

Crystals of 6-aminohexanoate-cyclic-dimer hydrolase were obtained by the sitting-drop vapour-diffusion method using sodium citrate as a precipitant. Diffraction data from native and mercury-derivative crystals were collected to resolutions of 1.90 and 2.06 Å, respectively.
Keywords: 6-aminohexanoate-cyclic-dimer hydrolase, biodegradation, nylon oligomers
Abstract
6-Aminohexanoate-cyclic-dimer hydrolase (EI) from Arthrobacter sp. KI72 was expressed in Escherichia coli and purified by anion-exchange chromatography. EI was crystallized by the sitting-drop vapour-diffusion method with sodium citrate as precipitant in imidazole buffer pH 8.0. The crystal is hexagonal, with unit-cell parameters a = b = 130.75, c = 58.23 Å. Diffraction data were collected from native and mercury(II) dichloride-derivative crystals to resolutions of 1.90 and 2.06 Å, respectively.
1. Introduction
Nylon-6 is produced by the polymerization of ∊-caprolactam and consists of more than 100 units of 6-aminohexanoate (Ahx). However, during the polymerization reaction some molecules fail to polymerize and remain as oligomers, while others undergo head-to-tail condensation to form cyclic oligomers. These nylon oligomers are byproducts of nylon-6 factories and thereby contribute to the release of industrial waste material into the environment. Recently, the biodegradation of xenobiotic compounds has been recognized as a useful way to eliminate environmental pollutants. In addition, the biodegradation of xenobiotic compounds provides us with a suitable system to study how the enzymes responsible for the degradation have evolved.
From previous studies, we have found that the nylon-oligomer-degrading bacterium Arthrobacter sp. KI72 possesses three enzymes responsible for the degradation: 6-aminohexanoate-cyclic-dimer hydrolase (EI; Kinoshita et al., 1977 ▶), exo-type 6-aminohexanoate-oligomer hydrolase (EII; Kinoshita et al., 1981 ▶) and endo-type 6-aminohexanoate-oligomer hydrolase (EIII; Kakudo et al., 1993 ▶; Negoro, 2000 ▶).
The EI gene (nylA) has been cloned (Negoro et al., 1983 ▶) and sequenced (Tsuchiya et al., 1989 ▶). A homology search against the protein database (UniProt/PDB/PRF) using the program BLAST (http://www.ddbj.nig.ac.jp/search/blast-j.html) extracted several proteins having low but significant homology [indole acetamide hydrolase from Burkholderia cepacia (35.4% amino-acid sequence identity; EMBL ID code emb|AF029344|) and two amidases, MupX (36.2%; EMBL ID code emb|AF318063|) and PcoX (36.7%; EMBL ID code emb|AY626157|), from Pseudomonas fluorescens]. These amide hydrolases release ammonia during the hydrolysis of amide compounds. However, analysis of the substrate specificity of EI has shown that EI is specific for Ahx cyclic dimer but has no detectable activity towards more than 60 types of amide compound, including Ahx linear dimer, ∊-caprolactam, various peptides and β-lactams (Kinoshita et al., 1977 ▶; Negoro, 2000 ▶). These results raise a question about the structural factors that determine the differences in the substrate specificities of these enzymes.
Knowledge of the crystal structure of the EI enzyme will allow us to study the catalytic mechanism and the evolution of the enzyme. However, little information is available on crystal structures of the amide hydrolase family. In this paper, we report the crystallization and preliminary crystallographic analysis of the EI protein.
2. Materials and methods
2.1. Overexpression and purification
A 1.6 kbp SalI–HindIII fragment containing the nylA gene and including mutations in the initiation codon and the Shine–Delgarno sequence was amplified by PCR using pNDH901 (template; Fig. 1 ▶), Pyrobest DNA polymerase (Takara Co.), forward primer FE1S-1 (5′-GGCGTCGACCAGCAGGAGGAGCGCGACAATGA-3′) and reverse primer RE1H-1 (5′-CAGTTGGGGACGAAAGCTTGGCCCG-3′) as illustrated in Fig. 1 ▶. The amplified fragment was digested with SalI and HindIII and the 1.6 kbp SalI–HindIII fragment was ligated with pUC18 (Sambrook & Russell, 2001 ▶), which had been digested with SalI and HindIII, to give hybrid plasmid pUEFA (Fig. 1 ▶). Escherichia coli JM109 (pUEFA) cells were grown at 298 K in 300 ml Terrific broth medium (12 g Bacto tryptone, 24 g Bacto yeast extract, 4.0 g glycerol, 2.3 g KH2PO4, 12.5 g K2HPO4, 1 l water pH 7.0) with ampicillin (100 mg ml−1) and isopropyl 1-thio-β-galactoside (1 mM). After 24 h of cultivation, cells were harvested by centrifugation at 5000g for 10 min, washed with buffer A (20 mM phosphate buffer containing 10% glycerol pH 7.3) and suspended in 50 ml buffer A. The cells were lysed by sonication (20 kHz, six 1 min blasts) and the lysate obtained by centrifugation at 35 000g for 30 min was used as crude enzyme solution. Precipitates obtained by the addition of (NH4)2SO4 (25% saturation) to the crude enzyme solution were initially removed and (NH4)2SO4 was then added to 55% saturation. EI fractions were collected as precipitate by centrifugation at 20 000g for 30 min. The protein was dissolved in 20 ml buffer A and desalted using a PD-10 desalting column (Amersham Biosciences). The fractions containing protein were loaded onto a Hi-Trap Q-Sepharose (Amersham Biosciences) column (5 ml) equilibrated with buffer A. After the column had been washed with buffer A, the enzyme was eluted by increasing the NaCl concentration from 0 to 0.25 M in 200 ml buffer A (total volume) at a flow rate of 2 ml min−1. 5 ml fractions were collected and the purity of protein in each fraction was checked by SDS–PAGE (12.5%). The EI protein purified to homogeneity was concentrated using a micro-concentrator (Centriprep YM-10; Millipore Inc.). All purification procedures were carried out at 277 K and 5 mg purified EI was obtained.
Figure 1.

Structure of the plasmid expressing the nylA gene. Plasmid pNDH901 is a hybrid plasmid in which a 2.2 kbp EcoRI/SalI fragment containing the EI gene (nylA) was expressed under a tac promoter in vector pKP1500 (Miki et al., 1987 ▶) . To increase the expression level, the unusual initiation codon (GTG in wild-type nylA) was replaced with the more common codon (ATG) and the ribosome-binding (Shine–Dalgarno) sequence (AGAAGG) was replaced with the consensus sequence in E. coli (AGGAGG) by PCR using two primers (FE1S-1 and RE1H-1). In the constructed hybrid plasmid pUEFA, the nylA gene cloned in a 1.6 kbp SalI/HindIII fragment was expressed under a lac promoter in vector pUC18.
2.2. Crystallization
Initial crystallization tests were performed by the sitting-drop vapour-diffusion method in 96-well plates using Wizard I and Wizard II screen kits (Emerald Biostructures). Droplets were prepared by mixing 2–3 µl purified EI solution (10 mg ml−1 protein in buffer A) and 2–3 µl reservoir solution and were equilibrated against 100 µl reservoir solution at 283 K. To obtain crystals suitable for X-ray diffraction, a survey for optimum conditions was made by varying the concentrations of sodium citrate (0.8, 0.9, 1.0 M) and glycerol (0–25%, 2.5% intervals) and the buffer solution (HEPES pH 7.0–8.0, 0.2 pH-unit intervals; Tris–maleate pH 6.5–8.5, 0.5 pH-unit intervals; imidazole pH 8.0).
2.3. Data collection and crystallographic analysis
For data collection, the native crystals were soaked in a cryoprotectant solution [1.0 M sodium citrate, 25%(v/v) glycerol and 0.1 M imidazole buffer pH 8.0] for 24 h prior to freezing in a nitrogen cold stream. For the preparation of the mercury(II) dichloride derivative, the crystals were soaked in cryoprotectant solution containing 0.1 mM mercury(II) dichloride for 24 h and then back-soaked in cryoprotectant solution for 1 h. X-ray diffraction data sets for native and mercury-derivative crystals were collected at 100 K with synchrotron radiation using a Rigaku Jupiter CCD detector system on the BL38B1 beamline at SPring-8 (Hyogo, Japan) and an ADSC Quantum 315 detector system on the BL-5A beamline at Photon Factory (Ibaraki, Japan), respectively. The wavelength used for the diffraction experiments was 1.0000 Å in both cases and the crystal-to-detector distances were maintained at 180 mm (Jupiter) and 300 mm (ADSC) with an oscillation range per image of 1°, covering a total oscillation range of 180°. The exposure times for native and derivative crystals were 20 and 10 s per image, respectively. Determination of the unit-cell parameters and integration of reflections were performed using the HKL-2000 program package (Otwinowski & Minor, 1997 ▶). The unit-cell parameters and crystal mosaicity were post-refined with SCALEPACK.
3. Results
The nylA gene, encoding a polypeptide of 493 amino acids, was cloned and expressed in E. coli and the EI protein was purified to homogeneity. The purity of the protein samples used for crystallization was confirmed by SDS–PAGE.
Of the 100 conditions (Wizard I and II) tested in crystallization screening, microcrystals were obtained using 0.1 M imidazole buffer pH 8.0 containing 1.0 M sodium citrate (Wizard I condition No. 36) as a precipitant after one week. Crystals of the highest quality were obtained using the above precipitant solution containing 12.5% glycerol. Hexagonal bipyramidal crystals grew to typical dimensions of 0.3 × 0.3 × 0.5 mm (Fig. 2 ▶).
Figure 2.

Crystal (0.3 × 0.3 × 0.5 mm) of EI obtained using a reservoir solution consisting of 1.0 M sodium citrate and 0.1 M imidazole buffer pH 8.0 containing 12.5% glycerol.
Diffraction data for the native and mercury chloride-derivative crystals were collected to 1.90 and 2.06 Å, respectively. Neither a dose-dependent increase in R merge nor a decrease in the signal-to-noise ratio [i.e. 〈I/σ(I)〉] per image was detected, indicating that there was no significant radiation damage during data collection. The Laue group symmetry was determined to be 6/m from data-scaling statistics. Systematic absences in the 00l reflections indicated the space group to be P62 or its enantiomorphic counterpart P64. The unit-cell parameters were a = b = 130.75, c = 58.23 Å for the native crystal and a = b = 131.39, c = 58.07 Å for the mercury(II) dichloride-derivative crystal. Crystal parameters and diffraction data statistics are summarized in Table 1 ▶. The Matthews volume V M (Matthews, 1968 ▶) is 2.76 Å3 Da−1, corresponding to a solvent content of 55.4%, if one monomer is assigned to the asymmetric unit of the crystal. Preliminary analysis shows that several Hg sites can be found and structure solution is in progress.
Table 1. Data-collection and phasing statistics.
Values in parentheses are for the outer shell.
| Data collection | Native | Mercury(II) chloride |
|---|---|---|
| Space group | P62 or P64 | P62 or P64 |
| Unit-cell parameters | ||
| a = b (Å) | 130.75 | 131.39 |
| c (Å) | 58.23 | 58.07 |
| Wavelength (Å) | 1.0000 | 1.0000 |
| Resolution (Å) | 30–1.90 (1.97–1.90) | 30–2.06 (2.13–2.06) |
| Total reflections | 485579 | 363039 |
| Unique reflections | 44300 (4021) | 34955 (2980) |
| Completeness (%) | 98.6 (90.4) | 97.2 (82.3) |
| Rmerge† (%) | 3.8 (30.4) | 5.8 (33.4) |
| 〈I/σ(I)〉 | 24.5 (5.2) | 16.5 (6.6) |
R
merge = 100 × 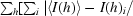
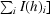 , where I(h)i is the ith observation of reflection h and 〈I(h)〉 is the mean intensity of all observations of h.
, where I(h)i is the ith observation of reflection h and 〈I(h)〉 is the mean intensity of all observations of h.
Acknowledgments
We would like to express our sincere thanks to Dr Y. Shiro of RIKEN SPring-8 Center for helpful discussions. This work was supported in part by grants from the 21st Century COE Program, the National Project on Protein Structural and Functional Analyses and the JAXA project.
References
- Kakudo, S., Negoro, S., Urabe, I. & Okada, H. (1993). Appl. Environ. Microbiol.59, 3978–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita, S., Negoro, S., Muramatsu, M., Bisaria, V. S., Sawada, S. & Okada, H. (1977). Eur. J. Biochem.80, 489–495. [DOI] [PubMed] [Google Scholar]
- Kinoshita, S., Terada, T., Taniguchi, T., Takene, Y., Masuda, S., Matsunaga, N. & Okada, H. (1981). Eur. J. Biochem.116, 547–551. [DOI] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Miki, T., Yasukochi, T., Nagatani, H., Furuno, M., Orita, T., Yamada, H., Imoto, T. & Horiuchi, T. (1987). Protein Eng.1, 327–332. [DOI] [PubMed] [Google Scholar]
- Negoro, S. (2000). Appl. Microbiol. Biotechnol.54, 461–466. [DOI] [PubMed] [Google Scholar]
- Negoro, S., Taniguchi, T., Kanaoka, M., Kimura, H. & Okada, H. (1983). J. Bacteriol.155, 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Sambrook, J. & Russell, D. W. (2001). Molecular Cloning: A Laboratory Manual, 3rd ed. Cold Spring Harbor, NY, USA: Cold Spring Harbor Laboratory Press.
- Tsuchiya, K., Fukuyama, S., Kanzaki, N., Kanagawa, K., Negoro, S. & Okada, H. (1989). J. Bacteriol.171, 3187–3191. [DOI] [PMC free article] [PubMed] [Google Scholar]