]
- Stoddard, B. L., Howell, P. L., Ringe, D. & Petsko, G. A. (1990). Biochemistry, 29, 8885–8893. [DOI] [PubMed] [Google Scholar]
- Volety, A. K. & Chu, F. L. (1995). Dev. Comput. Immunol.19, 135–142. [DOI] [PubMed]
- Wright, A., Ahmed, H., Gauthier, J., Silva, A. & Vasta, G. (2002). Mol. Biochem. Parasitol.123, 73–77. [DOI] [PubMed] [Google Scholar]
- Yamano, S. & Maruyama, T. (1999). J. Biochem. (Tokyo), 125, 186–193. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: PmSOD1, 2cw2, r2cw2sf
PDB reference: PmSOD2, 2cw3, r2cw3sf