

The structure of HLA-A*1101 in complex with a HBV peptide homologue is presented and discussed in the context of vaccine design.
Keywords: HLA-A*1101, major histocompatibility complex class I, hepatitis B, HBV, peptide decamer, vaccine development
Abstract
A high-resolution structure of the human MHC-I molecule HLA-A*1101 is presented in which it forms a complex with a sequence homologue of a peptide that occurs naturally in hepatitis B virus DNA polymerase. The sequence of the bound peptide is AIMPARFYPK, while that of the corresponding natural peptide is LIMPARFYPK. The peptide does not make efficient use of the middle E pocket for binding, which leads to a rather superficial and exposed binding mode for the central peptide residues. Despite this, the peptide binds with high affinity (IC50 of 31 nM).
1. Introduction
Major histocompatibility class I (MHC-I) molecules are essential for the control of infections by intracellular pathogens (Klein et al., 1993 ▶). During chaperone-assisted folding in the endoplasmic reticulum (Elliott & Williams, 2005 ▶), each MHC-I molecule binds a peptide from the cytoplasm and transports it to the cell surface, where the peptide–MHC complexes are scrutinized by cytotoxic T lymphocytes (CTL). This sampling of the intracellular protein environment allows CTLs to identify and eliminate infected cells (Townsend & Bodmer, 1989 ▶). In humans, MHC molecules are referred to as human leukocyte antigens (HLAs).
Peptides bind to MHC-I molecules in a special peptide-binding groove, where six binding pockets (A–F) determine the specificity of the MHC-I molecule (Garrett et al., 1989 ▶). Most peptides binding to MHC-I molecules consist of 8–11 residues, but binding of peptides up to 14 residues in length has been reported (Probst-Kepper et al., 2004 ▶). MHC-I molecules are highly polymorphic and more than 1000 different variants, each with its own binding specificity, exist in the human population (http://www.anthonynolan.org.uk/HIG/index.html). Studies of the many MHC-I alleles have allowed a grouping based on peptide-binding preferences and so far 12 different supertypes have been identified (Lund et al., 2004 ▶; Sette & Sidney, 1999 ▶). More than 99% of all humans carry MHC-I molecules belonging to at least one of these supertypes.
HLA-A*1101 is known to be important for the control of infections by many different pathogens including HIV (Culmann et al., 1991 ▶), Epstein–Barr virus (Gavioli et al., 1993 ▶) and hepatitis B virus (HBV; Achour et al., 1986 ▶). It is one of the most common MHC-I alleles and is present in up to 27% of some Asian populations (Bodmer et al., 1999 ▶). Furthermore, it belongs to the A3 supertype, the second most common supertype, found in 44% of the human population (Sette & Sidney, 1999 ▶). Thus, peptides targeting HLA-A*1101 are attractive for inclusion in peptide-based vaccines as they afford broad population coverage and understanding HLA-A*1101 will aid in identifying such peptides. Here, we present the 1.6 Å X-ray structure of a complex of HLA-A*1101 with an HBV peptide homologue.
2. Materials and methods
2.1. Protein production
The complex of HLA-A*1101 was prepared as described previously (Blicher et al., 2005 ▶; Pedersen et al., 1995 ▶; Sidney et al., 1996 ▶). Briefly, the α-chain of HLA-A*1101 and β2-microglobulin (β2m) were produced recombinantly in Escherichia coli and purified under denaturing conditions. β2m was refolded on its own, while the complex was refolded by dilution of denatured α-chain into a folding buffer containing excess (folded) β2m and peptide (200 µM α-chain, 1.0 mM β2m, 2.0 mM peptide). Following stepwise concentration and centrifugation to remove misfolded protein, excess peptide and β2m were removed using size-exclusion chromatography (Superdex 75 column) and the complexes were concentrated to a final concentration of 6.0 mg ml−1 in 0.1 M MES pH 6.5.
The peptide used for folding of the HLA-A*1101 complex was purchased from Schafer-N (Copenhagen) as a crude preparation and was found to be approximately 87% pure (see §3). The peptide was synthesized using FMOC chemistry.
The final complex consists of residues 1–275 of the α-chain, corresponding to the extracellular part, residues 1–99 of β2m, corresponding to the native protein, and the peptide.
2.2. Crystallization and data collection
Crystals of HLA-A*1101 were prepared by vapour-diffusion experiments using the hanging-drop technique. Crystallization experiments were set up at room temperature (∼298 K), with individual drops consisting of 2 µl protein solution (6.0 mg ml−1 in 0.1 M MES pH 6.5) and 2 µl crystallization buffer. Each drop was equilibrated over a reservoir containing 500 µl crystallization buffer. The crystals grew from a crystallization buffer containing 30% PEG 5000 MME and 0.2 M ammonium sulfate (Crystal Screen 2, Hampton Research), appearing as clusters of monoclinic plates within 4 d and reaching their final size of 0.3 × 0.1 × 0.01 mm in three weeks. Crystals were cryocooled in liquid nitrogen directly from the crystallization buffer. A data set to 1.60 Å resolution was collected at the ID14-1 beamline at ESRF (Grenoble, France) using a Quantum 4 CCD detector. The space group was P21, with unit-cell parameters a = 58.0, b = 79.7, c = 56.3 Å, β = 116.4° and one complex per asymmetric unit. The data set was processed with the HKL suite (Otwinowski & Minor, 1997 ▶), giving the data statistics outlined in Table 1 ▶.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Space group | P21 |
| Resolution range (Å) | 30–1.60 (1.66–1.60) |
| Completeness (%) | 99.7 (98.7) |
| Total observations | 205852 (18837) |
| Unique reflections | 60539 (5977) |
| Average I/σ(I) | 18.5 (2.5) |
| Rsym† (%) | 7.4 (36.7) |
| Wilson B factor (Å2) | 17 |
| Model refinement | |
| Rcryst‡ (%) | 14.8/14.0§ |
| Rfree¶ (%) | 21.2/20.0§ |
| DPI†† (Å) | 0.084 |
| No. of atoms/average B factor (Å2) | |
| Protein atoms | 3191/20 |
| Water molecules | 543/37 |
| Sulfate ions | 15/53 |
| Ramachandran statistics‡‡ | |
| Most favoured (%) | 92.5 |
| Disallowed (%) | 0.0 |
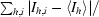
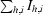
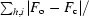
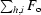
2.3. Structure determination and refinement
The structure was solved by molecular replacement (MR) with AMoRe (Navaza, 1994 ▶) using a structure of HLA-A*6801 without peptide or water molecules (PDB code 1hsb; Guo et al., 1992 ▶) as the model. The MR solution was subsequently fed into the program ARP/wARP (Perrakis et al., 2001 ▶) for removal of bias by iterative rebuilding of the entire molecule including the peptide. All subsequent refinement cycles included refinement of atomic positions and B factors and were carried out with SHELXL (Sheldrick & Schneider, 1997 ▶) with 5% of the data reserved for calculation of R free. Manual rebuilding and adjustments of the model during refinements were carried out using O (Jones et al., 1991 ▶). Details of the refinement process are listed in Table 2 ▶. The model was refined with riding H atoms and restrained anisotropic B factors for all atoms (not hydrogen).
Table 2. Refinement progress.
| Refinement step | Rcryst/Rfree (%) | H2O | Comment |
|---|---|---|---|
| First round | 22.5/26.1 | 284 | Input model from ARP/wARP |
| Second round | 20.2/24.4 | 281 | + riding H atoms |
| Third round | 16.9/22.1 | 349 | + restrained anisotropy |
| Final round(s) | 14.8/21.2 | 543 |
No B-factor cutoff was used during the last rounds of water refinement (previous rounds: 60 Å2). Rather, all water molecules with clear 2F o − F c density at 1σ and hydrogen-bonding partners within 3.5 Å were retained. The final model includes 274 of the 275 residues of the extracellular domain of the α-chain, all 99 residues of β2m, the HBV peptide homologue (AIMPARFYPK), three sulfate ions and 543 water molecules. The following 12 residues were modelled in double conformations: Ser2α, Ser11α, Arg35α, Asn86α, Met98α, Met189α, Thr216α, Gln226α, Lys243α, Thr4β, Val27β and Ser55β.
2.4. Peptide-binding measurements
Peptide-binding experiments were conducted according to standard protocols (Buus et al., 1995 ▶). Briefly, binding of peptide to the α-chain was conducted as a folding by dilution competition assay in the presence of peptide and β2m as previously described (Pedersen et al., 2001 ▶). A titration series of peptide was mixed with a fixed amount of α-chain, β2m and radioactive tracer peptide (125I-labelled). The reaction was incubated for at least 4 h at 291 K in order to obtain a steady state. The peptide binding was examined by Sephadex G25 spun column chromatography (Buus et al., 1995 ▶) and the radioactivity in the MHC complex was counted and compared with the total radioactivity. The two peptides (AIMPARFYPK and IMPARFYPK) used in peptide-binding experiments were purchased from Schafer-N (Copenhagen) as HPLC-purified preparations more than 95% pure.
2.5. Mass spectrometry
Liquid-chromatography mass spectrometry (LC–MS) was used to investigate the composition of the HBV peptide batch. The sample (1 mg ml−1 in water) was applied onto a C18 column controlled by a Hewlett Packard Series 1100 HPLC and eluted with a gradient of acetonitrile in water (0–50%) containing 0.02% trifluoroacetic acid (TFA). The eluting fractions were continuously monitored using electrospray ionization (ESI) mass spectrometry performed on a Bruker Esquire LC mass spectrometer.
3. Results and discussion
3.1. The AIMPARFYPK peptide
The HLA-A*1101 complex was formed in the presence of ∼87% pure IMPARFYPK peptide (see below) and crystallized. This peptide occurs naturally in hepatitis B virus DNA polymerase (residues 110–118) and binds to HLA-A*1101 (Sidney et al., 1996 ▶). Unexpectedly, it turned out that the crystals contained a peptide with an additional N-terminal alanine. The natural amino-acid residue in this position is a leucine (NCBI accession P03156), which makes the extended peptide a homologue of the natural sequence. The electron density of the peptide was very well defined and unambiguously showed the presence of the additional N-terminal residue (Fig. 1 ▶). Furthermore, the automatic and unbiased model-building program ARP/wARP implemented in CCP4i (Collaborative Computational Project, 1994 ▶; Potterton et al., 2003 ▶) assigned an extra residue to the N-terminus of the peptide, giving rise to the decamer peptide with sequence AIMPARFYPK. This finding prompted further investigation into the source of this peptide.
Figure 1.

Electron density of the AIMPARFYPK peptide. The electron density is contoured at 1σ and clearly shows density for all of the peptide including the additional N-terminal alanine (left). The peptide is coloured according to B values from blue to red (13–48 Å2). (a) Side view, (b) top view. All figures were prepared with PyMOL (DeLano, 2002 ▶).
The most likely source of contaminating peptide species is the peptide stock used to generate the HLA-A*1101 complex. To analyze for the presence of peptide impurities, the peptide stock was subjected to mass spectrometry. This analysis showed an 87% content of IMPARFYPK and identified several minor contaminants (data not shown), of which one matched the AIMPARFYPK peptide and comprised approximately 1.4% of the stock. This N-terminally extended peptide was synthesized and tested for its binding to HLA-A*1101 in a peptide-binding assay and compared with the binding of the original HBV peptide (re-synthesized). The results are summarized in Table 3 ▶ and demonstrate similar IC50 values for the two peptides.
Table 3. Peptide-binding data.
| Sequence | IC50 (nM) |
|---|---|
| IMPARFYPK | 26 |
| AIMPARFYPK | 31 |
As the IC50 values of the IMPARFYPK and AIMPARFYPK peptides are very similar, the equilibrium conditions during folding of the HLA-A*1101 complex imply a relative distribution of the individual peptide complexes with HLA-A*1101 corresponding to that of the peptide stock solution. The fact that the HLA-A*1101 crystallized with the extended peptide impurity could be a consequence of preferential crystallization of HLA-A*1101 in complex with the extended peptide. As the overall structure of MHC-I molecules is generally largely unaffected by changes in peptide identity, any special peptide preferences not dictated by the peptide–MHC interaction itself would have to arise from direct interactions between neighbouring complexes in the crystal. A few crystal-packing interactions between molecules in the present structure involve the outer edges of the peptide-binding groove, but only in the part holding the peptide C-terminus. Furthermore, only one weak interaction between the peptide (Phe7p) and a neighbouring β2m molecule (Lys19β) is found. A more likely explanation is therefore the occurrence of differential non-equilibrium stability under the crystallization and/or purification conditions, since the relative distribution of peptides in HLA-A*1101 would be affected if the complexes have different half-lives. In biological settings, similar non-equilibrium conditions occur at the cell surface and in general only long-lived complexes will be available in amounts sufficient for T-cell recognition.
3.2. The quality of the HLA-A*1101 structure
The structure of HLA-A*1101 was solved to a resolution of 1.60 Å with R cryst and R free values of 14.8 and 21.2%, respectively (see Tables 1 ▶ and 2 ▶ and Fig. 2 ▶). The protein complex is complete except for the C-terminal residue (Glu275α) of the α-chain, which could not be modelled owing to insufficient electron density. The largest positive peak in the F o − F c map is found in this region next to Trp274α and has a peak density of 0.44 e Å−3 (7.9σ, σ = 0.055 e Å−3). All other positive peaks are below 0.25 e Å−3. The largest negative peak (−0.42 e Å−3, −7.6σ) is centred on the S atom of sulfate ion 4002. This is most likely to be the consequence of an underestimation (hard restraint) of the anisotropic motion of this ion and/or incomplete occupancy. The occupancy was not refined for any of the sulfate ions as it couples very strongly with the B factor. In addition to the protein complex and three sulfate ions, the structure includes 543 water molecules.
Figure 2.

Schematic overview of the structure of HLA-A*1101 in complex with the AIMPARFYPK peptide. The AIMPARFYPK peptide (red; labelled ‘p’) is viewed from the N-terminal end. The α-chain is shown in grey with individual subdomains indicated; β2-microglobulin is in blue. Notice the relatively exposed position of the peptide in the binding groove.
3.3. The interactions of HLA-A*1101 with the peptide
The AIMPARFYPK peptide is bound to the peptide-binding groove of the α-chain through 25 hydrogen bonds, 12 of which are mediated by water molecules (Table 4 ▶). All hydrogen bonds are to the peptide backbone, except for the binding of the amino group of the C-terminal lysine side chain to the backbone carbonyl O atom of Asp116α. Not all potential hydrogen-bonding acceptors of the peptide residues are used for binding (see Table 4 ▶). In particular, the central residues Met3p, Pro4p, Ala5p and Arg6p exhibit large mobility as judged from the B values (Fig. 1 ▶).
Table 4. Peptide-binding interactions.
| Peptide | Hydrogen-bond partner† | ||||
|---|---|---|---|---|---|
| Residue | Atom | Residue | Atom | Distance (Å) | van der Waals contact residues‡ |
| Ala1p | N | Tyr7α | OH | 3.0 | Met5α, Tyr7α, Glu63α, Trp167α |
| N | Tyr171α | OH | 2.7 | ||
| O | Tyr159α | OH | 2.5 | ||
| Ile2p | N | Glu63α | O∊1 | 2.9 | Tyr7α, Tyr9α, Met45α, Glu63α, Asn66α, Val67α, Tyr99α, Tyr159α, W69 |
| O | W69 | OW | 3.0 | ||
| Met3p | N | Tyr99α | OH | 2.9 | Asn66α, Tyr99α, Arg114α, Gln156α, Tyr159α, (W215) |
| Pro4p | O | W384 | OW | 2.7 | Tyr159α, (SO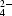 4003), (W179), (W340) 4003), (W179), (W340) |
| Ala5p | N | Asn66α | Oδ1 | 3.4 | (Asn66α), (W189), (W384) |
| Arg6p | N | W384 | OW | 3.1 | Gln155α, Tyr8p |
| O | W41 | OW | 3.5 | ||
| O | W167 | OW | 2.7 | ||
| O | (W206) | OW | 3.5 | ||
| N∊ | (Tyr8p) | OH | 3.0 | ||
| Nη2 | (Tyr8p) | OH | 3.0 | ||
| Nη1 | (W340) | OW | 2.7 | ||
| Phe7p | N | (W377) | OW | 2.9 | Thr73α, Val76α, Pro9p, (W41) |
| Tyr8p | N | W41 | OW | 2.9 | Trp147α, Trp147α, Ala150α, Ala152α, Gln155α, Arg6p, (W41), (W62), (W125), (W167), (W210) |
| O | (W41) | OW | 3.4 | ||
| O | (W45) | OW | 2.6 | ||
| O | (Trp147α) | N∊1 | 3.0 | ||
| Pro9p | O | (W62) | OW | 2.7 | Asp77α, Lys146α, Trp147α, Phe7p, W11, W62, W91, (W187), (W291), (W468) |
| O | (Trp147α) | N∊1 | 3.3 | ||
| Lys10p | N | Asp77α | Oδ1 | 3.0 | Asp77α, Leu81α, Ile95α, Thr143α, Trp147α, W45 |
| OXT | Tyr84α | OH | 2.6 | ||
| OXT | Thr143α | Oγ1 | 2.7 | ||
| O | Lys146α | Nζ | 2.7 | ||
| O | W11 | OW | 2.9 | ||
| Nζ | Asp116α | Oδ2 | 2.7 | ||
| Nζ | W2 | OW | 2.9 | ||
| Nζ | W6 | OW | 2.9 | ||
The cutoff distance for a hydrogen bond is 3.5 Å. Entities in parentheses form hydrogen bonds with suboptimal geometry (polar contact) or are judged not to be important for peptide binding.
The cutoff distance for a van der Waals contact is 4.0 Å. Residues having any atom within this distance are considered to be contact residues. Entities in parentheses are judged not to be important for peptide binding.
As would be expected (see Fig. 3 ▶), Ala1p binds in the A pocket, Ile2p in the B pocket, Met3p in the D pocket and Lys9p in the F pocket, with the common hydrogen-bonding patterns at the N- and C-termini. Although Ala5p points towards the edge of the C pocket, it only makes contact with Asn66α. Arg6p rests on top of Gln155α between the D and E pockets and only makes a superficial contact with the binding groove. Phe7p rests against a hydrophobic patch on the upper part of the region between the C and F pockets. Peptides binding to HLA-A*1101 and HLA-A*6801 have previously been reported to make effective use of the E binding pocket using either the sixth (nonamer peptides) or seventh (decamer peptides) residue of the peptide (Li & Bouvier, 2004 ▶), which allows a relatively deep position of the peptide in the groove. The AIMPARFYPK peptide is positioned rather high in the groove even when compared with other A3 supertype molecules with decameric peptides bound (PDB codes 1qvo and 1tmc; see Figs. 2 ▶ and 4 ▶). This is a consequence of the bulky nature of the side chains of this particular peptide (i.e. residues Arg6p, Phe7p and Tyr8p), especially Phe7p, which apparently does not fit into the E pocket. Instead, Tyr8p occupies the upper part of the E pocket, leaving the lower part filled with water molecules. Pro9p is positioned in the region between the C, E and F pockets and its side chain primarily interacts with the side chain of Phe7p. In the absence of strong interactions between the central peptide residues and the peptide-binding groove, Arg6p interacts with Tyr8p and Phe7p with Pro9p, thereby stabilizing the peptide conformation.
Figure 3.

A T-cell view of the bound AIMPARFYPK peptide of the HLA-A*1101 complex. Binding pockets are indicated with colours: A, green; B, cyan; C, red; D, yellow; E, orange; F, blue. The peptide and selected water molecules suggested to be important for peptide binding are shown in white (see also Table 4 ▶). Notice the large number of water molecules beneath the peptide and that Pro4p covers Met3p.
Figure 4.

The peptide positions in the binding groove after superimposition of MHC complex structures. The Cα atoms of the peptides have been connected and α-chain residues have been removed for clarity. (a) The AIMPARFYPK peptide (red, numbered) follows a relatively superficial path, whereas other decamer peptides bound by A3 supertype molecules [green, HLA-A*6801 complex (PDB code 1tmc; Collins et al., 1995 ▶); cyan, HLA-A*1101 complex (PDB code 1qvo; Li & Bouvier, 2004 ▶)] achieve a deeper position by placing residue 6 in the E pocket (indicated with an E). Complexes with nonamer peptides [yellow, HLA-A*1101 complex (PDB code 1q94; Li & Bouvier, 2004 ▶); green, HLA-A*1101 complex (PDB code 1x7q; Blicher et al., 2005 ▶)] utilize residue 5 for binding in the E pocket in a similar fashion. (b) Comparison of the AIMPARFYPK peptide (red) with two decamer peptides [purple, HLA-A*0201 complex (PDB code 1i4f; Hillig et al., 2001 ▶); orange, HLA-B*2705 (PDB code 2bss; Stewart-Jones, Di Gleria et al., 2005 ▶)] and two 11-residue peptides [blue, HLA-B*5703 (PDB code 2bvo; Stewart-Jones, Gillespie et al., 2005 ▶); green, HLA-B*3501 (PDB code 1zsd; Miles et al., 2005 ▶)] bound by other HLA alleles. Although the AIMPARFYPK peptide bulges out of the groove, it is not unusual for a decamer peptide. The orientation is the same as in (a).
Although the exposed bulging conformation of the AIMPARFYPK peptide in the binding groove approaches that of peptides of 11 residues (see Fig. 4 ▶ b), it is neither unusual for a decameric peptide nor does it constitute a general problem for immunogenicity (Burrows et al., 2006 ▶), since binding as well as TCR recognition of peptides up to 13 residues in length has been reported (Speir et al., 2001 ▶; Tynan, Borg et al., 2005 ▶; Tynan, Burrows et al., 2005 ▶). All residues from Pro4p to Pro9p are directly accessible for interaction with a T-cell receptor (see Fig. 3 ▶). Of note, the residue in position three (Met3p) is not accessible, a property also observed for the other A11 (e.g. PDB code 1qvo) and A68 (e.g. PDB code 1tmc) decamer complexes (Collins et al., 1995 ▶; Li & Bouvier, 2004 ▶). The N-terminal alanine residue is accessible for interaction, although very deeply buried in the groove. Molecular modelling indicates that there is plenty of room for this alanine to be replaced by a leucine without any significant rearrangements of either peptide or α-chain residues (not shown). Furthermore, T-cell reactivity data for HLA-A*0201 suggest that many T cells interact weakly or not at all with the first residue of the peptide (Hausmann et al., 1999 ▶; Lee et al., 2004 ▶). Taken together, this is likely to make the AIMPARFYPK peptide a good representative of the naturally occurring peptide LIMPARFYPK. In the context of vaccine design, the Met3p residue could be substituted with a polar residue such as glutamine or even a non-natural amino-acid residue to make more efficient use of the D pocket. This would enhance the overall binding properties of the peptide without affecting T-cell recognition and thus increase its potential as a HBV vaccine candidate.
Supplementary Material
PDB reference: HLA-A*1101 in complex with HBV peptide homologue, 2hn7, r2hn7sf
Acknowledgments
The authors would like to thank Anette Henriksen for assisting in data collection, Thomas Kofoed for assisting with the mass-spectrometric analysis, Kasper Lamberth for the peptide-binding measurements and Ole Kristensen and Osman Mirza for useful discussions and comments. This work was supported by Danish Medical Research Council grant 9601615, the Fifth Framework program of the European Commission (grant QLRT-1999-00173), the NIH (HHSN266200400025C) and the Danish Natural Science Research Foundation DANSYNC project.
References
- Achour, A., Begue, B., Gomard, E., Paul, P., Sayagh, B., Van Pel, A. & Levy, J. P. (1986). Eur. J. Immunol.16, 597–604. [DOI] [PubMed] [Google Scholar]
- Blicher, T., Kastrup, J. S., Buus, S. & Gajhede, M. (2005). Acta Cryst. D61, 1031–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodmer, J., Cambon-Thomsen, A., Hors, J., Piazza, A. & Sanchez-Mazas, A. (1999). In Proceedings of the Twelfth International Histocompatibility Workshop and Conference, edited by D. Charron. Paris: EDK.
- Burrows, S. R., Rossjohn, J. & McCluskey, J. (2006). Trends Immunol.27, 11–16. [DOI] [PubMed] [Google Scholar]
- Buus, S., Stryhn, A., Winther, K., Kirkby, N. & Pedersen, L. O. (1995). Biochim. Biophys. Acta, 1243, 453–460. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Collins, E. J., Garboczi, D. N., Karpusas, M. N. & Wiley, D. C. (1995). Proc. Natl Acad. Sci. USA, 92, 1218–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruickshank, D. W. (1999). Acta Cryst. D55, 583–601. [DOI] [PubMed] [Google Scholar]
- Culmann, B., Gomard, E., Kieny, M. P., Guy, B., Dreyfus, F., Saimot, A. G., Sereni, D., Sicard, D. & Levy, J. P. (1991). J. Immunol.146, 1560–1565. [PubMed] [Google Scholar]
- DeLano, W. L. (2002). The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA, USA.
- Elliott, T. & Williams, A. (2005). Immunol. Rev.207, 89–99. [DOI] [PubMed] [Google Scholar]
- Garrett, T. P., Saper, M. A., Bjorkman, P. J., Strominger, J. L. & Wiley, D. C. (1989). Nature (London), 342, 692–696. [DOI] [PubMed] [Google Scholar]
- Gavioli, R., Kurilla, M. G., Campos-Lima, P. O., Wallace, L. E., Dolcetti, R., Murray, R. J., Rickinson, A. B. & Masucci, M. G. (1993). J. Virol.67, 1572–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, H. C., Jardetzky, T. S., Garrett, T. P., Lane, W. S., Strominger, J. L. & Wiley, D. C. (1992). Nature (London), 360, 364–366. [DOI] [PubMed] [Google Scholar]
- Hausmann, S., Biddison, W. E., Smith, K. J., Ding, Y. H., Garboczi, D. N., Utz, U., Wiley, D. C. & Wucherpfennig, K. W. (1999). J. Immunol.162, 5389–5397. [PubMed] [Google Scholar]
- Hillig, R. C., Coulie, P. G., Stroobant, V., Saenger, W., Ziegler, A. & Hulsmeyer, M. (2001). J. Mol. Biol.310, 1167–1176. [DOI] [PubMed] [Google Scholar]
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed] [Google Scholar]
- Klein, J., Satta, Y., O’hUigin, C. & Takahata, N. (1993). Annu. Rev. Immunol.11, 269–295. [DOI] [PubMed] [Google Scholar]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291. [Google Scholar]
- Lee, J. K., Stewart-Jones, G., Dong, T., Harlos, K., Di Gleria, K., Dorrell, L., Douek, D. C., van der Merwe, P. A., Jones, E. Y. & McMichael, A. J. (2004). J. Exp. Med.200, 1455–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. & Bouvier, M. (2004). J. Immunol.172, 6175–6184. [DOI] [PubMed] [Google Scholar]
- Lund, O., Nielsen, M., Kesmir, C., Petersen, A. G., Lundegaard, C., Worning, P., Sylvester-Hvid, C., Lamberth, K., Roder, G., Justesen, S., Buus, S. & Brunak, S. (2004). Immunogenetics, 55, 797–810. [DOI] [PubMed] [Google Scholar]
- Miles, J. J., Elhassen, D., Borg, N. A., Silins, S. L., Tynan, F. E., Burrows, J. M., Purcell, A. W., Kjer-Nielsen, L., Rossjohn, J., Burrows, S. R. & McCluskey, J. (2005). J. Immunol.175, 3826–3834. [DOI] [PubMed] [Google Scholar]
- Navaza, J. (1994). Acta Cryst. A50, 157–163. [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Pedersen, L. O., Nissen, M. H., Hansen, N. J., Nielsen, L. L., Lauenmoller, S. L., Blicher, T., Nansen, A., Sylvester-Hvid, C., Thromsen, A. R. & Buus, S. (2001). Eur. J. Immunol.31, 2986–2996. [DOI] [PubMed] [Google Scholar]
- Pedersen, L. O., Stryhn, A., Holter, T. L., Etzerodt, M., Gerwien, J., Nissen, M. H., Thogersen, H. C. & Buus, S. (1995). Eur. J. Immunol.25, 1609–1616. [DOI] [PubMed] [Google Scholar]
- Perrakis, A., Harkiolaki, M., Wilson, K. S. & Lamzin, V. S. (2001). Acta Cryst. D57, 1445–1450. [DOI] [PubMed] [Google Scholar]
- Potterton, E., Briggs, P., Turkenburg, M. & Dodson, E. (2003). Acta Cryst. D59, 1131–1137. [DOI] [PubMed] [Google Scholar]
- Probst-Kepper, M., Hecht, H.-J., Herrmann, H., Janke, V., Ocklenburg, F., Klempnauer, J., van den Eynde, B. J. & Weiss, S. (2004). J. Immunol.173, 5610–5616. [DOI] [PubMed] [Google Scholar]
- Sette, A. & Sidney, J. (1999). Immunogenetics, 50, 201–212. [DOI] [PubMed] [Google Scholar]
- Sheldrick, G. M. & Schneider, T. R. (1997). Methods Enzymol.277, 319–343. [PubMed]
- Sidney, J., Grey, H. M., Southwood, S., Celis, E., Wentworth, P. A., del Guercio, M. F., Kubo, R. T., Chesnut, R. W. & Sette, A. (1996). Hum. Immunol.45, 79–93. [DOI] [PubMed] [Google Scholar]
- Speir, J. A., Stevens, J., Joly, E., Butcher, G. W. & Wilson, I. A. (2001). Immunity, 14, 81–92. [DOI] [PubMed] [Google Scholar]
- Stewart-Jones, G. B., Di Gleria, K., Kollnberger, S., McMichael, A. J., Jones, E. Y. & Bowness, P. (2005). Eur. J. Immunol.35, 341–351. [DOI] [PubMed] [Google Scholar]
- Stewart-Jones, G. B., Gillespie, G., Overton, I. M., Kaul, R., Roche, P., McMichael, A. J., Rowland-Jones, S. & Jones, E. Y. (2005). J. Immunol.175, 2459–2468. [DOI] [PubMed] [Google Scholar]
- Townsend, A. & Bodmer, H. (1989). Annu. Rev. Immunol.7, 601–624. [DOI] [PubMed] [Google Scholar]
- Tynan, F. E., Borg, N. A., Miles, J. J., Beddoe, T., El Hassen, D., Silins, S. L., van Zuylen, W. J., Purcell, A. W., Kjer-Nielsen, L., McCluskey, J., Burrows, S. R. & Rossjohn, J. (2005). J. Biol. Chem.280, 23900–23909. [DOI] [PubMed] [Google Scholar]
- Tynan, F. E., Burrows, S. R., Buckle, A. M., Clements, C. S., Borg, N. A., Miles, J. J., Beddoe, T., Whisstock, J. C., Wilce, M. C., Silins, S. L., Burrows, J. M., Kjer-Nielsen, L., Kostenko, L., Purcell, A. W., McCluskey, J. & Rossjohn, J. (2005). Nature Immunol.6, 1114–1122. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: HLA-A*1101 in complex with HBV peptide homologue, 2hn7, r2hn7sf