

Expression, crystallization and preliminary X-ray diffraction studies of a novel bifunctional N-acetylglutamate synthase/kinase from X. campestris homologous to vertebrate N-acetylglutamate synthase are reported.
Keywords: argA, argB, N-acetylglutamate synthase, N-acetylglutamate kinase, bifunctional enzymes, arginine biosynthesis
Abstract
A novel N-acetylglutamate synthase/kinase bifunctional enzyme of arginine biosynthesis that was homologous to vertebrate N-acetylglutamate synthases was identified in Xanthomonas campestris. The protein was overexpressed, purified and crystallized. The crystals belong to the hexagonal space group P6222, with unit-cell parameters a = b = 134.60, c = 192.11 Å, and diffract to about 3.0 Å resolution. Selenomethionine-substituted recombinant protein was produced and selenomethionine substitution was verified by mass spectroscopy. Multiple anomalous dispersion (MAD) data were collected at three wavelengths at SER-CAT, Advanced Photon Source, Argonne National Laboratory. Structure determination is under way using the MAD phasing method.
1. Introduction
N-Acetylglutamate synthase (NAGS) catalyzes the acetylation of l-glutamate by acetyl-CoA to initiate arginine biosynthesis in microbes and plants (Cunin et al., 1986 ▶; Slocum, 2005 ▶). However, in vertebrates NAGS has a different function, producing N-acetylglutamate (NAG), an essential allosteric activator of carbamyl phosphate synthetase I (CPSI), the first and rate-limiting enzyme of ureagenesis (Hall et al., 1958 ▶). NAGS deficiency in humans leads to hyperammonemia owing to the decreased activity of CPSI deprived of its cofactor NAG (Metzenberg et al., 1958 ▶; Caldovic et al., 2003 ▶; Haberle et al., 2003 ▶). NAGS genes and/or proteins have been identified in bacteria, yeast, plants and vertebrates, including mammals, and show surprising diversity across the phyla (Caldovic & Tuchman, 2003 ▶).
N-Acetylglutamate kinase (NAGK) phosphorylates the γ-carboxyl group of NAG in the obligatory second step of microbial and plant arginine biosynthesis (Cunin et al., 1986 ▶; Slocum, 2005 ▶). Mammals lack this enzyme and use a different pathway and the enzymes of the urea cycle to produce arginine (Wu & Morris, 1998 ▶). The crystal structures of NAGK from Escherichia coli, Mycobacterium tuberculosis, Pseudomonas aeruginosa and Thermotoga maritima have been determined (Ramon-Maiques et al., 2002 ▶, 2006 ▶).
When human and mouse NAGS were first identified and cloned, they were found to share limited sequence similarity to E. coli NAGK but lacked NAGK activity (Sonoda & Tatibana, 1983 ▶; Caldovic, Morizono, Panglao et al., 2002 ▶; Caldovic, Morizono, Yu et al., 2002 ▶). Additional NAGS genes have been identified from vertebrate phyla such as fish (zebrafish and fugu; Morizono et al., 2004 ▶), but surprisingly some bacterial species (Xylella fastidiosa, Xanthomonas campestris and Xanthomonas axonopodis) also have a vertebrate-like NAGS with an amino-acid sequence that differs significantly from those of other classic bacterial NAGS. Interestingly, the NAGS from X. campestris is a bifunctional enzyme (NAGS/K) that displays NAGS and NAGK activity in vitro and thus probably catalyzes both the first and the second reactions of arginine biosynthesis. In most other bacteria NAGS catalyzes only the first reaction in the pathway (Rajagopal et al., 1998 ▶), while the second reaction is catalyzed by a separate kinase enzyme (Gil et al., 1999 ▶).
Although there is considerable sequence similarity (∼42%) between X. campestris and mammalian NAGS, the effect of arginine on enzyme activity is disparate. In X. campestris, as in other bacteria and yeast, arginine has an inhibitory effect on the enzyme, while in mammals it has a stimulatory effect (Caldovic et al., 2006 ▶). No structures have been reported for NAGS proteins, most of which are relatively insoluble, unstable and not easily amenable to crystallization. Crystallization of X. campestris NAGS/K therefore provides an opportunity to obtain the first structural information for NAGS and to begin to understand its structure–function relationships and regulation by arginine. This structure will also provide a structural model for human NAGS and enable better understanding of the molecular basis of NAGS deficiency.
In this report, we provide preliminary X-ray diffraction data for the novel bifunctional NAGS/K from X. campestris.
2. Experimental
2.1. Cloning and protein expression and purification
A gene (gi:21231683) annotated as argB, but identified by homology to vertebrate NAGS genes, encoding a protein with 447 amino-acid residues was cloned from X. campestris genomic DNA. The gene (now termed argA/B based on its assigned function below) was PCR-amplified from X. campestris genomic DNA (ATCC 33913), using Hot-start Turbo Pfu DNA polymerase and the primers 5′-TCCCATATGTCCCTTCCTGCACAGCCCCAC-3′ and 5′-AACGGATCCTTATTATCACCCCAGCAAGGTGGGTTGACG-3′, which introduce NdeI and BamHI sites (bold) at the initiator ATG and downstream of the stop codon. The PCR products were cloned into pCR4Blunt-Topo vector using the Zero-blunt Topo cloning kit (Invitrogen). The plasmid with the correct insert was identified, isolated and digested with NdeI and BamHI. The fragments were ligated into pET15b (Novagen) using T4 DNA ligase (New England BioLabs) and transformed into E. coli DH5α cells (Invitrogen). Plasmids were isolated and those containing the correct insert were identified by restriction-enzyme analysis, verified by DNA sequencing and then transformed into E. coli BL21(DE3) cells (Invitrogen) for expression. We found this two-step cloning strategy to be more efficient than the traditional procedures for cloning PCR products directly into expression vectors. The protein was produced using the Overnight Express Auto-induction System 1 (Novagen) according to the manufacturer’s protocol. The target protein was purified using an AKTA FPLC system, initially on a His-trap Ni-affinity column (GE Healthcare) and then on a Hightrap DEAE column (GE Healthcare), following the procedure described previously (Shi et al., 2005 ▶). The His6 tag and linker were cleaved from the target protein by incubating with thrombin at 277 K for 24 h. Protein purity was verified by SDS–PAGE (12% polyacrylamide gel) followed by Coomassie staining; a single band with the expected molecular weight was observed. Protein concentration was determined by the Bradford method using the BioRad protein-assay dye reagent with bovine serum albumin as a standard (Bradford, 1976 ▶).
2.2. Enzymatic assay
NAGS enzyme activity was measured with a liquid-chromatography mass-spectrometry (LC–MS) system that quantifies the production of NAG as described previously (Morizono et al., 2004 ▶; Caldovic et al., 2006 ▶). Enzymatic activity was assayed in 100 µl 12 mM Tris buffer pH 9.0 containing 2.5 mM acetyl-CoA, 50 mM l-glutamate and ∼2 µg enzyme at 303 K. The reaction was quenched with 100 µl 30% trichloroacetic acid after 5 min. Precipitated protein was removed by micro-centrifugation for 5 min. 10 µl of clear reaction solution was injected into a reverse-phase LC–MS system under isocratic conditions using a mobile phase consisting of 93% solvent A (1% trifluoroacetic acid) and 7% solvent B (1 ml trifluoroacetic acid in 1 l of 1:9 water:acetonitrile) with a flow rate of 0.6 ml min−1. Detection was performed in positive-ion mode by selected ion monitoring at m/z 148 for l-glutamate and m/z 190 for NAG.
NAGK activity was measured using a colorimetric assay which detects the formation of acetylglutamyl hydroxamate from acetylglutamate phosphate in the presence of ammonium hydroxamate (Haas & Leisinger, 1975 ▶). The 100 µl incubation volume contained 100 mM MES buffer pH 6.0, 20 mM ATP, 100 mM NAG, 400 mM NH2OH.HCl, 40 mM MgCl2 and ∼2 µg of protein. The reaction mixture was incubated for 20 min at 303 K and terminated by the addition of 500 µl 5%(w/v) ferric chloride solution (FeCl3·6H2O) containing 8%(w/v) trichloroacetic acid and 0.3 M HCl. The absorbance of the hydroxamate–Fe3+ complex was measured at 540 nm.
2.3. Preparation and verification of selenomethionine-substituted NAGS/K protein
Selenomethionine-substituted NAGS/K protein was prepared using the Overnight Express Auto-induction System 2 (Novagen) as described previously (Shi et al., 2005 ▶). In brief, the expression plasmid was transformed into a metE − host strain B834(DE3) (Novagen). The clone was inoculated into 1 l sterile deionized water supplemented with the chemicals provided in the kit, 125 mg l-selenomethionine and 100 mg ampicillin. Vitamin B12 (cyanocobalamin) was added to a final concentration of 100 nM and the culture was incubated at 303 K for 16 h. After reaching the stationary phase, the cells were harvested and the protein was purified as described above for the native protein. Approximately 10 µg purified native and selenomethionine-substituted protein were digested overnight at 310 K using Promega sequencing-grade trypsin [1:50(w:w) enzyme:protein ratio] in 50 mM ammonium bicarbonate pH 7.4. After desalting, 0.3 µl of the resulting peptide solution was mixed with 0.3 µl saturated α-hydroxycinnamic acid and spotted onto the MALDI plate. Analysis was preformed on a 4700 ABI TOF/TOF mass spectrometer (Applied Biosystem) operated in reflection positive-ion mode.
2.4. Site-directed mutagenesis and reductive methylation of surface lysines
Site-directed mutant genes of X. campestris NAGS were created by utilizing primers containing the desired mutations (Table 1 ▶) and the QuickChange Mutagenesis Kit according to the manufacturer’s protocol (Strategene). Initially, a thermal cycle was applied to denature the double-stranded X. campestris NAGS wild-type plasmid that was previously cloned into the pET15b expression vector and appropriate mutagenic primers were then annealed to it. After annealing, Pfu Turbo DNA polymerase from the QuickChange kit was used to extend primers without primer displacement and to seal nicks. The product was treated with DpnI to digest parental plasmids, which are susceptible because of their methylated DNA. Transformation into XL10-Gold ultracompetent cells then allowed the conversion of mutated single-stranded DNA to double-stranded plasmid DNA. Finally, the sequences of the mutant DNA were verified by DNA sequencing (Beckman) using the commercially available primer pair annealed to the plasmid promoter and terminator regions.
Table 1. Primers used for mutagenesis to replace polar surface residues of NAGS/K.
The codons mutated are highlighted.
| Mutant | Primer sequence |
|---|---|
| K279A/E280A | 5′-CGGCGGATCTGGCCGCGGCACTGTTCACCCACAAG-3′ |
| E386A/E387A | 5′-TGGAACGTGATGCGCGCAGCAACCCCGCAGCTGTTV-3′ |
| K26A/E27A | 5′-AGCATGGCCAGCGCGGCAGCAATCAGCCAGTACC-3′ |
| E94A/K95A | 5′-ATCGGCCGCCGGCATCGCAGCGCAGACCGTCAACGG-3′ |
| E416A/K417A/K419A | 5′-GGCTGCATCAAGCAGGCAGCGTGGGCGGTGTTCTGGTATGGGC-3′ |
Methylation of surface lysine residues was performed using the JBScreen Methylation kit according to the manufacturer’s protocol (Jena Bioscience). Methylation was verified by TOF–TOF mass spectrometry using a procedure identical to that for selenomethionine-substituted proteins.
2.5. Crystallization
The purified protein was concentrated to 14 mg ml−1 with an Amicon-YM30 membrane concentrator (Millipore). Screening for crystallization conditions was performed using sitting-drop vapor diffusion in 96-well plates (Hampton Research) at 291 K by mixing 2 µl protein solution with 2 µl reagent solution from the sparse-matrix Crystal Screens 1 and 2 and Index Screen (Hampton Research). Initial screening indicated that crystals can be grown under many conditions to give similar hexagonal bipyramidal crystals (Fig. 1 ▶). Further optimizations for the crystallization conditions were conducted using the hanging-drop vapor-diffusion method.
Figure 1.

A typical hexagonal bipyramidal crystal. The largest dimension was approximately 0.4 mm.
2.6. Data collection
Prior to data collection, the crystals were transferred on the cover slip on which they were grown to a well solution containing 1 ml 9.0 M sodium formate overnight for dehydration. Since 9.0 M sodium formate is a good cryoprotectant, the crystal was then frozen by direct immersion into liquid nitrogen. Data sets for selenomethionine-substituted proteins were collected at the selenium absorption edge, the inflection point and a remote position using a synchrotron source (SER-CAT, Advanced Photon Source). Data sets for the native crystals were collected to about 3.0 Å on an R-AXIS IV X-ray generator. All data were processed using the HKL-2000 package (Otwinowski & Minor, 1997 ▶); statistics are summarized in Table 2 ▶.
Table 2. Summary of crystallographic data for native protein and Se-MAD data.
Values in parentheses are for the highest resolution shell.
| Native | Se edge | Se inflection | Se remote | |
|---|---|---|---|---|
| Wavelength (Å) | 1.5417 | 0.9793 | 0.9794 | 0.9747 |
| Resolution (Å) | 50–3.0 (3.1–3.0) | 50–2.9 (3.0–2.9) | 50–3.0 (3.1–3.0) | 50–3.0 (3.0–2.9) |
| Unit-cell parameters (Å, °) | a = 134.60, b = 134.60, c = 192.11, β = 120 | a = 133.38, b = 133.38, c = 191.89, β = 120 | a = 133.23, b = 133.23, c = 192.28, β = 120 | a = 133.28, b = 133.28, c = 192.14, β = 120 |
| No. of measurements | 228866 | 124206 | 122564 | 114030 |
| No. of unique reflections | 18521 (844) | 18122 (695) | 16548 (335) | 17492 (617) |
| Redundancy | 12.4 (9.5) | 6.9 (4.4) | 7.4 (4.7) | 6.5 (3.9) |
| Completeness (%) | 86.6 (40.9) | 78.9 (31.2) | 79.5 (16.7) | 75.9 (27.6) |
| Rmerge† | 0.124 (0.722) | 0.072 (0.301) | 0.077 (0.386) | 0.072 (0.349) |
| 〈I/σ(I)〉 | 16.7 (1.9) | 23.8 (2.9) | 16.8 (2.3) | 23.1 (2.4) |
R
merge = 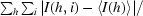
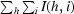 , where I(h, i) is the intensity of the ith observation of reflection h and 〈I(h)〉 is the average intensity of redundant measurements of reflection h.
, where I(h, i) is the intensity of the ith observation of reflection h and 〈I(h)〉 is the average intensity of redundant measurements of reflection h.
3. Results and discussion
Activity assays indicated that the NAGS-like gene from X. campestris encodes a novel bifunctional protein with the catalytic activities of both NAGS and NAGK (a detailed report on the biochemical characterization of this protein is currently in preparation). Hence, the gene should be annotated argA/B rather than the current annotation argB in databases and since there is no evidence for a separate argB gene, the argA/B-encoded protein (NAGS/K) is likely to catalyze the first two reactions of arginine biosynthesis in vivo. In contrast, the homologous vertebrate proteins only display NAGS activity even though they have a domain resembling NAGK (Caldovic, Morizono, Panglao et al., 2002 ▶; Caldovic, Morizono, Yu et al., 2002 ▶).
The selenomethionine-substituted NAGS/K protein could be prepared easily in large amounts using the Overnight Express Auto-induction System 2 (Novagen) and was verified to have ∼80% of its methionines replaced by selenomethionines. The fluorescence scan of the selenomethionine-substituted crystals showed a strong signal at the selenium absorption edge.
Wild-type and selenomethionine-substituted NAGS/K from X. campestris could easily be crystallized under many conditions, but screening of crystals on an X-ray diffractometer demonstrated that all the crystals belong to the same space group with similar unit cells and diffract to a similar limited resolution of ∼4.5 Å. Extensive efforts were made to optimize the crystallization conditions in order to improve crystal quality. We first tried to cocrystallize with various ligands such as ATP, ADP, acetyl-CoA, CoA, NAG, the substrate analog AMPPNP and arginine. However, none improved the resolution of the diffraction pattern. We then tried to reduce the entropy of protein surface residues (Derewenda, 2004 ▶). Five sets of mutants (K26A/E27A, E94A/K95A, K279A/W280A, E386A/E387A and E416/K417A/K419A) were produced and purified. Surprisingly, all these mutants crystallized in a similar unit cell and diffracted to similar resolution. We also tried chemical modification using reductive methylation of surface lysine residues to improve the quality of crystals, as proposed by Rypniewski et al. (1993 ▶). MALDI–TOF/TOF mass-spectrometric analysis confirmed that all NAGS/K lysines were methylated. The methylated protein was amenable to crystallization; however, the crystals belong to a similar unit cell and diffract to a resolution similar to that of the wild-type protein.
The best crystals were obtained using the osmolytes trimethylamine-N-oxide (TMAO) and proline in the protein medium to stabilize the protein (Bolen, 2004 ▶) and 9.0 M sodium formate to dehydrate the crystals. Crystals prepared in this way diffracted to ∼3.0 Å resolution along the c axis and to 3.5 Å along the a and b axes (Fig. 2 ▶).
Figure 2.

The diffraction pattern of Se-substituted NAGS/K obtained from a flash-frozen crystal at the SER-CAT Advanced Photon Source, Argonne National Laboratory. The exposure time was 4 s, with an oscillation range of 0.5° and a crystal-to-detector distance of 350 mm. The crystal diffracted X-rays anisotropically to ∼3.0 Å along the c axis and to ∼3.5 Å along the a and b axes.
Crystals used to obtain data had a maximal dimension of 0.4 mm and grew within a week at 291 K from protein supplemented with 10 mM proline and 20 mM TMAO and a well solution containing 1 ml 0.67 M sodium formate pH 7.0. Selenomethionine-substituted crystals were grown under identical conditions.
Initial indexing identified that the crystals belong to a hexagonal Bravais lattice with unit-cell parameters a = b = 134.60, c = 192.11 Å, β = 120°. Inspection of the systematic absences revealed (00l) = 3n, consistent with the hexagonal space group P6222 or P6422. A packing-density calculation (Matthews, 1968 ▶) for a monomer weight of 49 kDa suggested the presence of two monomers in the asymmetric unit (V M = 2.43 Å3 Da−1, corresponding to a solvent content of 48.9%. However, preliminary analysis of the MAD data indicated only one molecule in the asymmetrical unit, corresponding to 74.5% solvent content. High solvent content may be one of the reasons for the low resolution of the diffraction pattern, which was improved by dehydration of the crystal.
Although the MAD data extend to about 3.0 Å, five of the six possible selenium positions were easily identified using the program SHELXD (Usón & Sheldrick, 1999 ▶). Phasing and density modification with SHELXE (Schneider & Sheldrick, 2002 ▶) gave a pseudo-free correlation coefficient (CC) of 76.14% in space group P6222, where a value above 70% indicates an interpretable map, in contrast to the pseudo-free CC of 67.68% in the enantiomorphic space group P6422. The resulting electron-density map viewed using the graphics program Coot (Emsley & Cowtan, 2004 ▶) had a clear protein–solvent boundary, confirming the structural solution, and model building is currently in progress.
Acknowledgments
This work was supported by Public Health Service grant DK-064913 (MT) and DK-067935 (DS) from the National Institute of Diabetes, Digestive and Kidney Diseases and HD-32652 (MT) from the National Institute of Child Health and Human Development. We thank Dr David Davies for facilitating the use of the diffraction equipment in the Molecular Structure Section of the National Institute of Health and Dr Fred Dyda for help in data collection. Data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID (or 22-BM) beamline at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at http://www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. W-31-109-Eng-38.
References
- Bolen, D. W. (2004). Methods, 34, 312–322. [DOI] [PubMed] [Google Scholar]
- Bradford, M. (1976). Anal. Biochem.72, 1219–1223. [DOI] [PubMed]
- Caldovic, L., Lopez, G. Y., Haskins, N., Panglao, M., Shi, D., Morizono, H. & Tuchman, M. (2006). Mol. Genet. Metab.87, 226–232. [DOI] [PubMed] [Google Scholar]
- Caldovic, L., Morizono, H., Panglao, M. G., Gallegos, R., Cheng, S. F., Pacman, S. & Tuchman, M. (2003). Hum. Genet.112, 364–368. [DOI] [PubMed] [Google Scholar]
- Caldovic, L., Morizono, H., Panglao, M. G., Gallegos, R., Yu, X., Shi, D., Malamy, M. H., Allewell, N. M. & Tuchman, M. (2002). Biochem. Biophys. Res. Commun.299, 581–586. [DOI] [PubMed] [Google Scholar]
- Caldovic, L., Morizono, H., Yu, X., Thompson, M., Shi, D., Gallegos, R., Allewell, N. M., Malamy, M. H. & Tuchman, M. (2002). Biochem. J.364, 825–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldovic, L. & Tuchman, M. (2003). Biochem. J.372, 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunin, R., Glansdorff, N., Pierard, A. & Stalon, V. (1986). Microbiol. Rev.50, 314–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derewenda, Z. S. (2004). Structure, 12, 529–535. [DOI] [PubMed] [Google Scholar]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Gil, F., Ramon-Maiques, S., Marina, A., Fita, I. & Rubio, V. (1999). Acta Cryst. D55, 1350–1352. [DOI] [PubMed] [Google Scholar]
- Haas, D. & Leisinger, T. (1975). Eur. J. Biochem.52, 365–375. [DOI] [PubMed] [Google Scholar]
- Haberle, J., Schmidt, E., Pauli, S., Kreuder, J. G., Plecko, B., Galler, A., Wermuth, B., Harms, E. & Koch, H. G. (2003). Hum. Mutat.21, 593–597. [DOI] [PubMed] [Google Scholar]
- Hall, L. M., Metzenberg, R. L. & Cohen, P. P. (1958). J. Biol. Chem.230, 1013–1321. [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Metzenberg, R. L., Marshall, M. & Cohen, P. P. (1958). J. Biol. Chem.233, 102–105. [PubMed] [Google Scholar]
- Morizono, H., Caldovic, L., Shi, D. & Tuchman M. (2004). Mol. Genet. Metab.81, S4–S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Rajagopal, B. S., DePonte, J. III, Tuchman, M. & Malamy, M. H. (1998). Appl. Environ. Microbiol.64, 1805–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramon-Maiques, S., Fernandez-Murga, M. L., Gil-Ortiz, F., Vagin, A., Fita, I. & Rubio, V. (2006). J. Mol. Biol.356, 695–713. [DOI] [PubMed] [Google Scholar]
- Ramon-Maiques, S., Marina, A., Gil-Ortiz, F., Fita, I. & Rubio, V. (2002). Structure, 10, 329–342. [DOI] [PubMed] [Google Scholar]
- Rypniewski, W. R., Holden, H. M. & Rayment, I. (1993). Biochemistry, 32, 9851–9858. [DOI] [PubMed] [Google Scholar]
- Schneider, T. R. & Sheldrick, G. M. (2002). Acta Cryst. D58, 1772–1779. [DOI] [PubMed] [Google Scholar]
- Shi, D., Yu, X., Roth, L., Morizono, H., Hathout, Y., Allewell, N. M. & Tuchman, M. (2005). Acta Cryst. F61, 676–679. [DOI] [PMC free article] [PubMed]
- Slocum, R. D. (2005). Plant Physiol. Biochem.43, 729–745. [DOI] [PubMed] [Google Scholar]
- Sonoda, T. & Tatibana, M. (1983). J. Biol. Chem.258, 9839–9844. [PubMed] [Google Scholar]
- Usón, I. & Sheldrick, G. M. (1999). Curr. Opin. Struct. Biol.9, 643–648. [DOI] [PubMed] [Google Scholar]
- Wu, G. & Morris, S. M. Jr (1998). Biochem. J.336, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]