

Abstract
The human androgen receptor (AR) is a ligand activated nuclear transcription factor and mediates the induction of genes involved in the development of the male phenotype and male secondary sex characteristics, as well as the normal and abnormal growth of the prostate. We have identified the pair of hydroxysteroid dehydrogenases (HSDs) that regulate ligand access to the AR in human prostate. We find that type 3 3α-HSD (aldo-keto reductase (AKR)1C2) catalyzes the NADPH dependent reduction of the potent androgen 5α-dihydrotestosterone (5α-DHT) to yield the inactive androgen 3α-androstanediol (3α-diol). We also find that RoDH like 3α-HSD (RL-HSD) catalyzes the NAD+ dependent oxidation of 3α-diol to yield 5α-DHT. Together these enzymes are involved in the pre-receptor regulation of androgen action. Inhibition of AKR1C2 would be desirable in cases of androgen insufficiency and inhibition of RL-HSD might be desirable in benign prostatic hyperplasia.
Keywords: Hydroxysteroid dehydrogenase, aldo-keto reductase, short chain dehydrogenase, benign prostatic hyperplasia, prostate cancer
1. Pre-receptor regulation of steroid hormone action
Steroid hormone receptors [androgen receptor (AR), estrogen receptor, progesterone receptor, glucocorticoid receptor (GR), and mineralocorticoid receptor (MR)] are ligand-activated nuclear receptors (Carcon-Jurica et al., 1990; Evans, 2005). The activated receptors bind to hormone response elements on hormone responsive genes resulting in gene expression in target tissues (Truss and Beato, 1993). Ligand specific conformational changes on the receptor determine the recruitment of co-activators and co-repressors in a tissue specific manner, and as a result tissue specific effects on gene transcription can be observed (Shibata et al., 1997). This is the pharmacological basis for the mechanism of action of selective steroid receptor modulators (especially partial agonists). Often the co-activators and co-repressors are involved in chromatin remodeling and act as histone-acetyl transferases and histone-deacetylases (Liu et al., 1999). Thus multiprotein complexes determine changes in gene transcription mediated by nuclear receptor ligands and the rational design of therapeutics targeting these complexes is challenging.
Another level of steroid receptor regulation involves the control of steroid ligands for their respective nuclear receptors. This pre-receptor regulation for steroid hormone action was first documented with the type 2 and type 1 11β-hydroxysteroid dehydrogenases (HSDs) (Funder et al., 1988; Whorwood and Stewart, 1996; White, 2001), Figure 1. In the human kidney, the type 2 11β-HSD protects the MR from mineralocorticoid excess by oxidizing cortisol (a glucocorticoid with high affinity for the MR) to cortisone (Krozowski et al., 1995). Licorice based inhibitors of this enzyme or inherited deficiencies cause apparent mineralocorticoid excess, attesting to the importance of this enzyme in protecting the MR from ligand excess (White, 2001). Thus, mineralocorticoid specificity is HSD rather than receptor mediated (Funder et al., 1988; White, 2001). Conversely, the type 1 11β-HSD can amplify the glucocorticoid signal by reducing cortisone to cortisol. This increase in GR signaling results in increased gluconeogenesis, glucose intolerance, insulin resistance, and may contribute to type 2 diabetes and metabolic syndrome (Seckl and Walker, 2001). Consequently, major drug companies are targeting the type 1 11β-HSD for metabolic syndrome. This example illustrates how isoforms of HSDs can govern ligand access to nuclear receptors at the pre-receptor level with important physiological consequences.
Figure 1.

Pre-receptor regulation of hormone action mediated by HSDs. The example of 11β-HSDs. Regulation of the mineralocorticoid receptor (MR) and glucocorticoid receptor (GR) by 11β-hydroxysteroid dehydrogenase type 2 and type 1, respectively (A); The example of 3α-HSDs. Regulation of the androgen receptor (AR) by 3α-hydroxysteroid dehydrogenase isoforms (AKR1C2 and RL-HSD), (B)
We have been proponents that pre-receptor regulation of steroid hormone action is a general concept that can be extended to all steroid hormone receptors (Penning, 1997; 2003). Furthermore, we and others have proposed and shown that HSDs are intimately involved in this mechanism (Labrie et al., 1995; 1997). HSDs interconvert alcohols and ketones in a positional and stereochemically selective manner on the steroid nucleus and side chain by working preferentially as NAD+ specific oxidases or NADPH specific reductases. In catalyzing these reactions HSDs act as molecular switches for hormone action by converting potent steroid hormones into their cognate inactive metabolites and vice-versa. HSDs that catalyze these reactions belong to two gene superfamilies, the short-chain dehydrogenases/reductases (SDRs) and the aldo-keto reductases (AKRs) (Jornvall et al., 1995; Penning, 1997; and Hyndman et al., 2003). Often SDRs and AKRs work in pairs as oxidases and reductases, respectively thereby controlling hormone response in a tissue specific manner. This article will elaborate this point for the AR in human prostate.
The most potent natural ligand for the AR is 5α-dihydrotestosterone (5α-DHT) which has a Kd = 10−11 M for this receptor. We will show that this hormone is eliminated by human type 3 3α-HSD (AKR1C2) which catalyzes the reduction of 5α-DHT to yield the inactive androgen 3α-androstanediol (3α-diol) which has a Kd for the AR = 10−6 M. Thus, this simple conversion of a 3-ketosteroid to a 3α-hydroxysteroid results in a five-order of magnitude decrease in the affinity of the steroid for the AR. Once formed 3α-diol is conjugated and excreted. By contrast RoDH like 3α-HSD (RL-HSD) (HSD17B6 an SDR) oxidizes circulating 3α-diol back to 5α-DHT (Rizner et. al., 2003; Bauman et al., 2006b), leading to activation of the AR, Figure 1. Inhibition of AKR1C2 would be important in androgen insufficiency while inhibitors of RL-HSD may be important in benign prostatic hyperplasia. We will now review the evidence that these two enzymes govern ligand access to the AR in human prostate.
2. Androgen biosynthesis and metabolism in the prostate
In adult males (18–55 yr), testosterone from the Leydig cells of the testis is converted in the prostate to the more potent androgen 5α-DHT by type 2 5α-reductase (SRD5A2), thereby amplifying the androgen signal (Russell and Wilson, 1994). The importance of type 2 5α-reductase comes from studies on 5α-reductase deficiency. Individuals with this disease present with apparent female genitalia at birth but develop a penis at age 12. Throughout life these individuals exhibit no facial hair, do not display male pattern baldness, and the prostate remains underdeveloped or atrophied [Imperato-McGinley, 1974, 1992]. Because of the importance of 5α-DHT in maintaining prostate growth the levels of this intraprostatic hormone are tightly regulated by 3α/3β-HSDs. These enzymes reduce 5α-DHT to yield either 3α-diol, which has poor affinity for the AR, or 3β-androstanediol which is potent ligand for ERβ (Weihua et al, 2002; Guerini et al., 2005). Studies in the rat, dog, marsupial and human also indicate that circulating 3α-diol can be oxidized back to 5α-DHT (Orlowski et al., 1983; Walsh and Wilson, 1976; DeKlerk et al., 1979; Jacobi et al., 1977; Liehy et al., 2001; Shaw et al., 2000;). In fact 50% of an administered dose of 3α-diol in humans was converted to 5α-DHT within the prostate, indicating that the back conversion is significant in androgen action (Horst et al., 1975; Kinouchi and Horton 1974). Backdoor pathways to 5α-DHT using 3α-diol as a precursor have been proposed in which the production of DHT “by-passes” the formation of Δ4-androstene-3,17-dione and testosterone, (Auchus, 2004).
Two prevalent diseases of the aging male which are androgen dependent are benign prostatic hyperplasia (BPH) and prostate cancer (CaP). Both these diseases occur in men in the age group of 50–80 years of age in which production of Leydig cell testosterone is compromised (“andropause”). In this age group there is increased dependency on circulating adrenal androgens e.g. dehydroepiandrosterone (DHEA) for the intraprostatic formation of 5α-DHT (Labrie et al., 1995;), Figure 2B. This pathway involves conversion of DHEA into Δ4–androstene-3,17-dione catalyzed by type 2 3β-HSD/ketosteroid isomerase (HSD3B2); reduction of Δ4-androstene-3,17-dione to testosterone catalyzed by type 5 17β-HSD (AKR1C3); followed by conversion of testosterone to 5α-DHT catalyzed by type 2 5α-reductase (SRD5A2). These observations raise the issue as to which enzymes are responsible for the elimination of 5α-DHT in the prostate, and which are responsible for the back conversion of 3α-diol to 5α-DHT. These enzymes will control ligand access to the AR in the normal and diseased gland.
Figure 2.

Sources of testosterone and 3α-androstanediol in adult male. Adult male (A) and andropause (B) The solid box shows steroidogenesis in Leydig cells, and the dotted-box shows steroidogenesis in the adrenal gland. The pathway in gray shows a backdoor pathway to 5α-DHT via 3α-diol which does not involve the intermediates Δ4-androstene-3,17-dione or testosterone. Enzymes involved in the individual steps are italicized.
3. Aldo-keto reductases and androgen metabolism
3. 1. Human AKR1C Isoforms
Humans express four AKIC isoforms, which show different ratios of 3-, 17- and 20-ketosteroid reductase activities that may regulate ligand access to steroid hormone receptors. Each of these enzymes has been purified to homogeneity as a recombinant protein. The enzymes are soluble monomeric 37 kDa proteins, they share > 86% sequence identity at the amino acid level, and their steroid specificity has been measured (Penning et al., 2000). In addition, crystal structures exist for each of these enzymes. These studies revealed that AKR1C1 functions predominately as a 20α-HSD and will convert progesterone into 20α-hydroxyprogesterone; AKR1C2 will preferentially function as 3α-HSD and will convert 5α-DHT into 3α-diol; AKR1C3 will function as a 17β-HSD and will convert Δ4-androstene-3,17-dione to testosterone and estrone to 17β-estradiol; and AKR1C4 will function as a liver specific 3α-HSD, where it is involved in steroid hormone metabolism and bile-acid biosynthesis.
Recently, the 3-ketosteroid reductase activities of the four human AKR1C isoforms were shown to produce both 3α-diol and 3β-diol, in different ratios indicating that they lacked stereospecificty (Steckelbroeck et al., 2004). Ratios of the specific activities for 3α-diol: 3β-diol formation at saturating 5α-DHT concentrations showed that AKR1C2 favored 3α-diol formation 80-fold over AKR1C1 which instead favored 3β-diol production, Table 1.
Table 1.
Kinetic Properties of AKR and SDR Isoforms Involved in Androgen Metabolism.
| AKR Specific Activities [nmoles min−1mg−1]1 | ||||
|---|---|---|---|---|
| Product | AKR1C1 | AKR1C2 | AKR1C3 | AKR1C4 |
| 3α-Diol | 3.9 | 76.1 | 3.7 | 119.0 |
| 3β-Diol | 16.1 | 3.8 | 2.5 | 32.8 |
| 3α-diol: 3β-diol | 0.25 | 20.1 | 1.55 | 3.61 |
|
| ||||
| SDR Steady State Kinetic Parameters2 | ||||
|
| ||||
| Enzyme | Reaction | Vmax (nmoles) min−1 mg−1 | Km (μM) | Vmax/Km (nmoles min−1 mg−1/μM) |
|
| ||||
| RL-HSD | 3α-Diol ± NAD+ | 5.9 ± 0.26 | 0.4 ± 0.04 | 14.8 |
| RODH5 | 3α-Diol ± NAD+ | 18.5 ± 4.1 | 39.6 ± 3.3 | 0.5 |
| RODH4 | 3α-Diol ± NAD+ | 8.8 ± 0.19 | 0.27 ± 0.03 | 32.4 |
Assayed with 40 μM 5α-DHT plus 180 μM NADPH at 37 °C. From Steckelbroeck et al, 2004
Adapted from Bauman et al, 2006b.
Available crystal structures for the AKR1C1•NADP+•20α-hydroxyprogesterone complex (PDB entry1MRQ) (Couture et al., 2003) and the AKR1C2•NADP+•ursodeoxycholate complex (PDB entry 1H1) (Jin et al., 2001), and molecular modeling studies explain this difference in stereochemical preference for 5α-DHT reduction (Jin and Penning, 2006a). These two enzymes differ by only a single amino acid at the active site. For example L54 in AKR1C1 is substituted by V54 in AKR1C2. This change in the AKR1C2 active site allows 5α-DHT to contact one side of the steroid pocket. Consequently, the β-face of the steroid is in close proximity to the 4-pro-R-hydride of NADPH which is transferred from the A-face of the cofactor to the β-face of the steroid to form the 3α-alcohol. In the case of AKR1C1 the reverse is true, since L54 repels 5α-DHT from this side of the pocket, the α-face of the steroid is presented to NADPH and the stereochemistry of the reaction is inverted, Figure 3.
Figure 3.
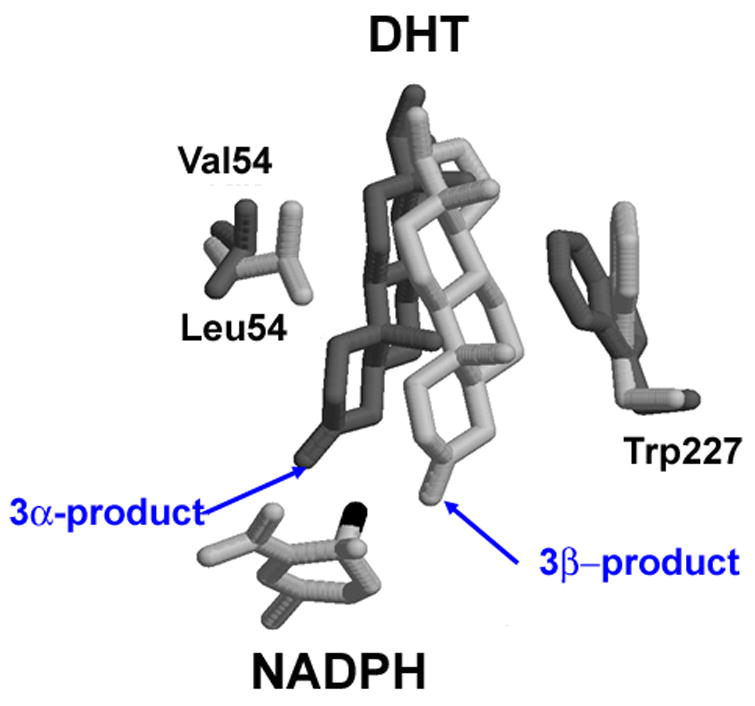
Molecular modeling explains why AKR1C1 and AKR1C2 preferentially reduce 5α-DHT to yield 3β-diol and 3α-diol, respectively. DHT is docked into the active site of AKR1C1 (light gray) and AKR1C2 (dark gray). Adapted from Jin and Penning 2006a.
AKR1C isoform specific RT-PCR was used to measure expression in nine human tissues. AKR1C1-3 were all expressed in prostate (Penning et al., 2000). Using pooled prostatic RNA from 22 Caucasian males AKR1C1-AKR1C3 expression in the adult gland was confirmed (Bauman et al., 2006). Based on the high specific activity and preference to form 3α-diol, AKR1C2 was identified as the isoform responsible for the elimination of 5α-DHT in human prostate.
3. 2. AKR1C2 and 5α-DHT Reduction
In vitro, AKR1C2 functions as an NAD(P)(H)-dependent oxidoreductase and interconverts 5α-DHT with 3α-diol, freely. However, to address its preferred direction, detailed steady-state and transient state kinetics on the recombinant enzyme (Jin and Penning, 2006b) as well as mammalian cell transfection studies were performed (Rizner et al., 2003).
All AKRs, including AKR1C2 catalyze a sequential ordered bi bi mechanism. In this mechanism the binding of NADPH is obligatory before steroid hormone can bind (Askonas et al., 1991; Trauger et al., 2002). Once the ternary complex is formed the “bond-making and breaking events” which define the chemical transformation occur. The products 3α-diol and NADP+ are then released in that order. The steady state kinetic parameters for 5α-DHT reduction by AKR1C2 were kcat = 0.033 s−1, Km = 2.9 μM, and kcat/Km = 1.1 × 104 M−1 s−1; and for 3α-diol oxidation the steady state parameters were kcat = 0.008 s−1, Km = 3.1 μM, and kcat/Km = 2.6 × 103 M−1 s−1. The Keq for the reaction calculated from the kinetic Haldane gave a value of 8.0 indicating that the reduction of 5α-DHT is favored. Although NAD(H) can substitute for NADP(H), the latter cofactor pair is easily preferred since they display nanomolar affinity for the enzyme as compared to high micromolar affinity for NAD(H).
To determine the rate-determining step in the elimination of 5α-DHT the reaction was dissected by transient kinetics using stopped-flow spectroscopy. The binding of NADP(H) measured by quenching the intrinsic protein tryptophan fluorescence showed that a tight E**•NADP(H) complex was formed as a result of two slow conformational changes that occur on the protein. This results in a 80-fold increase in affinity for NADPH where the Kd decreases from 9.6 μM to 120 nM and a 219-fold increase in affinity for NADP+ where the Kd decreases from 46 μM to 210 nM. These high affinities for NADP(H) have metabolic consequences for the enzyme (addressed later).
The rate-determining step in several AKRs is the rate of NADP+ release (Grimshaw et al., 1995). In AKR1C2 the smallest microscopic rate constant (k = 0.66 s−1) measured governs NADP+ release but was 20-times greater than kcat, suggesting that other steps also contribute to rate determination. Multiple turnover experiments showed burst-phase kinetics, which indicated that slow product release steps occurred. Global fitting (DynaFit) of the transient kinetic data to the minimal equation for an ordered bi bi mechanism predicted that three slow-events of similar magnitude accounted for the low kcat = 0.033 s−1. They are the chemical event (0.12 s−1), the release of 3α-diol (0.081 s−1), and the release of NADP+ (0.21 s−1) (Jin and Penning, 2006b).
To further dissect out the preferred direction of AKR1C2, its cDNA was transiently transfected into COS-1 cells and stably transfected into LNCaP (androgen dependent prostate cancer) cells for studies on androgen metabolism (Rizner et al., 2003). In both cell types the expressed enzyme is forced to use the prevailing concentrations of cofactor that exists. In COS-1 cell lysates, AKR1C2 catalyzed the NADPH-dependent reduction of 5α-DHT and the NAD+ dependent oxidation of 3α-diol. However, when intact cells were used only 5α-DHT reduction could be observed and a similar result was observed in LNCaP cells.
To elucidate the reason why the NADPH dependent reduction of 3-ketosteroids is favored in intact cells the effect of redox-status on this reaction was examined. Using the recombinant enzyme in vitro we found that the NADPH-reduction of 5α-DHT to 3α-diol catalyzed by AKR1C2 occurred unimpeded even in the presence of 1 mM NAD+ (Rizner et al., 2003). By contrast we found that the NAD+ dependent oxidation of 3α-diol to 5α-DHT was potently inhibited by low micromolar concentrations of NADPH. Thus the high affinity of NADPH displayed by the enzyme prevents the oxidative reaction due to potent product inhibition. In summary, the favorable Keq for the reduction reaction coupled with the potent inhibition of the oxidative reaction, combine to make AKR1C2 predominately a 3-ketosteroid reductase in intact cells. This property likely applies to all HSDs which are AKRs. Thus AKR1C2 will only account for the elimination of 5α-DHT in the prostate gland.
4. RL-HSD and the oxidation of 3α-diol in the prostate
The identity of the oxidative enzyme that catalyzes the conversion of circulating 3α-diol back to DHT has remained elusive. One source of circulating 3α-diol for the prostate is the liver where the concerted action of type 1 5α-reductase type and AKR1C4 will convert Δ4-androstene-3,17-dione or testosterone to 3α-diol, Figure 2A. Other pathways to 3α-diol are possible which involve formation of 17α-hydroxyprogesterone by CYP17 (CYP17A1) followed by sequential reduction by type 1 5α-reductase and AKR1C enzymes to yield 5α-pregnane-3α,17α-diol-20-one. CYP17 via its 17,20-lyase activity then removes the side-chain to yield androsterone. For this pathway to occur, CYP17, type 1 5α-reductase and AKR1C isoforms would have to exist in the adrenal. Evidence that this pathway exists especially under conditions in which 17α-hydroxyprogesterone accumulates has been presented (Auchus, 2004).
Five short-chain dehydrogenase/reductases (SDRs) can catalyze the conversion of 3α-diol to 5α-DHT and each have been implicated in this reaction in human prostate. However, the ability of the individual enzymes to perform this reaction has not been directly compared and their expression levels in the prostate have not been examined. The enzymes of interest are: L-3-hydroxyacyl coenzyme A dehydrogenase (endoplasmic reticulum amyloid β-peptide binding protein (ERAB; HADH2 alias HADSC)(He et al., 1999; 2000); RL-HSD (Biswas and Russell 1997); 11-cis-retinol dehydrogenase (RoDH 5) (Wang et al., 1999; Huang and Luu-The, 2001); novel type of human microsomal 3α-HSD (NT-3α-HSD) (DHRS9; Chetyrkin et al., 2001); and retinol dehydrogenase 4 (RoDH-4) (Jurukovski et al. 1999; and Gough et al. 1998); all these enzymes are microsomal in localization. The cDNA for each enzyme was cloned into a bis-cistronic construct to yield (pcDNA3-3α-HSD-Lac-Z) where a CMV promoter drives the expression of the 3α-HSD of interest plus β-galactosidase as a single transcript. The presences of an IRES (internal ribosomal entry sequence) permits the single transcript to be processed as two proteins. Thus the expression of 3α-HSD can be normalized to β-galactosidase (internal standard) in the absence of antibodies for each enzyme (Bauman et al., 2006b).
Transient transfection into COS-1 cells followed by measurement of the conversion of 0.1 μM 3α-diol to 5α-DHT showed that three enzymes (RoDH4, RoDH5, and RL-HSD) converted 80% of DHT into steroid product within 30 min, whereas NT-3α-HSD and ERAB converted less than 5–10% of this substrate over the same time frame. Transfection studies showed that these enzymes were unable to reduce 5α-DHT to 3α-diol. In fact the mock-transfected cells were superior in performing this reaction suggesting that each of these SDRs preferentially function as 3α-hydroxysteroid oxidases. Steady state kinetic parameters for RoDH4, RoDH5 and RL-HSD were then compared in the COS-1 cell lysates for the NAD+ dependent oxidation of 3α-diol. It was found that RoDH4 and RL-HSD were high affinity low capacity enzymes for the oxidation reaction, Table 1.
To determine whether the oxidative 3α-HSDs (RoDH4, RoDH5 and RL-HSD) were necessary and sufficient to convert 3α-diol to 5α-DHT to cause trans-activation of the AR, reporter gene assays were performed. COS-1 cells were co-transfected with AR, a p(androgen response element)2-tk-CAT reporter gene construct in the absence or presence of the oxidative 3α-HSD of interest and exposed to fixed concentrations of 3α-diol over the range of 10−12 to 10−6 M. Oxidative 3α-HSDs that regulate ligand occupancy of the AR should convert 3α-diol to 5α-DHT and increase reporter gene activity. It was found that the EC50 value for 3α-diol in mock transfected cells was 1 × 10−7 M, however, in the presence of co-transfected RoDH4, RoDH5 and RL-HSD the EC50 value for 3α-diol was reduced to 1 × 10−9 M consistent with its conversion to 5α-DHT, Figure 4.
Figure 4.

Trans-activation of the androgen receptor by 3α-androstanediol in mammalian cells. Trans-activation of the AR by 3α-diol in the presence of oxidative 3α-HSDs. (A), Activation of the (ARE)2-tk-CAT reporter gene by the AR in the presence of co-transfected HSDs versus the concentration of 3α-diol (10−12 to 10−6 M); (B) the calculated EC50 values to reach a 100% trans-activation; where 100% trans-activation is the maximal response seen with 5α-DHT. The fold increase in chloramphenicol acetyl transferase (CAT) activity seen at maximal response was 30-fold; and (C) The cellular basis of the assay. Abbreviations, T = testosterone. Adapted from Bauman et al., 2006b.
Our cell-based approach indicated that three candidate oxidative 3α-HSDs may be responsible for the back-reaction in human prostate: RoDH4, RoDH5, and RL-HSD, however, to identify which enzyme may be the most relevant in the prostate, expression studies were required. We established validated real-time RT-PCR protocols to measure the expression of these enzymes in prostate RNA pooled from 22 Caucasian males. It was found that only ERAB and RL-HSD were abundantly expressed; of these only RL-HSD is able to oxidize 3α-diol to 5α-DHT efficiently, and in sufficient quantities to trans-activate AR. Thus RL-HSD is the isoform identified as the 3α-HSD responsible for the back reaction in human prostate (Bauman et al., 2006b).
5. 3α-HSD (AKR1C2 and RL-HSD) expression in the prostate by cell–type
Our studies suggest that the two enzymes that regulate ligand occupancy of the AR in human prostate are the reductive 3α-HSD (AKR1C2) and the oxidative 3α-HSD (RL-HSD). To determine whether these enzymes co-exist in the same cell type and whether their expression levels are altered by disease status we conducted real-time PCR in primary cultures of human prostate stromal and epithelial cells from normal males, patients with BPH, and patients with CaP (Bauman et al., 2006a). We found that AKR1C2 was predominately expressed in epithelial cells and while no significant differences in levels of expression were found in cells from normal patients and patients with either BPH or CaP. A trend of increased expression in disease was noted but did not reach significance. By contrast levels of the RL-HSD transcript were 50–100 fold less than AKR1C2 in this cell type. The expression of RL-HSD was a mirror image of that observed with AKR1C2. We found that RL-HSD was predominately expressed in stromal cells but was absent from epithelial cells and a significant increase in expression was noted in stromal cells from BPH patients (p value < 0.005) Figure 7. Real-time PCR detection of the AR in the same cell types indicated that it was primarily expressed in the stromal cells and that it was also elevated in BPH (p value < 0.001). The inability to detect high expression of AR in these primary epithelial cells reflect their basal rather than luminal origin. Stolz and coworkers have shown that AKR1C2 is lost during prostate cancer and may be responsible for an increase intraprostatic 5α-DHT levels [Ji et al., 2007]. However, these studies were performed by conducting real-time PCR measurements of RNA isolated from prostate tumor samples where the cell-type was heterogeneous.
In summary, it is proposed that 5α-DHT is inactivated in the epithelial cells by AKR1C2 to form 3α-diol. In stromal cells 3α-diol is reactivated to 5α-DHT by RL-HSD and this effect is more pronounced in BPH, where the 5α-DHT formed could bind to the stromal cell AR which is also upregulated. Based on these findings RL-HSD becomes a target for adjuvant therapy of BPH.
6. Conclusions
AKR1C2 and RL-HSD govern the pre-receptor regulation of the AR in human prostate. This is based on substrate specificity, directionality, tissue and cellular distribution within normal and diseased prostate. A case can be made that RL-HSD may represent an alternative target to type 2 5α-reductase to block abnormal growth of the prostate gland in BPH. Whether the same AKR/SDR enzyme pair governs ligand access to the AR in other androgen target tissues e.g., bone, muscle and kidney remains to be determined. Identification of the discrete enzyme pair involved in regulating ligand access to the AR in different target tissues provides an attractive approach to achieve tissue specific androgen and anti-androgen responses that differs from the use of selective androgen receptor modulators.
Figure 5.

Expression of AKR1C2, RL-3α-HSD and AR in prostate stromal and epithelial cells. Representative scatter box plots of transcripts of enzymes (AKR1C2 and RL-HSD) that regulate ligand access to the androgen receptor (AR) in epithelial and stromal cells from normal and diseased patients (CaP and BPH) using real-time RT-PCR panels A–D. Expression of the AR in the same cell types panels E–F. One μg of total RNA was reverse-transcribed to cDNA from the epithelial and stromal cells and 50 ng of cDNA was added to each real-time PCR experiment that was performed in triplicate with the mean shown for each sample. Data is normalized to the housekeeping gene PBGD and is expressed as fg of each transcript per ng of total cDNA. Normal (n=14), CaP (n=14) and BPH (n=6) epithelial cells and normal (n=15), CaP (n=16) and BPH (n=21) stromal cells were used for the study, adapted from Bauman et al., 2006a.
Acknowledgments
This work was supported by grants R01-CA90744, R01-DK-47015 and P30-ES013508 awarded by the National Institutes of Health to T.M.P.
Abbreviations
- AKR
aldo-keto reductase
- AKR1C1
20α (3α)-hydroxysteroid dehydrogenase
- AKR1C2
human type 3 3α-hydroxysteroid dehydrogenase
- AKR1C3
human type 2 3α-hydroxysteroiud dehydrogenase, type 5 17β-hydroxytsreoid dehydrogenase
- AKR1C4
human type 1 3α-hydroxysteroid dehydrogenase
- AR
androgen receptor
- BPH
benign prostatic hyperplasia
- CaP
cancer of the prostate
- 5α-DHT
5α-dihydrotestosterone
- 3α-diol
5α-androstane-3α,17β-diol
- ERAB
endoplasmic reticulum β-peptide binding protein
- GR
glucocorticoid receptor
- HSD
hydroxysteroid dehydrogenase
- MR
mineralocorticoid receptor
- NT-3α-HSD
novel type of human microsomal 3α-HSD
- RL-HSD
RoDH like 3α-HSD
- RoDH
retinol dehydrogenase
- SDR
short-chain dehydrogenase/reductase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Askonas LJ, Ricigliano JW, Penning TM. The kinetic mechanism catalysed by homogeneous rat liver 3α-hydroxysteroid dehydrogenase. Evidence for binary and ternary dead-end complexes containing non-steroidal anti-inflammatory drugs. Biochem J. 1991;278:835–841. doi: 10.1042/bj2780835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auchus RJ. The backdoor pathway to dihydrotestosterone. Trends in Endocrinology & Metabolism. 2004;15:432–438. doi: 10.1016/j.tem.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Bauman DR, Steckelbroeck S, Peehl DM, Penning TM. Transcript profiling of the androgen signal in normal prostate, benign prostatic hyperplasia, and prostate cancer. Endocrinology. 2006a;147:5806–5816. doi: 10.1210/en.2006-0627. [DOI] [PubMed] [Google Scholar]
- Bauman DR, Steckelbroeck S, Williams MV, Peehl DM, Penning TM. Identification of the major oxidative 3α-hydroxysteroid dehydrogenase in human prostate that converts 5α-androstane-3α,17β-diol to 5α-dihydrotestosterone: A potential therapeutic target for androgen dependent disease. Mol Endocrinol. 2006b;20:444–458. doi: 10.1210/me.2005-0287. [DOI] [PubMed] [Google Scholar]
- Biswas MG, Russell DW. Expression cloning and characterization of oxidative 17β- and 3α-hydroxysteroid dehydrogenases from rat and human prostate. J Biol Chem. 1997;272:15959–15966. doi: 10.1074/jbc.272.25.15959. [DOI] [PubMed] [Google Scholar]
- Carcon-Jurica MA, Schrader WT, O’Malley B. Steroid receptor family: structure and functions. Endocrine Rev. 1990;11:201–220. doi: 10.1210/edrv-11-2-201. [DOI] [PubMed] [Google Scholar]
- Chetyrkin SV, Belyaeva OV, Gough WH, Kedishvili NY. Characterization of a novel type of human microsomal 3α-hydroxysteroid dehydrogenase: unique tissue distribution and catalytic properties. J Biol Chem. 2001;276:22278–22286. doi: 10.1074/jbc.M102076200. [DOI] [PubMed] [Google Scholar]
- Couture JF, Legrand O, Cantin L, Luu-The V, Labrie F, Breton R. Human 20α-hydroxysteroid dehydrogenase: crystallographic and site-directed mutagenesis studies lead to the identification of an alternative binding site for C-21 steroids. J Mol Biol. 2003;331:593–604. doi: 10.1016/s0022-2836(03)00762-9. [DOI] [PubMed] [Google Scholar]
- DeKlerk DP, Coffey DS, Ewing LL, McDermott IR, Reiner WG, Robinson CH, Scott WW, Strandberg JD, Talalay P. Comparison of spontaneous and experimentally induced canine prostatic hyperplasia. J Clin Invest. 1979;64:842–849. doi: 10.1172/JCI109532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RM. The nuclear receptor superfamily: a Rosetta Stone for physiology. Mol Endocrinol. 2005;19:1429–1438. doi: 10.1210/me.2005-0046. [DOI] [PubMed] [Google Scholar]
- Funder JW, Pearce PT, Smith R, Smith AI. Mineralocorticoid action: target tissue specificity is enzyme, not receptor, mediated. Science. 1988;242:583–5. doi: 10.1126/science.2845584. [DOI] [PubMed] [Google Scholar]
- Gough WH, Van Ooteghem S, Sint T, Kedishvili NY. cDNA cloning and characterization of a new human microsomal NAD+-dependent dehydrogenase that oxidizes all trans-retinol and 3α-hydroxysteroids. J Biol Chem. 1998;273:19778–19785. doi: 10.1074/jbc.273.31.19778. [DOI] [PubMed] [Google Scholar]
- Grimshaw CE, Bohren KM, Lai CJ, Gabbay KH. Human aldose reductase: rate constants for a mechanism including interconversion of ternary complexes by recombinant wild-type enzyme. Biochemistry. 1995;34:14356–14365. doi: 10.1021/bi00044a012. [DOI] [PubMed] [Google Scholar]
- Guerini V, Sau D, Scaccianoce E, Rusmini P, Ciana P, Maggi A, Martini PG, Katzenellenbogen BS, Martini L, Motta M, Poletti A. The androgen derivative 5α-androstane-3β,17β-diol inhibits prostate cancer cell migration through activation of the estrogen receptor β subtype. Cancer Res. 2005;65:5445–5453. doi: 10.1158/0008-5472.CAN-04-1941. [DOI] [PubMed] [Google Scholar]
- He XY, Merz G, Mehta P, Schultz H, Yang SY. Human brain short chain L-3-hydroxyacyl coenzyme A dehydrogenase is a single-domain multifunctional enzyme. Characterization of a novel 17β-hydroxysteroid dehydrogenase. J Biol Chem. 1999;274:15014–15919. doi: 10.1074/jbc.274.21.15014. [DOI] [PubMed] [Google Scholar]
- He XY, Merz G, Yang YZ, Pullakart R, Mehta P, Schulz H, Yang SY. Function of human brain short chain L-3-hydroxyacyl coenzyme A dehydrogenase in androgen metabolism. Biochim Biophys Acta. 2000;1484:267–277. doi: 10.1016/s1388-1981(00)00014-7. [DOI] [PubMed] [Google Scholar]
- Horst HJ, Denis M, Kaufmann J, Voigt KD. In vivo uptake and metabolism of [3H]-5α-androstane-3α,17β-diol and of [3H]-5α-androstane-3β,17β-diol by human prostatic hypertrophy. Acta Endocrinol. 1975;79:394–402. [PubMed] [Google Scholar]
- Huang XF, Luu-The V. Characterization of the oxidative 3α-hydroxysteroid dehydrogenase activity of human recombinant 11-cis retinal dehydrogenase. Biochim Biophys Acta. 2001;1547:351–358. doi: 10.1016/s0167-4838(01)00200-x. [DOI] [PubMed] [Google Scholar]
- Hyndman DR, Bauman DR, Heredia VV, Penning TM. The aldo-keto reductase superfamily homepage. Chem-Biol Inter. 2003:143–144. 621–623. doi: 10.1016/s0009-2797(02)00193-x. [DOI] [PubMed] [Google Scholar]
- Imperato-McGinley J, Guerrero L, Gautier T, Peterson RE. Steroid 5a-reductase deficiency in man: an inherited form of pseduohermaphroditism. Science. 1974;186:1213–1215. doi: 10.1126/science.186.4170.1213. [DOI] [PubMed] [Google Scholar]
- Imperato-McGinley J, Gautier T, Zirinsky K, Hom T, Paloma O, Stein E, Vaughan ED, Markisz JA, Ramirez de Arellano E, Kazam E. Prostate visualization in males homozygous and heterozygous for 5α-reductase deficiency. J Clin Endocrinol Metabo. 75:1022–1026. doi: 10.1210/jcem.75.4.1400866. [DOI] [PubMed] [Google Scholar]
- Jacobi GH, Moore RJ, Wilson JD. Characterization of the 3α-hydroxysteroid dehydrogenase of dog prostate. J Steroid Biochem. 1977;8:719–723. doi: 10.1016/0022-4731(77)90002-4. [DOI] [PubMed] [Google Scholar]
- Ji Q, Chang L, Stanczyk FZ, Ookhtens M, Sherrod A, Stolz A. Impaired dihydrotestosterone catabolism in human prostate cancer. Critical role of AKR1C2 as a pre-receptor regulator of androgen signaling. Cancer Res. 2007;67:1361–1369. doi: 10.1158/0008-5472.CAN-06-1593. [DOI] [PubMed] [Google Scholar]
- Jin Y, Penning TM. Molecular docking simulations of steroid substrates into human cytosolic hydroxysteroid dehydrogenases (AKR1C1 and AKR1C2): Insights into positional and stereochemical preferences. Steroids. 2006a;71:380–391. doi: 10.1016/j.steroids.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Jin Y, Penning TM. Multiple steps determine the overall rate of the reduction of 5α-dihydrotestosterone catalyzed by human type 3 3α-hydroxysteroid dehydrogenase: implications for the elimination of androgens. Biochemistry. 2006b;45:13054–13063. doi: 10.1021/bi060591r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Stayrook SE, Albert RH, Palackal NT, Penning TM, Lewis M. Crystal structure of human type III 3α-hydroxysteroid dehydrogenase/bile-acid binding protein complexed with NADP+ and ursodeoxycholate. Biochemistry. 2001;40:10161–10168. doi: 10.1021/bi010919a. [DOI] [PubMed] [Google Scholar]
- Jornvall H, Persson B, Krook M, Atrian S, Gonzalez-Duarte R, Jeffery J, Ghosh D. Short-chain dehydrogenases/reductases (SDR) Biochemistry. 1995;34:6003–6013. doi: 10.1021/bi00018a001. [DOI] [PubMed] [Google Scholar]
- Jurukovski V, Markova NG, Karaman-Jurukovska N, Randolph RK, Su J, Napoli JL, Simon M. Cloning and characterization of retinol dehydrogenase transcripts expressed in human keratinocytes. Mol Genet Metabol. 1999;67:62–73. doi: 10.1006/mgme.1999.2840. [DOI] [PubMed] [Google Scholar]
- Kinouchi T, Horton R. 3α-Androstanediol kinetics in man. J Clin Invest. 1974;54:646–653. doi: 10.1172/JCI107802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krozowski Z, MaGuire JA, Stein-Oakley AN, Dowling J, Smith RE, Andrews RK. Immunohistochemical localization of the 11β-hydroxysteroid dehydrogenase type II enzyme in human kidney and placenta. J Clin Endocrinol Metab. 1995:2203–2209. doi: 10.1210/jcem.80.7.7608280. [DOI] [PubMed] [Google Scholar]
- Labrie F, Belanger A, Simard J. Intracrinology. Autonomy and freedom of peripheral tissues. Annuals Endocrinology. 1995;56:23–29. [PubMed] [Google Scholar]
- Labrie F, Luu-The V, Lin SX, Labrie C, Simard J, Breton R, Belanger A. The key role of 17β-hydroxysteroid dehydrogenases in sex steroid biology. Steroids. 1997;62:148–158. doi: 10.1016/s0039-128x(96)00174-2. [DOI] [PubMed] [Google Scholar]
- Leihy MW, Shaw G, Wilson JD, Renfree MB. Virilization of the urogenital sinus of the tammar wallaby is not unique to 5α-androstane-3α,17β-diol. Mol Cell Endocrinol. 2001;181:111–115. doi: 10.1016/s0303-7207(01)00527-5. [DOI] [PubMed] [Google Scholar]
- Liu Z, Wong J, Tsai SY, Tsai MJ, O’Malley BW. Steroid receptor coactivator-1 (SRC-1) enhances ligand-dependent and receptor -dependent cell-free transcription of chromatin. Proc Natl Acad Sci USA. 1999;96:9485–9490. doi: 10.1073/pnas.96.17.9485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlowski J, Bird CE, Clark AF. Androgen 5α-reductase and 3α-hydroxysteroid dehydrogenase activities in ventral prostate epithelial and stromal cells from immature and mature rats. J Endocrinol. 1983;99:131–139. doi: 10.1677/joe.0.0990131. [DOI] [PubMed] [Google Scholar]
- Penning TM. Molecular endocrinology of hydroxysteroid dehydrogenases. Endocrine Reviews. 1997;18:281–305. doi: 10.1210/edrv.18.3.0302. [DOI] [PubMed] [Google Scholar]
- Penning TM, Burczynski ME, Jez JM, Hung CF, Lin HK, Ma H, Moore M, Palackal N, Ratnam K. Human 3α-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem J. 2000;351:67–77. doi: 10.1042/0264-6021:3510067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penning TM. Hydroxysteroid dehydrogenases and pre-receptor regulation of steroid hormone action. Human Reproduction Update. 2003;9:193–205. doi: 10.1093/humupd/dmg022. [DOI] [PubMed] [Google Scholar]
- Rizner T, Lin HK, Peehl DM, Steckelbroeck S, Bauman DR, Penning TM. Human type 3 3α-hydroxysteroid dehydrogenase (AKR1C2) and androgen metabolism in prostate cells. Endocrinology. 2003;144:2922–2932. doi: 10.1210/en.2002-0032. [DOI] [PubMed] [Google Scholar]
- Russell DW, Wilson JD. Steroid 5α-reductase: two genes/two enzymes. Annu Rev Biochem. 1994;63:25–61. doi: 10.1146/annurev.bi.63.070194.000325. [DOI] [PubMed] [Google Scholar]
- Seckl JR, Walker BR. Minireview: 11β-Hydroxysteroid dehydrogenase type 1 a tissue-specific amplifier of glucocorticoid action. Endocrinology. 2001;142:1371–1376. doi: 10.1210/endo.142.4.8114. [DOI] [PubMed] [Google Scholar]
- Shaw G, Renfree MB, Leihy MW, Shackleton CH, Roitman E, Wilson JD. Prostate formation in a marsupial is mediated by the testicular androgen 5α–androstane-3α,17β-diol. Proc Natl Acad Sci USA. 2000;97:12256–12259. doi: 10.1073/pnas.220412297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata H, Spencer TE, Onate SA, Jenster G, Tsai SY, Tsai MJ, O’Malley BW. Role of co-activators and co-repressors in the mechanisms of steroid/thyroid receptor action. Recent Progress in Hormone Res. 1997;52:141–164. [PubMed] [Google Scholar]
- Steckelbroeck S, Jin Y, Gopishetty S, Oyesanmi B, Penning TM. Human cytosolic 3α-hydroxysteroid dehydrogenases of the aldo-keto reductase superfamily display significant 3β-hydroxysteroid dehydrogenase activity: implications for steroid hormone metabolism and action. J Biol Chem. 2004;279:10784–10795. doi: 10.1074/jbc.M313308200. [DOI] [PubMed] [Google Scholar]
- Trauger JW, Jiang A, Sterans BA, LoGrasso PV. Kinetics of allopregnanolone formation catalyzed by human 3α-hydroxysteroid dehydrogenase type III (AKR1C2) Biochemistry. 2002;41:13451–13459. doi: 10.1021/bi026109w. [DOI] [PubMed] [Google Scholar]
- Truss M, Beato M. Steroid hormone receptors: interaction with deoxyribonucleic acid and transcription factors. Endocrine Rev. 1993;14:459–479. doi: 10.1210/edrv-14-4-459. [DOI] [PubMed] [Google Scholar]
- Walsh PC, Wilson JD. The induction of prostatic hypertrophy in the dog with androstanediol. J Clin Invest. 1976;57:1093–1097. doi: 10.1172/JCI108353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Chai X, Eriksson U, Napoli JL. Activity of human 11-cis-retinol dehydrogenase (Rdh5) with steroids and retinoids and expression of its mRNA in extra-ocular human tissue. Biochem J. 1999;338:23–27. [PMC free article] [PubMed] [Google Scholar]
- Weihua Z, Lathe R, Warner M, Gustafsson JA. An endocrine pathway in the prostate ERβ, AR, 5α-androstane-3β,17β-diol and CYP7B1 regulates prostate growth. Proc Natl Acad Sci USA. 2002;99:13589–13594. doi: 10.1073/pnas.162477299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White PC. 11β-Hydroxysteroid dehydrogenase and its role in the syndrome of apparent mineralocorticoid excess. Am J Med Sci. 2001;322:308–315. doi: 10.1097/00000441-200112000-00003. [DOI] [PubMed] [Google Scholar]
- Whorwood CB, Stewart PM. Human hypertension caused by mutations in the 11β-hydroxysteroid dehydrogenase gene: a molecular analysis of apparent mineralocorticoid excess. Hypertens Suppl. 1996:S19–24. [PubMed] [Google Scholar]