

The Kunitz-type trypsin/chymotrypsin inhibitor isolated from C. papaya latex has been crystallized using the hanging-drop vapour-diffusion method. Two different crystal forms are observed, diffracting to 2.6 and 1.7 Å.
Keywords: protease inhibitors, detwinning
Abstract
A Kunitz-type protease inhibitor purified from the latex of green papaya (Carica papaya) fruits was crystallized in the presence and absence of divalent metal ions. Crystal form I, which is devoid of divalent cations, diffracts to a resolution of 2.6 Å and belongs to space group P31 or P32. This crystal form is a merohedral twin with two molecules in the asymmetric unit and unit-cell parameters a = b = 74.70, c = 78.97 Å. Crystal form II, which was grown in the presence of Co2+, diffracts to a resolution of 1.7 Å and belongs to space group P212121, with unit-cell parameters a = 44.26, b = 81.99, c = 140.89 Å.
1. Introduction
Proteolytic enzymes or proteases catalyse the cleavage of peptide bonds in other proteins. Some proteases are involved in intracellular and extracellular protein digestion, but most of them function in much more specialized biological processes such as the activation of zymogens, the release of hormones and physiologically active peptides from precursors, the translocation of polypeptides through membranes, the trimming of assembling proteins or the activation of receptors (Neurath, 1984 ▶). Despite their life-giving functions, enzymes that break down proteins are potentially hazardous to their environment; therefore, their activities must be kept strictly controlled. This control is achieved by regulated expression/secretion, by activation of the pro-proteases, by specific degradation of the mature enzymes and by blockage of their proteolytic activity through inhibition (Bode & Huber, 2000 ▶; Khan & James, 1998 ▶). The latter regulation mode plays a key role in many cells, tissues and organisms. Almost all known naturally occurring protease inhibitors directed towards endogenous proteases are proteins; only some microorganisms secrete nonproteinaceous compounds that block the host protease activity (Krowarsch et al., 2003 ▶).
The study of protease inhibitors is nearly as old as that of the proteases themselves. They are divided into families according to the class of proteolytic enzymes inhibited, the sequence and structural similarity, the location of disulfide bridges and the reactive site (Laskowski & Kato, 1980 ▶). The Kunitz-type inhibitors of serine proteases are subdivided into two families: the bovine pancreatic trypsin inhibitor (BPTI) family and the soybean trypsin inhibitor (STI) family (Rawlings et al., 2004 ▶).
The STI-type inhibitors consist of a β-trefoil fold (Murzin et al., 1992 ▶) with an approximate threefold internal symmetry. This fold also has been repeatedly observed in other protein superfamilies (Lacy et al., 1998 ▶; Transue et al., 1997 ▶; Zhu et al., 1991 ▶). The reactive site (Arg63-Ile64 in STI) is located at one end of the structure on a protruding loop that possesses a characteristic canonical conformation. Unlike other protease inhibitors, the reactive loop of STI-like inhibitors is not constrained by secondary-structure elements or disulfide bridges that could limit its conformational freedom (Song & Suh, 1998 ▶). For this family, the reactive loop is stabilized by the side chain of a conserved Asn residue and a network of hydrogen bonds (Ravichandran et al., 2001 ▶).
Here, we describe the crystallization of a Kunitz-type serine protease inhibitor isolated from the latex of green Carica papaya fruits (PPI). PPI has a molecular weight of 23 kDa and two disulfide bridges. The protein is active against bovine trypsin and α-chymotrypsin. Inhibition was stoichiometric for both proteases, with 1 mol of enzyme being inhibited by 1 mol of PPI. However, PPI previously incubated with an equimolar amount of trypsin weakly inhibits α-chymotrypsin and when pre-incubated with α-chymotrypsin strongly inhibits trypsin (Odani et al., 1996 ▶). PPI (SWISS-PROT accession No. P80691) has only 28% sequence identity with STI, the archetypal member of this protein family. Moreover, the sequence of the loop of STI that binds into the active site of trypsin is not conserved. Therefore, crystal structure determination of PPI may reveal novel features that are relevant for the mode of action of STI-type protease inhibitors.
2. Materials and methods
2.1. Purification of PPI
The protein was purified as described elsewhere (Azarkan et al., 2004 ▶, 2006 ▶). Dried latex collected from green papaya fruits was resuspended in water in the presence of S-methyl methanethiolsulfonate. This suspension was dialyzed against water at 277 K and centrifuged to remove debris. The supernatant was applied onto a cation-exchange SP-Sepharose column equilibrated with 100 mM sodium acetate pH 5.0. After washing extensively, bound proteins were eluted with a linear concentration gradient from 100 to 800 mM sodium acetate pH 5.0. PPI-rich fractions were pooled together, concentrated and dialyzed against water at 277 K. Sodium acetate was dissolved in the PPI-rich pool to a final concentration of 4 M and this solution was applied onto a Fractogel EMD Propyl 650 (S) column pre-equilibrated in 2 M ammonium sulfate, 50 mM Tris pH 8.0. Elution of the bound protein was carried out by decreasing the concentration of ammonium sulfate from 2 to 0 M. Again, PPI-rich fractions were pooled together, concentrated and dialyzed against water at 277 K. Sodium acetate (4 M final concentration) was then added to the protein sample, which was subsequently passed a second time over the Fractogel EMD Propyl 650 (S) column (again pre-equilibrated in 2 M ammonium sulfate, 50 mM Tris pH 8.0). The PPI-containing fractions collected after this second pass were subsequently loaded onto a thiophilic gel prepared by activation of Biogel A 0.5M with divinyl sulfone and subsequent treatment with 2-mercaptoethanol (Hansen et al., 1998 ▶). The chromatographic conditions in this last purification step are identical to those used to perform the hydrophobic interaction chromatography on Fractogel EMD Propyl 650 (S). This resulted in highly pure PPI solution suitable for crystallization experiments. Prior to crystallization, the protein was exhaustively dialyzed against ultrapure water and concentrated to 26.0 mg ml−1 using a 5000 MWCO Vivaspin 15R concentrator (Sartorius). The protein concentration was determined spectroscopically using an absorption coefficient of 26 270 M −1 cm−1 at 280 nm calculated according to Gill & von Hippel (1989 ▶) using the published amino-acid sequence (Odani et al., 1996 ▶).
2.2. Activity assay
Inhibitory activity was assayed by following the hydrolysis of dl-BAPNA by bovine trypsin in the presence of various amounts of PPI. 10 µl bovine trypsin (35 µM stock solution in 1 mM HCl) and various amounts of PPI were diluted in 50 mM Tris–HCl buffer pH 8.2 containing 20 mM Ca2+ and 0.0–0.7 µM PPI. This reaction mixture was incubated at 310 K and the reaction was stopped at different time points by the addition of 30% acetic acid. The hydrolysis of dl-BAPNA was determined spectrophotometrically by measuring the release of p-nitroaniline at 410 nm.
2.3. Crystallization
Crystallization conditions were screened by the hanging-drop vapour-diffusion method using Hampton Research Crystal Screen and Crystal Screen II (Jancarik & Kim, 1991 ▶). For all crystallization experiments, PPI was dialyzed against deionized water. For the initial crystallization trials, the protein was concentrated to 15 mg ml−1. For crystallization in the presence of Co2+, PPI was used at 7 mg ml−1. Drops consisting of 1.5 µl protein solution and 1.5 µl precipitant solution were equilibrated against 500 µl precipitant solution at 293 K (for initial screens as well as for optimization experiments). Promising conditions were further optimized by varying the precipitant concentration, temperature, buffer pH and the ratio of protein to precipitant solution in the drops. Crystals of the metal-free inhibitor diffracted only to 2.6 Å; however, the addition of a divalent cation to the mother liquor allowed extension of the data collection to 1.7 Å.
For optimization experiments by microseeding, 5–15 crystals were typically transferred to 15 µl precipitant solution and mechanically crushed by pipetting and vortexing. This freshly prepared seed solution was then diluted 10 000–1 000 000-fold and used immediately. In a typical experiment, a drop containing 1.5 µl protein solution was mixed with 1.5 µl precipitant solution and 0.5 µl seeds and equilibrated against 0.5 ml precipitant solution in a hanging drop setup.
2.4. Data collection and analysis
All crystals were either cryocooled directly in the X-ray beam after the addition of a suitable cryoprotectant or mounted in glass capillaries for data collection at room temperature. X-ray data were collected at EMBL stations BW7A and X13 of the DESY synchrotron (Hamburg, Germany) using a MAR CCD detector and at station ID14-1 of the ESRF synchrotron (Grenoble, France) using an ADSC Quantum Q4 CCD detector. All data were indexed and processed with the HKL suite of programs (Otwinowski & Minor, 1997 ▶). Intensities were converted to structure-factor amplitudes using the CCP4 program TRUNCATE (Collaborative Computational Project, Number 4, 1994 ▶).
The bulk of the crystals from the metal-free inhibitor were perfect merohedral twins. These data were examined for possible twinning by inspecting the acentric moments of the intensity distributions and were detwinned using the CCP4 program DETWIN (Collaborative Computational Project, Number 4, 1994 ▶).
3. Results and discussion
PPI was purified to high homogeneity using a combination of different chromatographic techniques: cation-exchange, hydrophobic interaction and thiophilic interaction chromatographies (Azarkan et al., 2004 ▶, 2006 ▶). The purity of the sample was confirmed by the presence of a single band in a silver-stained SDS–PAGE and a single band in a Coomassie-blue stained isoelectric focusing gel. The protein inhibited bovine trypsin in a 1:1 ratio with a dissociation constant of 3 × 10−7 M, estimated by determining the amount of free trypsin at the enzyme–inhibitor equivalent (Odani et al., 1996 ▶).
Prismatic crystals (crystal form I) were obtained after one week of incubation in 2.0 M ammonium sulfate. These crystals remained quite small (largest dimension 0.1 mm) and did not diffract. However, after several weeks crystals of a similar shape also appeared in 2.0 M sodium formate, 0.2 M sodium acetate pH 4.6 (drop consisting of 1.5 µl protein solution + 1.5 µl reservoir solution, reservoir volume 0.5 ml, 293 K). Although nucleation was slow, the growth of these crystals was highly reproducible, with the largest crystals (0.3 × 0.3 × 0.2 mm) appearing in 1.5–1.75 M sodium formate, 0.1 M sodium acetate pH 4.5–4.9 (Fig. 1 ▶ a). These crystals were tested for X-ray diffraction on beamlines X13 and BW7A of the DESY synchrotron (Hamburg, Germany) as well as beamline ID14-1 of the ESRF (Grenoble, France). They belong to space group P31, with unit-cell parameters a = b = 74.70, c = 78.97 Å, and diffracted to 2.6 Å. Initially, the crystals were frozen directly in the cold stream after soaking them in reservoir solution enriched with 30%(v/v) glycerol for 1 min. This led to relatively high mosaicities (1.2°). During an attempt to prepare a bromide derivative, it was found that a more effective cryoprotectant consists of 0.1 M sodium formate, 0.07 M sodium acetate pH 4.5, 1.2 M sodium bromide and 30% glycerol. When frozen after a 1 min incubation soak in this solution, the mosaicities fell to 0.2–0.5°, comparable with those observed with room-temperature data.
Figure 1.

Crystals of PPI. (a) Crystal form I grown in 2.0 M sodium formate, 20 mM sodium acetate pH 4.6. (b) Original crystals of crystal form II grown at 293 K in 1.8 M ammonium sulfate, 100 mM MES pH 6.5, 10 mM cobalt chloride. (c) Typical crystals of crystal form II of PPI grown at 283 K after optimization. The clusters of needles are only loosely attached to each other and the large single crystal in the middle was picked out for data collection.
Data from crystal form I were initially processed in Laue group 321 (space groups P321, P3121 or P3221). During a search for heavy-atom derivatives, a crystal was observed that scaled only in Laue group 3 (space groups P3, P31 or P32). This led us to re-examine the data for possible twinning by looking at the acentric moments of the intensity distributions and by analysing the data using the Crystal Twinning Server (http://www.doe-mbi.ucla.edu/Services/Twinning). From this analysis, it was concluded that the bulk of the crystals were perfect merohedral twins (Chandra et al., 1999 ▶) belonging to one of the lower symmetry space groups. A single data set indicated a twinning fraction of 0.22 and was subsequently detwinned and used for the structure determination.
The addition of divalent cations, especially Co2+, triggers a burst of nucleation of PPI microcrystals and small crystals (morphologically different from crystal form I). The most promising condition observed in the crystal screen was 1.8 M ammonium sulfate, 0.1 M MES pH 6.5 and 0.01 M CoCl2 (drops consisting of 1.5 µl protein solution + 1.5 µl reservoir solution, reservoir volume 0.5 ml, 293 K; Fig. 1 ▶ b). In order to reduce the nucleation and optimize crystal growth, the concentration of ammonium sulfate was reduced to 1.4 M and the crystallization experiments were set up at 283 K. Under these conditions, nucleation was severely reduced and the use of microseeds prepared from the original crystals led to the growth of large crystals (Fig. 1 ▶ c). These crystals (crystal form II) belong to space group P212121, with unit-cell parameters a = 44.26, b = 81.99, c = 140.89 Å, and diffract to 1.6 Å on beamline X13 of the DESY synchrotron, Hamburg, Germany. A full data set from a form II PPI crystal was collected to a resolution of 1.70 Å. For data collection, these crystals were soaked in a cryoprotectant solution (1.4 M ammonium sulfate, 0.1 M MES, 0.01 M CoCl2 pH 6.5 and 20% glycerol) and frozen directly in the cryostream.
The statistics of data collection for both crystal forms are given in Table 1 ▶. The unit-cell volumes allow the presence of one, two or three molecules in the asymmetric unit, with two being the most likely in both cases [Matthews coefficient of 2.9 Å3 Da−1 (Matthews, 1968 ▶), corresponding to a solvent content of 59.4% for the first crystal form and 59.6% for the second].
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Crystal form I | Crystal form II | |
|---|---|---|
| Detector | MAR CCD | MAR CCD |
| Beamline | BW7A | X13 |
| Data-collection temperature (K) | 100 | 100 |
| Wavelength (Å) | 0.9150 | 0.8073 |
| Unit-cell parameters (Å) | a = b = 74.70, c = 78.97 | a = 44.26, b = 81.99, c = 140.89 |
| Space group | P31 or P32 | P212121 |
| Content of the asymmetric unit | 2 monomers | 2 monomers |
| Resolution range (Å) | 15.0–2.60 (2.69–2.60) | 20–1.70 (1.76–1.70) |
| No. of observed reflections | 178588 (20260) | 318867 (22384) |
| No. of unique reflections | 15027 (1809) | 51851 (5596) |
| Rmerge† | 0.110 (0.763) | 0.065 (0.165) |
| Completeness | 100 (100) | 90.4 (82.2) |
| I/σ(I) | 24.38 (2.27) | 17.35 (6.13) |
| Redundancy | 11.88 (11.20) | 6.15 (4.00) |
R
merge = 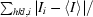
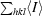 .
.
PPI belongs to the Kunitz family of serine protease inhibitors, with soybean trypsin inhibitor (STI) being the model protein of the family. The large representation of protease inhibitors in Leguminosae plants suggests a possible role in protecting plant tissue from the action of proteases secreted by pathogens (Casaretto & Corcuera, 1995 ▶). This is in agreement with the lack of detectable serine protease activity in papaya latex and the observation that PPI accumulates upon repeated wounding of papaya fruits, which led to the proposition of a similar role for PPI (Azarkan et al., 2004 ▶). Structural studies on several members of this family have provided a detailed picture of their inhibition mechanism (Dattagupta et al., 1996 ▶; Heussen et al., 1984 ▶; Kouzuma et al., 1997 ▶; Song & Suh, 1998 ▶; Vallee et al., 1998 ▶; Walter & Bode, 1983 ▶). The inhibitor prevents the access of substrates to the active site of the enzyme through steric hindrance. The exposed reactive loop inserts into the active-site cleft in a substrate-like manner, blocking the catalytic Ser Oγ in a near-Michaelis-like geometry (Bode & Huber, 2000 ▶; Krowarsch et al., 2003 ▶; Song & Suh, 1998 ▶). The intermolecular and intramolecular interactions involving the reactive loop, the inhibitor core and the enzyme-binding site are so tight that dissociation rarely occurs (Bode & Huber, 2000 ▶).
In contrast to the reactive loops of the Kunitz-BPTI, Kazal, squash and PI-1 families, which are heavily constrained by disulfide bridges (Antonini et al., 1983 ▶; Betzel et al., 1993 ▶; Bode et al., 1989 ▶), in the Kunitz STI-type family a conserved asparagine residue (Asn13 in STI) is the crucial piece in the hydrogen-bond network that stabilizes the reactive loop (Ravichandran et al., 2001 ▶). PPI, however, has the amide group of the conserved asparagine substituted by the bulky side chain of a tyrosine, yet the protein remains a potent inhibitor of trypsin and α-chymotrypsin (Azarkan et al., 2006 ▶; Odani et al., 1996 ▶). Moreover, the study of this molecule at atomic resolution will help us to understand how PPI is still capable of strongly inhibiting trypsin after previous incubation with an equimolar amount of α-chymotrypsin and vice versa (Odani et al., 1996 ▶) and the apparent need for divalent metal ions for its inhibitory activity (Azarkan et al., 2006 ▶).
Acknowledgments
This work was supported by the Vlaams Interuniversitair Instituut voor Biotechnologie (VIB), the Onderzoeksraad of the VUB and the Fonds voor Wetenschappelijk Onderzoek Vlaanderen (FWO). The authors acknowledge the use of synchrotron beamtime at the EMBL beamlines at the DORIS (Hamburg, Germany) and ESRF (Grenoble, France) storage rings.
References
- Antonini, E., Ascenzi, P., Bolognesi, M., Gatti, G., Guarneri, M. & Menegatti, E. (1983). J. Mol. Biol.165, 543–558. [DOI] [PubMed] [Google Scholar]
- Azarkan, M., Dibiani, R., Goormaghtigh, E., Raussens, V. & Baeyens-Volant, D. (2006). Biochim. Biophys. Acta, 1764, 1063–1072. [DOI] [PubMed] [Google Scholar]
- Azarkan, M., Wintjens, R., Looze, Y. & Baeyens-Volant, D. (2004). Phytochemistry, 65, 525–534. [DOI] [PubMed] [Google Scholar]
- Betzel, C., Dauter, Z., Genov, N., Lamzin, V., Navaza, J., Schnebli, H. P., Visanji, M. & Wilson, K. S. (1993). FEBS. Lett.317, 185–188. [DOI] [PubMed] [Google Scholar]
- Bode, W., Greyling, H. J., Huber, R., Otlewski, J. & Wilusz, T. (1989). FEBS Lett.242, 285–292. [DOI] [PubMed] [Google Scholar]
- Bode, W. & Huber, R. (2000). Biochim. Biophys. Acta, 1477, 241–252. [DOI] [PubMed] [Google Scholar]
- Casaretto, J. A. & Corcuera, L. J. (1995). Biol. Res.28, 239–249. [PubMed] [Google Scholar]
- Chandra, N., Acharya, K. R. & Moody, P. C. (1999). Acta Cryst. D55, 1750–1758. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Dattagupta, J. K., Podder, A., Chakrabarti, C., Sen, U., Dutta, S. K. & Singh, M. (1996). Acta Cryst. D52, 521–528. [DOI] [PubMed] [Google Scholar]
- Gill, S. C. & von Hippel, P. H. (1989). Anal. Biochem.182, 319–333. [DOI] [PubMed] [Google Scholar]
- Hansen, P., Scoble, J. A., Hanson, B. & Hoogenraad, N. J. (1998). J. Immunol. Methods, 215, 1–7. [DOI] [PubMed] [Google Scholar]
- Heussen, C., Joubert, F. & Dowdle, E. B. (1984). J. Biol. Chem.259, 11635–11638. [PubMed] [Google Scholar]
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst.24, 409–411. [Google Scholar]
- Khan, A. R. & James, M. N. (1998). Protein Sci.7, 815–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzuma, Y., Yamasaki, N. & Kimura, M. (1997). J. Biochem. (Tokyo), 121, 456–463. [DOI] [PubMed] [Google Scholar]
- Krowarsch, D., Cierpicki, T., Jelen, F. & Otlewski, J. (2003). Cell. Mol. Life Sci.60, 2427–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy, D. B., Tepp, W., Cohen, A. C., DasGupta, B. R. & Stevens, R. C. (1998). Nature Struct. Biol.5, 898–902. [DOI] [PubMed] [Google Scholar]
- Laskowski, M. Jr & Kato, I. (1980). Annu. Rev. Biochem.49, 593–626. [DOI] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Murzin, A. G., Lesk, A. M. & Chothia, C. (1992). J. Mol. Biol.223, 531–543. [DOI] [PubMed] [Google Scholar]
- Neurath, H. (1984). Science, 224, 350–357. [DOI] [PubMed] [Google Scholar]
- Odani, S., Yokokawa, Y., Takeda, H. & Abe, S. (1996). Eur. J. Biochem.241, 77–82. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Ravichandran, S., Dasgupta, J., Chakrabarti, C., Ghosh, S., Singh, M. & Dattagupta, J. K. (2001). Protein Eng.14, 349–357. [DOI] [PubMed] [Google Scholar]
- Rawlings, N. D., Tolle, D. P. & Barrett, A. J. (2004). Biochem. J.378, 705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, H. K. & Suh, S. W. (1998). J. Mol. Biol.275, 347–363. [DOI] [PubMed] [Google Scholar]
- Transue, T. R., Smith, A. K., Mo, H., Goldstein, I. J. & Saper, M. A. (1997). Nature Struct. Biol.4, 779–783. [DOI] [PubMed] [Google Scholar]
- Vallee, F., Kadziola, A., Bourne, Y., Juy, M., Rodenburg, K. W., Svensson, B. & Haser, R. (1998). Structure, 6, 649–659. [DOI] [PubMed] [Google Scholar]
- Walter, J. & Bode, W. (1983). Hoppe-Seyler’s Z. Physiol. Chem.364, 949–959. [DOI] [PubMed] [Google Scholar]
- Zhu, X., Komiya, H., Chirino, A., Faham, S., Fox, G. M., Arakawa, T., Hsu, B. T. & Rees, D. C. (1991). Science, 251, 90–93. [DOI] [PubMed] [Google Scholar]