

Abstract
Accumulating evidence suggests that the reactive oxygen and nitrogen species are generated in cardiomyocytes and endothelial cells during myocardial ischemia/reperfusion injury, various forms of heart failure or cardiomyopathies, circulatory shock, cardiovascular aging, diabetic complications, myocardial hypertrophy, atherosclerosis, and vascular remodeling following injury. These reactive species induce oxidative DNA damage and consequent activation of the nuclear enzyme poly(ADP-ribose) polymerase 1 (PARP-1), the most abundant isoform of the PARP enzyme family. PARP overactivation, on the one hand, depletes its substrate, NAD+, slowing the rate of glycolysis, electron transport, and ATP formation, eventually leading to the functional impairment or death of the endothelial cells and cardiomyocytes. On the other hand, PARP activation modulates important inflammatory pathways, and PARP-1 activity can also be modulated by several endogenous factors such as various kinases, purines, vitamin D, thyroid hormones, polyamines, and estrogens, just to mention a few. Recent studies have demonstrated that pharmacological inhibition of PARP provides significant benefits in animal models of cardiovascular disorders, and novel PARP inhibitors have entered clinical development for various cardiovascular indications. Because PARP inhibitors can enhance the effect of anticancer drugs and decrease angiogenesis, their therapeutic potential is also being explored for cancer treatment. This review discusses the therapeutic effects of PARP inhibitors in myocardial ischemia/reperfusion injury, various forms of heart failure, cardiomyopathies, circulatory shock, cardiovascular aging, diabetic cardiovascular complications, myocardial hypertrophy, atherosclerosis, vascular remodeling following injury, angiogenesis, and also summarizes our knowledge obtained from the use of PARP-1 knockout mice in the various preclinical models of cardiovascular diseases.
Keywords: Angiogenesis, Apoptosis, Cardiomyopathy, Diabetes, DNA repair, Heart failure, Inflammation, Necrosis, Nitric oxide, Peroxynitrite, Poly(ADP-ribose) polymerase, Vascular remodeling
INTRODUCTION
The nuclear enzyme poly(ADP-ribose) polymerase 1 (PARP-1, EC 2.4.2.30) is the most abundant isoform of the PARP enzyme family, which is continuously undergoing expansion (Jagtap and Szabo 2005; Schreiber et al. 2006; Szabo et al. 2006a; Virag 2005; Virag and Szabo 2002). PARP-1, is a 116-kDa protein consisting of three main domains: the N-terminal DNA-binding domain containing two zinc fingers, the automodification domain, and the C-terminal catalytic domain. The primary structure of the enzyme is highly conserved in eukaryotes, with the catalytic domain showing the highest degree of homology between different species. PARP-1 functions as a DNA damage sensor and signaling molecule binding to both single- and double-stranded DNA breaks. Upon binding to damaged DNA, PARP-1 forms homodimers and catalyzes the cleavage of NAD+ into nicotinamide and ADP-ribose to form long branches of ADP-ribose polymers on glutamic acid residues of a number of target proteins including histones and PARP-1 (automodification domain) itself. Poly(ADP-ribosylation) deliberates negative charge to histones leading to electrostatic repulsion among histones and DNA, a process implicated in chromatin remodeling, DNA repair, and transcriptional regulation. Numerous transcription factors, DNA replication factors, and signaling molecules have also been shown to become poly(ADP-ribosylated) by PARP-1. The effects of PARP-1 on the function of these proteins is achieved by noncovalent protein–protein interactions or by covalent poly(ADP-ribosyl)ation. Poly(ADP-ribosyl)ation) is a fast dynamic process, which is also indicated by the short (<1 min) in vivo half-life of the polymer, and determined by two catabolic enzymes poly(ADP-ribose) glycohydrolase (PARG) and ADP-ribosyl protein lyase (Virag and Szabo 2002).
Until recently, it was thought that the regulation of PARP happens primarily at the level of DNA breakage: Recognition of DNA breaks was considered to be the primary regulator (activator) or the catalytic activity of PARP (Jagtap and Szabo 2005; Schreiber et al. 2006; Szabo et al. 2006a; Virag 2005; Virag and Szabo 2002). Excitedly, recent studies have provided evidence that PARP-1 activity can also be modulated by several endogenous and exogenous factors, including various kinases, estrogen, thyroid hormones, active forms of vitamin D, polyamines, purines, and caffeine metabolites (Szabo et al. 2006a). Poly(ADP-ribosyl)ation is involved in the regulation of multiple physiological and pathophysiological cellular functions such as DNA repair, gene transcription, cell cycle progression, cell death, chromatin function, and genomic stability, the discussion of which is a subject of several recent detailed overviews (Jagtap and Szabo 2005; Schreiber et al. 2006; Szabo et al. 2006a; Virag 2005; Virag and Szabo 2002), and is beyond the scope of this review article. In this review we will highlight the main mechanisms and pathways that underlie the rationale for the development of various PARP inhibitors for diverse cardiovascular indications.
PARP-1 AND ITS INHIBITORS
Triggers, Consequences, and Inhibitors of PARP-1 Activation
According to the unifying concept (Virag and Szabo 2002) cells exposed to DNA damaging agents can enter three major pathways based on the intensity of the trigger. Moderate genotoxic stimuli facilitate PARP-1 activation leading to DNA repair by signaling cell cycle arrest and by interacting with DNA repair enzymes such as XRCC1 and DNA-PK. Consequently, DNA damage is restored and cells survive without the risk of passing on mutated genes in this pathway. This scenario may occur in cells that are exposed to certain genotoxic agents (e.g., antitumor drugs), when the damage caused by these agents is largely dependent on the activation of PARP; under these circumstances the inhibition of PARP augments cytotoxicity, which can be exploited for anticancer therapy. More severe DNA damage triggers the second apoptotic cell death pathway during which caspases (the main executor enzymes of the apoptotic machinery) inactivate PARP-1 by cleaving it into two fragments by destroying its ability to respond to DNA strand breaks, thereby preventing the loss of cellular ATP associated with PARP activation and allowing the maintenance of the cellular energy essential for the execution of apoptosis. This route is intended to prevent cells from the pathological consequence of the third pathway mentioned later in which cells die by necrosis, a less controlled mechanism also posing a risk for neighboring cells. As such, PARP cleavage has been proposed to function as a molecular switch between apoptotic and necrotic modes of cell death (Boulares et al. 1999; Levrand et al. 2006; Virag and Szabo 2002). Extensive oxidative and/or nitrosative stress triggers the third pathway by inducing extensive DNA breakage, overactivation of PARP, and consequent depletion of the cellular stores of its substrate NAD+, impairing glycolysis, Krebs cycle, and mitochondrial electron transport, and eventually resulting in ATP depletion and consequent cell dysfunction and death by necrosis. In this case, pharmacological inhibition of PARP or genetic deletion of the PARP-1 preserves cellular NAD+ and ATP pools in oxidatively and/or nitrosatively stressed cardiomyocytes, endothelial or other cell types, thereby allowing them to function normally, or, if the apoptotic process has initiated, to utilize the apoptotic machinery and die by apoptosis instead of necrosis (Bhatnagar 1997; Bowes et al. 1998a, 1999; Fiorillo et al. 2006; Gilad et al. 1997; Levrand et al. 2006). The inhibition of this third pathway by PARP inhibitors may offer tremendous therapeutic benefit; for instance, in severe cardiovascular conditions (e.g., during the myocardial ischemia reperfusion following myocardial infarction, bypass surgery, cardiac transplantation, cardiac arrest, aortic reconstructive surgery, just to mention a few) by preventing acute cell death. In addition to its previously mentioned functions in cell death, two recently discovered roles of PARP have been described, which are crucial from the therapeutic perspective of most cardiovascular disorders to be described.
The first additional role of PARP-1 is its involvement of regulating the mitochondria-to-nucleus translocation of apoptosis-inducing-factor (AIF), a 67-kDa mitochondrial death-promoting protein, which induces DNA fragmentation by initiating the activation of a yet unidentified nuclease (Susin et al. 1999). PARP-1 activity appears to be essential for AIF to translocate to the nucleus in cells exposed to oxidative stress, a process most likely mediated by small PAR fragments signaling into the mitochondria (Andrabi et al. 2006; Yu et al. 2002, 2006). As such, AIF is currently believed to play an important role in PARP-1-dependent cell death (Andrabi et al. 2006; Dawson and Dawson 2004; Yu et al. 2002, 2006), supporting the hypothesis that a nuclear mitochondrial crosstalk dependent on poly(ADP-ribosylation) is critical in determining the fate of oxidatively injured cells. Interestingly, this crosstalk may also involve a PARP-1-dependent activation of the MAP kinase JNK1 via a pathway using members of the TNF signaling cascade (RIP1 and TRAF2) (Xu et al. 2006). Further studies are required to clarify this intriguing aspect of PARP-1 biology.
The second additional role of PARP-1 is its involvement in the regulation of the expression of various proteins implicated in the inflammation at the transcriptional level [(e.g., inducible nitric oxide synthase (iNOS), intercellular adhesion molecule-1 (ICAM-1), [COX-2, and major histocompatibility complex class II (MHC Class II)], which is of particular importance. The absence of functional PARP-1 (either genetic or pharmacological) decreased the expression of a host of proinflammatory mediators, including cytokines, chemokines, adhesion molecules, and enzymes (e.g., iNOS), and it also reduced tissue infiltration with activated phagocytes in experimental models of inflammation, circulatory shock, and ischemia reperfusion (Szabo 2006). NF-κB is a key transcription factor in the regulation of these proteins and PARP has been shown to act as a co-activator in the NF-κB-mediated transcription (Oliver et al. 1999). Poly(ADP-ribosylation) can loosen up chromatin structure and thereby make genes more accessible for the transcriptional machinery. These seminal observations have been extended to show that PARP-1 further participates in the activation of other essential proinflammatory signaling cascades, including JNK (Zingarelli et al. 2004a, 2004b) and p38 MAP-kinases (Ha et al. 2002), as well as the transcription factors activator-protein-1 (AP-1), stimulating factor-1 (Sp-1), octamer-binding transcription factor-1 (Oct-1), Yin Yang-1 (YY-1), and signal transducer and activator of transcription-1 (STAT-1) (Ha et al. 2002). The ability of PARP inhibitors to suppress the expression of proinflammatory genes may be further exploited in various cardiovascular disorders associated with acute (e.g., myocardial infarction, coronary bypass and aortic reconstructive surgeries and septic shock) and/or chronic inflammation (e.g., atherosclerosis, cardiovascular aging), inflammatory diseases, and various forms of cancer. Some of the key pathophysiological roles of PARP are shown in a simplified scheme (Fig. 1).
FIG. 1.
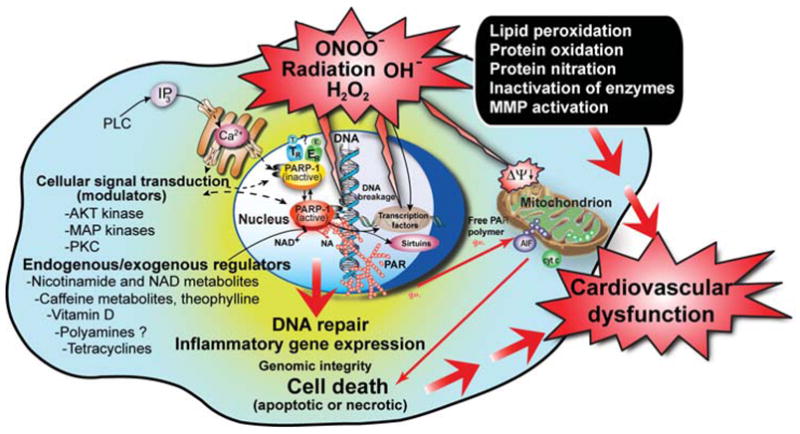
Triggers, mechanisms of PARP-mediated cell death, and exogenous/endogenous regulators/modulators of PARP activity. Reactive oxygen and nitrogen species (e.g., peroxynitrite)-dependent cytotoxicity in various cardiovascular diseases is mediated by a multitude of effects including lipid peroxidation, protein nitration and oxidation, DNA oxidative damage, activation of matrix metalloproteinases (MMPs), and inactivation of a series of enzymes. Mitochondrial enzymes are particularly vulnerable to attacks by peroxynitrite, leading to reduced ATP formation and induction of mitochondrial permeability transition by opening of the permeability transition pore, which dissipates the mitochondrial membrane potential (Δψm). These events lead to cessation of electron transport and ATP formation, mitochondrial swelling, and permeabilization of the outer mitochondrial membrane, allowing the efflux of several proapoptotic molecules, including cytochrome or C and apoptosis-inducing factor (AIF). In turn, cytochrome or C and AIF activate a series of downstream effectors, which eventually result in the fragmentation of nuclear DNA. In addition to its damaging effects on mitochondria, peroxynitrite inflicts more or less severe oxidative injury to DNA, resulting in DNA strand breakage, which in turn activates the nuclear enzyme poly(ADP-ribose) polymerase (PARP). Activated PARP consumes NAD to build up poly(ADP-ribose) polymers (PAR), which are themselves rapidly metabolized by the activity of poly(ADP-ribose) glycohydrolase (PARG). Some free PAR may exit the nucleus and travel to the mitochondria, where they amplify the mitochondrial efflux of AIF (nuclear to mitochondria crosstalk). Depending on the severity of the initial insult by peroxynitrite or other oxidants, the injured cell may either recover or die. In the latter case, the cell may be executed by apoptosis in the case of moderate PTP opening and PARP activation with preservation of cellular ATP, or by necrosis in case of widespread PTP opening and PARP overactivation, leading to massive NAD consumption and collapse of cellular ATP. Various endogenous factors can influence PARP activity either by inhibiting the binding of its substrate NAD+ to the active site of the enzyme or by forming a complex with PARP. An example for the latter may include estrogen (E) and thyroid hormones (T) and for the former nicotinamide (NA), NAD+ metabolites, caffeine metabolites, and vitamin D. PARP activity can also be modulated by various kinases by phosphorylation (e.g., MAP kinases and PKC), and PARP can modulate kinase (e.g., AKT) activity. Various exogenous factors such as caffeine and its endogenously formed metabolites, theophylline, and tetracycline antibiotics may also modulate PARP activity. Overall, PARP appears to be a subject of multiple lines of endogenous regulators, and it is conceivable that the processes regulated by PARP (e.g., DNA repair and cellular NAD homeostasis) are under a similarly dynamic control by a multitude of factors and influences.
Many pharmacological inhibitors of PARP have been developed over the last two decades. Some of the prototypical PARP inhibitor structures are shown in Figure 2. The medicinal chemistry and structure–activity relationships of these compounds are beyond the scope of this paper and have been overviewed in a number of recent expert reviews (Donawho et al. 2007; Jagtap et al. 2002, 2004, 2005; Ratnam and Low 2007; Southan and Szabo 2003; Thomas et al. 2007; Virag and Szabo 2002; Woon and Threadgill 2005). Please note that the inhibitors of lower potency tend to be less specific, as they also exert nonspecific antioxidant effects, as well as effects on many (if not all) of the various isoforms of the PARP family (Jagtap and Szabo 2005; Virag and Szabo 2002).
FIG. 2.
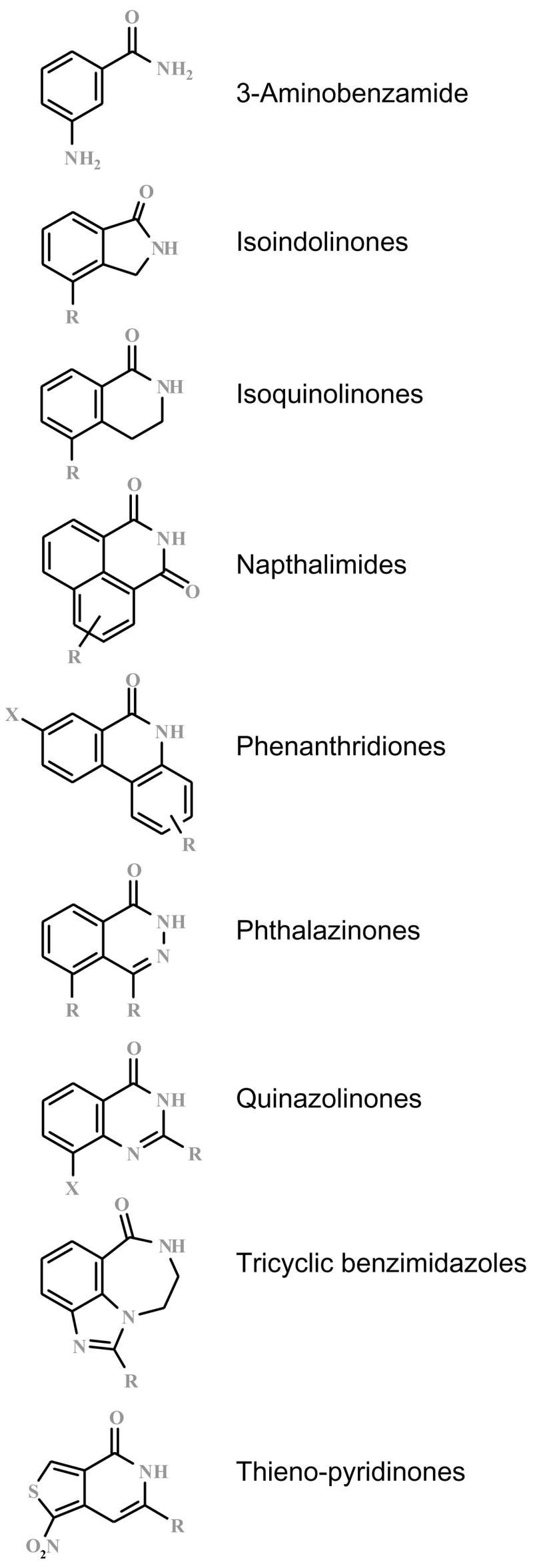
Some prototypical PARP inhibitor structures.
An area worthy of separate discussion is the relationship of PARP inhibition with sirtuin activation. The latter enzyme(s) are NAD+-dependent deacetylases and it is likely that changes in NAD+ metabolism due to PARP activation or inhibition could impact sirtuin function (Chong et al. 2005; Frye 1999). Activation of sirtuins has been shown to be protective in a number of aging related disorders and these pathways likely interact (Hassa et al. 2006). Furthermore, the PARP-1-dependent cardiac myocyte cell death during heart failure may also be mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity (Pillai et al. 2005a).
Due to confidentiality reasons, the structures and the potencies of many of the PARP inhibitors that have entered clinical trials (see separate section below) have not been disclosed in the scientific literature, but they all appear to be highly potent (presumably in the nanomolar range, when tested in the isolated PARP-1 enzyme).
Role of PARP-1 Activation in Various forms of Myocardial Ischemia Reperfusion Injury: Effects of PARP Inhibitors
Reperfusion injury (triggered by the transient disruption of the normal blood supply to target organs followed by reperfusion) is the principal cause of tissue damage occurring in conditions such as myocardial infarction, cardiopulmonary bypass, and aortic reconstructive surgeries, stroke, organ transplantation, and as well as a major mechanism of end-organ damage complicating the course of circulatory shock of various etiologies. The definitive treatment to reduce myocardial damage of ischemic myocardium is reperfusion; however, reperfusion itself leads to additional tissue injury mediated by a multitude of factors including reactive oxygen (superoxide anion, hydrogen peroxide, and hydroxyl radical) and reactive nitrogen species (e.g., peroxynitrite and nitrogen dioxide) upon reperfusion, as well as to the rapid transcriptional activation of an array of proinflammatory genes (Ferdinandy and Schulz 2003; Pacher et al. 2005b, 2006b, 2007; Turko and Murad 2002; Ungvari et al. 2005). Immediate consequences are the local sequestration and activation of polymorphonuclear leukocytes, leading to a rapid amplification of the initial inflammatory response and ROS generation, so-called “respiratory burst” (Lucchesi 1990). The sources of reactive oxygen species in reperfusion injury can be multiple such as mitochondria, xanthine oxidase, NAD(P)H oxidases, cyclooxygenase, and NOS (Griendling et al. 2000; Pacher et al. 2005b, 2006b, 2007; Ungvari et al. 2005). The burst of reactive oxygen and nitrogen species immediately upon reperfusion initiates a chain of deleterious cellular responses eventually leading to coronary endothelial dysfunction; adherence of neutrophils to endothelium, transendothelial migration, and the release of inflammatory mediators; transient impairment of left ventricular systolic contractile function or “myocardial stunning”; acute diastolic dysfunction; cellular calcium overload; re-energization-induced myocyte hypercontracture; arrhythmia; and cell death (Pacher et al. 2007; Ungvari et al. 2005).
There is marked overactivation of PARP in the reperfused myocardium, which parallels with the decline of the contractile function and myocardial NAD+ and ATP contents in preclinical models of myocardial infarction and cardiopulmonary bypass. Consequently, the pharmacological inhibition of PARP with various inhibitors (3-AB, nicotinamide, ISQ, 5-AIQ, IQD, BGP-15, GPI 6150, PJ-34, and INO-1001) or genetic deletion of PARP-1 in mice (Grupp et al. 1999; Pieper et al. 2000; Yang et al. 2000; Zhou et al. 2006; Zingarelli et al. 1998, 2003, 2004b), rats (Bowes et al. 1999; Docherty et al. 1999; Farivar et al. 2005; Fiorillo et al. 2002, 2003; Halmosi et al. 2001; Liaudet et al. 2001a; Pieper et al. 2000; Szabados et al. 2000; Szabo et al. 2002b, 2005b, 2006b, 2006c; Thiemermann et al. 1997; Wayman et al. 2001; Xiao et al. 2004; Zingarelli et al. 1997), rabbits (Thiemermann et al. 1997), dogs (Szabo et al. 2004b, 2004c), and pigs (Bowes et al. 1998b; Faro et al. 2002; Hauser et al. 2006) markedly improves the outcome of myocardial ischemia-reperfusion damage (in all in vitro or ex vivo models) associated with hypoxia/reoxygenation, coronary artery occlusion/reocclusion, cardiopulmonary bypass, and cardiac transplantation, which is also a subject of numerous recent overviews (Pacher et al. 2005b, 2007; Szabo 2005a; Szabo and Bahrle 2005; Ungvari et al. 2005). The favorable effects of PARP inhibitors in these preclinical models involve improvement in myocardial intracellular energy status and myocardial contractility; attenuation of the proinflammatory gene/mediator expression and neutrophil infiltration into the reperfused myocardium; and decrease of cardiomyocyte and endothelial cell necrosis (Tables 1 and 2) (Bowes et al. 1998b, 1999; Docherty et al. 1999; Farivar et al. 2005; Faro et al. 2002; Fiorillo et al. 2002, 2003; Grupp et al. 1999; Halmosi et al. 2001; Hauser et al. 2006; Liaudet et al. 2001a; Pacher et al. 2005b, 2007; Pieper et al. 2000; Szabados et al. 2000; Szabo 2005a; Szabo and Bahrle 2005; Szabo et al. 2002b, 2004b, 2004c, 2005c, 2006b, 2006c; Thiemermann et al. 1997; Ungvari et al. 2005; Wayman et al. 2001; Xiao et al. 2004; Yang et al. 2000; Zhou et al. 2006; Zingarelli et al. 1997).
TABLE 1.
Therapeutic effects of genetic deletion of PARP-1 in cardiovascular diseases
| Disease/trigger | Experimental model | PARP-1 neutralization | Key findings | References |
|---|---|---|---|---|
| Myocardial ischemia reperfusion (I/R) or ischemic preconditioning (IPC) | ||||
| Global I/R | Mouse heart | PARP-1−/− phenotype | Improved LV function, reduction of NAD+ consumption, improved mitochondrial function | Grupp et al. 1999; Pieper et al. 2000; Zhou et al. 2006 |
| Regional I/R | Mouse | PARP-1−/− phenotype | Decreased infarct size, neutrophil infiltration, and circulating TNFalpha, IL-10 and nitrate; reduced of ICAM-1/P-selectin expression | Yang et al. 2000; Zingarelli et al. 1998 |
| Regional I/R | Mouse | PARP-1−/− phenotype | Reduced myocardial damage and apoptosis, reduced NF-kappa B activation | Zingarelli et al. 2003 |
| Regional I/R | Mouse | PARP-1−/− phenotype | Decreased infarct size, reduction of the phosphorylative activity of JNK, reduced AP-1 activation, increased HSF-1 and HSP70 activation | Zingarelli et al. 2004b |
| Ischemic preconditioning | Mouse | PARP-1−/− phenotype | Suppression of the benefit of IPC | Liaudet et al. 2001b |
| Heart failure and myocardial hypertrophy | ||||
| Doxorubicin | Mouse | PARP-1−/− phenotype | Improved contractile function, reduced myocyte death, decreased mortality | Pacher et al. 2002b |
| Aortic banding | Mouse | PARP-1−/− phenotype | Decreased hypertrophy, myocardial collagen formation, and the mitochondrial-to-nuclear translocation of the cell death factor apoptosis-inducing factor (AIF) | Pillai et al. 2005b; Xiao et al. 2005 |
| Angiotensin II infusion | Mouse | PARP-1−/− phenotype | Protected from angiotensin II-induced cardiac hypertrophy | Pillai et al. 2006 |
| Circulatory shock | ||||
| Endotoxic and septic shock | Mouse | PARP-1−/− phenotype | Improved contractile function, decreased mortality and tissue injury | Oliver et al. 1999; Pacher et al. 2002a; Soriano et al. 2002 |
| Hemorrhagic shock | Mouse | PARP-1−/− phenotype | Protection from the rapid decrease in blood pressure after resuscitation, increased survival time, and decreased tissue injury | Liaudet et al. 2000 |
TABLE 2.
Therapeutic effects of PARP inhibitors in cardiovascular disease
| Disease/trigger | Experimental model | PARP inhibitor | Key findings | References |
|---|---|---|---|---|
| Oxidant-or hypoxia reoxygenation-induced cell death in vitro | ||||
| H2O2, peroxynitrite, hypoxia reoxygenation | Rat and human cardiomyoblasts | 3-AB, nicotinamide, ISQ, PJ-34 | Improved mitochondrial respiration, reduced cell death, protection against the reduction of action potential duration, improved cellular ATP levels | Bhatnagar 1997; Bowes et al. 1998a 1999; Fiorillo et al. 2006; Gilad et al. 1997 |
| Angiogenesis | ||||
| VEGF and bFGF | Human umbilical vein endothelial cells(HUVECs), aortic ring explants | 3-AB, PJ-34, 5-AIQ, IQD | Dose-dependent reduction of VEGF- or bFGF-induced proliferation, migration, and tube formation of human umbilical vein endothelial cells in vitro, and prevention of the sprouting of rat aortic ring explants | Rajesh et al. 2006a, 2006b |
| Myocardial ischemia reperfusion, ischemic preconditioning | ||||
| Global I/R | Rat heart | 3-AB, BGP-15, ISQ | Improvement of LV dysfunction, reduction of ATP and NAD+ catabolism, improved ATP and CK recovery | Docherty et al. 1999; Halmosi et al. 2001; Szabados et al. 2000 |
| Regional I/R | Rat heart | 3-AB | Reduction of infarct size | Bowes et al. 1999 |
| Regional I/R | Rat | 3-AB, GPI 6150 | Reduction of infarct size and LV dysfunction, preservation of myocardial ATP stores, reduction of neutrophil infiltration | Liaudet et al. 2001a; Pieper et al. 2000; Zingarelli et al. 1997 Wayman et al. 2001 |
| Regional I/R | Rat | 5-AIQ | Reduction of infarct size | |
| Regional I/R in diabetic animals | Rat | INO-1001 | Improved left ventricular function, reduced infarct size and mitochondrial-to-nuclear translocation of the cell death effector apoptosis-inducing factor (AIF) in myocardial infarction | Xiao et al. 2004 |
| Global and regional I/R | Rabbit, rabbit heart | 3-AB, nicotinamide, ISQ | Reduction of infarct size and improvement of left ventricular function | Thiemermann et al. 1997 |
| Cardiopulmonary bypass, crystalloid cardioplegia and extracorporal circulation | Dog | PJ-34, INO-1001 | Better recovery of left and right ventricular systolic function, increased coronary blood flow, improved vascular and pulmonary function | Szabo et al. 2004b; Szabo et al. 2004c |
| Regional I/R | Pig | 3-AB, PJ-34, INO-1001 | Reduction of infarct size and improvement of left ventricular function | Bowes et al. 1998b; Faro et al. 2002; Hauser et al. 2006 |
| Cardiac transplantation | Rat | 3-AB, 5-AIQ, PJ-34, INO-1001 | Decreased myocardial damage, reduced oxidative stress, improved cellular NAD+ and ATP levels, improved contractile and endothelial function, reduced ICAM-1 and P-selectin expression; decreased allograft rejection | Farivar et al. 2005; Fiorillo et al. 2002, 2003; Szabo et al. 2006b; Szabo et al. 2002b; Szabo et al. 2006c; Szabo et al. 2005 |
| Ischemic preconditioning | Rat | 3-AB | Suppression of the benefit of IPC | Liaudet et al. 2001b |
| Segment-elevated myocardial infarction followed by revascularization | Human | INO-1001 | Reduction in plasma levels of C-reactive protein; tendency for reduced plasma levels of IL6 | Morrow et al. 2007 |
| Circulatory shock | ||||
| Endotoxic shock | Mouse, rat | 3-AB, PJ-34, INO-1001 | Improved myocardial contractility, vascular function and survival, decreased inflammation and tissue injury | Jagtap et al. 2002; Pacher et al. 2002a, 2006a; Szabo et al. 1997; Tasatargil et al. 2005; Veres et al. 2003 |
| Hemorrhagic shock | Rat, pig | 3-AB, 5-iodo-6-amino-1,2-benzopyrone | Improved hemodynamics, prolonged survival | Szabo et al. 1998; Szabo 1998; Szabo and Billiar 1999 |
| Heart failure, myocardial hypertrophy, and hypertension | ||||
| Doxorubicin | Mouse | PJ-34, INO-1001 | Improved contractile function, reduced myocyte death, decreased mortality | Pacher et al. 2002b, 2006a |
| Chronic myocardial ischemia | Rat | PJ-34, INO-1001 | Improved left ventricular and vascular function, reduced hypertrophy and remodeling | Pacher et al. 2002c, 2006a |
| Chronic heart failure following isoproterenol-induced myocardial infarction | Rat | L-2286 | Reduced the progression of postinfarction heart failure, attenuated cardiac hypertrophy and interstitial fibrosis, and preserved the integrity of mitochondrial respiratory complexes. L-2286 also repressed the hypertrophy-associated increased phosphorylation of panPKC, PKC alpha/betaII, PKC delta, and PKC epsilon, which could be responsible for the activation of the antihypertrophic GSK-3beta. | Palfi et al. 2006 |
| Aortic banding | Mouse | INO-1001 | Decreased hypertrophy, myocardial collagen formation, and the mitochondrial-to-nuclear translocation of the cell death factor apoptosis-inducing factor (AIF) | Xiao et al. 2005 |
| Aortic banding, exercise | Mouse | 5-AIQ | Decreased pathological hypertrophy, myocardial collagen formation, improved cardiac function, without affecting physiological hypertrophy | Balakumar and Singh 2006 |
| Hypertension | ||||
| Genetic hypertension | Spontaneously hypertensive rats | PJ-34 | Improved endothelial function | Pacher et al. 2002f |
| Angiotensin II | Mouse, rat | PJ-34, INO-1001 | Improved endothelial function | Szabo et al. 2004a |
| Cardiovascular aging | ||||
| Advanced aging | Rat | PJ-34, INO-1001 | Improved cardiac and vascular function | Pacher et al. 2002e, 2002f, 2004b |
| Atherosclerosis, restenosis | ||||
| High fat diet | Apolipoprotein E (ApoE)-deficient mice | INO-1001 | Chronic treatment with the PARP inhibitor INO-100 reduced the degree of the endothelial dysfunction | Benko et al. 2004 |
| High fat diet | ApoE-deficient mice | Thieno[2,3-c]isoquinolin-5-one | Reduced plaque number and size and altered structural composition of plaques promoting its stability | Oumouna-Benachour et al. 2007 |
| Endarterectomy | Rat | INO-1001 | Reduced neointima hyperplasia following vascular injury | Beller et al. 2006 |
| Diabetic cardiovascular complications | ||||
| NOD diabetes | Mouse | PJ-34 | Improved contractile and vascular function | Pacher et al. 2002d |
| STZ diabetes | Mouse, rat | PJ-34 | Improved contractile function | Garcia Soriano et al. 2001; Pacher et al. 2002d; Soriano et al. 2001 |
Abbreviations: 3-aminobenzamide (3-AB); 1,5-dehydroxyisoquinoline (ISQ); 5-aminoisoquinolinone (5-AIQ); 1,5-isoquinolinediol (IQD); N-(6-oxo-5,6-dihydro-phenanthridin-2-yl) -N,N-dimethylacetamide (designated as PJ-34), vascular endothelial growth factor (VEGF); basic fibroblast growth factor (bFGF).
There are numerous excellent recent overviews on the oxidative-nitrosative stress-PARP pathway (Pacher et al. 2007; Szabo 2003; Szabo et al. 2006a; Virag and Szabo, 2002; Virag et al. 2003) and its role in various cardiovascular pathologies such as circulatory shock (Evgenov and Liaudet 2005; Szabo 2006, 2007), myocardial ischemia reperfusion (Szabo 2005a; Szabo and Bahrle 2005), heart failure (Pacher et al. 2005b; Ungvari et al. 2005), hypertension (Escobales and Crespo 2005), diabetic endothelial dysfunction and cardiomyopathy (Mabley and Soriano 2005; Pacher et al. 2005a; Pacher and Szabo 2005, 2006; Szabo 2005c), cardiovascular aging (Csiszar et al. 2005), and on the development of various PARP inhibitors for therapeutic indications (Donawho et al. 2007; Graziani and Szabo 2005; Jagtap and Szabo 2005; Ratnam and Low 2007; Southan and Szabo 2003; Szabo 2005b; Tentori and Graziani 2005; Thomas et al. 2007; Virag and Szabo 2002).
Several recent human studies have investigated the role of peroxynitrite (a reactive nitrogen species formed from the diffusion-limited reaction of nitric oxide and superoxide anion) also known to be an obligatory trigger of oxidative DNA damage and consequent PARP activation, in myocardial ischemia/reperfusion (I/R) in patients undergoing open heart surgery (Hayashi et al. 2001, 2003; Mehlhorn et al. 2003). These studies analyzed myocardial nitrotyrosine immunoreactivity (footprint of peroxynitrite formation and nitrative stress) from left ventricular biopsy specimen (Mehlhorn et al. 2003) or plasma nitrotyrosine levels from coronary sinus effluent and/or arterial blood (Hayashi et al. 2001, 2003) before and at the end of cardiopulmonary bypass. They found that the difference between plasma nitrotyrosine level from coronary sinus effluent and arterial blood (index of myocardium-derived peroxynitrite generation) peaked at 5 minutes following reperfusion, and was significantly correlated with the peak coronary sinus effluent and arterial blood difference in plasma malondialdehyde concentrations (index of myocardial oxidative stress and lipid peroxidation), and with postoperative maximum creatinine kinase level (index of myocardial injury) (Hayashi et al. 2001). The cardioplegia-induced myocardial I/R was also accompanied by increased iNOS expression, nitrotyrosine and 9-isoprostane formation (Mehlhorn et al. 2003), further supporting the role of both peroxynitrite and reactive oxygen species (ROS) in mediating the myocardial injury. Increased immunostaining for nitrotyrosine and iNOS were also demonstrated from the left ventricular biopsy specimens of patients with hibernating myocardium, a state of chronic contractile dysfunction present at rest in a territory subtended by a stenosed coronary artery that recovers following revascularization, most likely originated from repetitive episodes of transient ischemia (Baker et al. 2002), and in human coronary arteries of patients with human transplant coronary artery disease, a major cause of late mortality after cardiac transplantation (Ravalli et al. 1998), and during cardiac allograft rejection (Szabolcs et al. 1998). At present, human studies have not yet been conducted to investigate if PARP is activated in human cardiomyocytes and endothelial cells during the myocardial reperfusion injury. Nevertheless, a recent study (Toth-Zsamboki et al. 2006) investigated multiple aspects of human myocardial ischemia/reperfusion-related pathology by analyzing serum, plasma, and isolated peripheral leukocyte samples from cardiovascular patients with acute ST-segment elevation myocardial infarction and successful primary angioplastic intervention and provided evidence for: (a) oxidative/peroxidative imbalance (increased total plasma peroxide concentration and nitrotyrosine production), (b) angioplasty-triggered DNA damage (substantiated by increased levels of serum 8-hydroxy-2′ deoxyguanosine [8OHdG]), (c) rapid activation of PARP-1 in circulating human peripheral leukocytes (demonstrated by immunohistochemistry and Western blotting) following revascularization of the occluded coronary artery, and (d) translocation of the AIF from mitochondria to nuclei (which may well be a consequence of PARP activation). These observations further support the theory that recanalization of an occluded blood vessel triggers oxidative/nitrosative stress in humans, and demonstrate that local myocardial I/R triggered by percutaneous interventions in acute myocardial infarction is capable of generating systemic oxidative responses (Toth-Zsamboki et al. 2006).
Role of PARP-1 Activation in Various forms of Heart Failure, Cardiomyopathies, and Myocardial Hypertrophy
Multiple lines of evidence support the increased superoxide generation by various enzymatic other sources (e.g., xanthine oxidase, NADPH oxidases, cyclooxygenases, and the mitochondria) coupled with increased NO abundance (presumably from iNOS and/or nNOS overexpression) in various preclinical models of heart failure, favoring the generation of reactive oxidant peroxynitrite, which coupled with ROS may impair the cardiovascular function by various mechanisms (Pacher et al. 2005b, 2007; Schulz 2007; Turko and Murad 2002), one of which is ultimately oxidative DNA damage and PARP activation. Augmented myocardial nitrotyrosine formation, iNOS expression, and matrix metalloproteinase (MMP-2) and/or PARP activation were reported in acute and chronic mouse models of doxorubicin-induced heart failure (Bai et al. 2004; Mihm et al. 2002; Pacher et al. 2003; Pacher et al. 2002b; Pacher et al. 2006a; Szenczi et al. 2005; Weinstein et al. 2000), in heart failure induced by permanent left anterior coronary artery ligation in mice (Feng et al. 2001) and rats (Mihm et al. 2001; Pacher et al. 2002c, 2006a) or by pacing in dogs (Cesselli et al. 2001). Moreover, a novel peroxynitrite decomposition catalyst FP15 (Pacher et al. 2003) and PARP inhibitors PJ-34 (Pacher et al. 2002b) or INO-1001 (Pacher et al. 2006a) attenuated the development of cardiac dysfunction, myocardial nitrotyrosine formation, and increased the survival in doxorubicin-induced mouse cardiomyopathy models (Bai et al. 2004; Pacher et al. 2002b, 2003b, 2006a; Szenczi et al. 2005), and also in a rat model of chronic heart failure induced by permanent left anterior coronary artery ligation in rats (Pacher et al. 2002c, 2006a). In the latter model PARP inhibition with PJ-34 or INO-1001 was also associated with improved heart failure-associated decreased endothelial function and decreased myocardial hypertrophy and adverse remodeling (Pacher et al. 2002c, 2006a). Importantly, recent studies have also demonstrated overexpression of PARP-1 or increased activity in biopsies from human subjects with heart failure (Molnar et al. 2006; Pillai et al. 2005b).
In chronic heart failure associated with advanced aging or hypertension in rats and/or mice, increased ROS formation and nitrosative stress and increased poly(ADP-ribosyl)ation were also reported both in cardiomyocytes and endothelial cells (Booz 2007; Csiszar et al. 2005; Escobales and Crespo 2005; Pacher et al. 2005b, 2007; Turko and Murad 2002; Ungvari et al. 2005). Pharmacological inhibition of PARP attenuated the myocardial hypertrophy and improved endothelial function in these animal models of diseases (Csiszar et al. 2005; Pacher et al. 2002e, 2002f, 2004a, 2004b; Radovits et al. 2007) (Tables 1 and 2).
Recently, overexpression of the PARP-1 and increased poly(ADP-ribosyl)ation in myocardium of mice with aortic banding-induced CHF were also demonstrated (Pillai et al. 2005b; Xiao et al. 2005). In addition, genetic deletion of PARP-1 or pharmacological inhibition protected against hypertrophy response, heart failure, and cardiovascular dysfunction induced by aortic banding or angiotensin II infusion, and prevented the mitochondrial-to-nuclear translocation of the cell death and apoptosis-inducing factors (Balakumar and Singh 2006; Pillai et al. 2005b, 2006; Szabo et al. 2004a; Xiao et al. 2005) (Tables 1 and 2).
In mouse and rat models of diabetic cardiomyopathies the depression of myocardial systolic and diastolic function is also associated with a significant increase in protein nitration and poly(ADP-ribosyl)ation in the cardiac myocytes and endothelial cells and impaired vascular endothelial relaxation, which is remarkably improved by PARP inhibitors (Garcia Soriano et al. 2001; Mabley and Soriano 2005; Pacher et al. 2002d; Pacher and Szabo 2005, 2006; Soriano et al. 2001; Szabo 2005c). Importantly, the vascular nitrotyrosine and PAR content is increased in type-2 prediabetic and diabetic patients and is the predictor of impaired endothelial function (Szabo et al. 2002a).
PARP-1 knockout mice are resistant to cardiovascular collapse associated with endotoxic, septic, or hemorrhagic shock, and PARP inhibitors exert beneficial effects in these conditions by numerous complex interrelated mechanisms discussed in several recent overviews (Evgenov and Liaudet 2005; Szabo, 2006, 2007). Furthermore, both nitrotyrosine and PARP activity were found to be increased in myocardial biopsy specimens of human subjects with sepsis (Kooy et al. 1997; Soriano et al. 2006).
Role of PARP-1 Activation in Endothelial Dysfunction, Atherosclerosis, Vascular Remodeling, and Angiogenesis
Increasing evidence supports the view that the endothelial dysfunction associated with diabetes, hypertension, heart failure, and atherosclerosis is related to the local formation of reactive oxygen and nitrogen species in the vicinity of the vascular endothelium (Csiszar et al. 2005; Griendling et al. 2000; Pacher et al. 2007; Ungvari et al. 2005). Peroxynitrite may contribute to the vascular dysfunction by various complex interrelated mechanisms overviewed recently (Pacher et al. 2007), which may involve upregulation of adhesion molecules in endothelial cells, endothelial glycocalyx disruption, enhancement of neutrophil adhesion, inhibition of voltage-gated K+ (Kv) and Ca2+-activated K+ channels in coronary arterioles and vascular prostacyclin synthase, and apoptosis and/or PARP-dependent cell death in endothelial and vascular smooth muscle cells (Pacher et al. 2007), among many others. Indeed, PARP activation appears to be involved in the vascular dysfunction associated with circulatory shock (Evgenov and Liaudet 2005; Szabo 2006, 2007), myocardial ischemia reperfusion injury (Szabo 2005a; Szabo and Bahrle 2005), heart failure (Pacher et al. 2005b; Ungvari et al. 2005), hypertension (Escobales and Crespo 2005), diabetes (Mabley and Soriano 2005; Pacher et al. 2005a; Pacher and Szabo 2005, 2006; Szabo 2005c), and cardiovascular aging (Csiszar et al. 2005), both in experimental models and in patients, and the pharmacological inhibition of PARP improves endothelium-dependent relaxation in these pathological conditions (Tables 1 and 2, and see also above).
Experimental, clinical, and epidemiological studies have unraveled the significance of the cross-talk between inflammation, generation of reactive oxygen and nitrogen species, and lipid metabolism in the pathogenesis of atherosclerosis and vascular remodeling following injury (reviewed in Harrison et al. 2003; Pacher et al. 2007; Rubbo and O’Donnell 2005). Additional evidence suggests that atherosclerosis is not only associated with decreased NO bioavailability, but also with alterations in signal-transduction components downstream of NO, including among others, the NO receptor sGC, particularly in neointima (Evgenov et al. 2006). The classic hypothesis envisions reactive oxygen and nitrogen species oxidatively damage LDL trapped in the arterial intima forming oxidized LDL, which in turn initiates numerous events (e.g., foam cell formation, monocyte recruitment, and adhesion to the endothelium, inhibition of macrophage motility, smooth muscle cell proliferation, promotion of cytotoxicity, and attenuation of vascular reactivity) facilitating the development of atherosclerotic lesions.
Numerous studies have demonstrated increased 3-nitrotyrosine and iNOS expression in human atherosclerotic tissue (Pacher et al. 2007), which correlated with plaque instability in patients, supporting the pathogenetic role of peroxynitrite in atherosclerosis (Pacher et al. 2007). Consistently, elevated levels of oxidative DNA damage and DNA repair enzymes (e.g., PARP-1) were found in human atherosclerotic plaques (Martinet et al. 2002). In an ApoE mouse model of atherosclerosis fed on a high-fat diet PARP inhibition improved endothelial function (Benko et al. 2004), reduced atherosclerotic plaque size, and promoted factors of plaque stability (Oumouna-Benachour et al. 2007), presumably by reduction of inflammatory factors and cellular changes related to plaque dynamics.
Accumulating evidence suggests that reactive oxygen and nitrogen species and downstream effector pathways (e.g., PARP-1) play an important role in the pathogenesis of restenosis following vascular injury (Azevedo et al. 2000; Beller et al. 2006; Jacobson et al. 2003; Muscoli et al. 2004). Various studies demonstrated increased 3-nitrotyrosine immunoreactivity and/or iNOS overexpression in media and neointima following ballon injury (a model of restenosis), and increased 3-nitrotyrosine/tyrosine ratio in the serum of patients following stent implantation (Azevedo et al. 2000; Beller et al. 2006; Inoue et al. 2006; Jacobson et al. 2003; Muscoli et al. 2004). The serum 3-nitrotyrosine/tyrosine ratio appears to be an independent predictor of angiographic late lumen loss in patients (Inoue et al. 2006).
PARP inhibitors are being developed for the treatment for cancer, both in monotherapy as well as in combination with radiation and chemotherapeutic agents in humans, but the discussion of this subject, which is covered by several excellent recent overviews, is beyond the scope of this synopsis. Until recently it was thought that PARP inhibitors enhance the death of the cancer cells primarily by the interference with DNA repair at various levels (reviewed in Graziani and Szabo 2005; Jagtap and Szabo 2005; Ratnam and Low 2007; Tentori and Graziani 2005). Recent studies have established a novel concept that PARP inhibitors may decrease angiogenesis, either by inhibiting growth factor expression or by inhibiting growth factor-induced cellular proliferative responses (Obrosova et al. 2004; Rajesh et al. 2006a, 2006b). Several structurally distinct PARP inhibitors (3-aminobenzamide, PJ-34, 5-aminoisoquinolinone-hydrochloride, and 1,5-isoquinolinediol) showed antiangiogenic effects by decreasing VEGF- and FGF-induced proliferation, migration, and tube formation of human umbilical vein endothelial cells, and also in an ex vivo rat aortic ring assay of angiogenesis. These findings might also have implications to the mode of PARP inhibitors’ anticancer effects in vivo.
PARP Inhibitors in Clinical Trials
A number of PARP inhibitors have entered the stage of clinical testing, and many of these clinical candidates focus on cancer therapy, which is overviewed in more detail by Graziani and Szabo (2005), Haince et al. (2005), Plummer (2006), Ratnam and Low (2007), and Tentori and Graziani (2005). Based on murine data (Thomas et al. 2007) generated in cancer models using Agouron/Pfizer’s AG-014699, a phase I study was conducted to evaluate the safety of i.v. AG014699, when administered with temozolomide in solid tumors. The compound exhibited no dose-limiting toxicities (Ratnam and Low 2007). A subsequent phase II trial was conducted, which involved 40 evaluable patients with metastatic malignant melanoma. In this study, 18% of the study subjects demonstrated partial responses, with notable side effects (temozolomide-related myelosuppression and one toxic death) (Plummer et al. 2006; Ratnam and Low 2007).
KuDOS’/AstraZeneca’s oral PARP inhibitor KU-0059436 is currently in a phase I trial in patients with advanced tumors in the UK and the Netherlands (Fong et al. 2006; Ratnam and Low 2007). To date, the available data are only of a pharmacokinetic nature. However, anecdotal reports indicate a partial response in a patient with ovarian cancer, and stabilization of the disease for 24 weeks in a patient with metastatic soft tissue sarcoma.
Inotek
INO-1001. Inotek (Beverly, MA) in partnership with Genentech (South San Francisco, CA) is developing INO-1001, both for cardiovascular indications, as well as for cancer. For cardiovascular indications, it has been granted orphan drug status by the U.S. Food and Drug Administration for the prevention of postoperative aortic aneurysm repair complications and according to a 2005 review (Jagtap and Szabo 2005) is considered for several phase II trials for various cardiovascular indications. The first human clinical study with a PARP inhibitor in a cardiovascular indication has been conducted by Inotek. In this phase II prospective, single-blind, multi-center, dose escalation study of a single dose of intravenous INO-1001 (200 mg, 400 mg, or 800 mg) administered to 30 patients between the ages of 48 and 63 years presenting with acute ST-segment elevation myocardial infarction (STEMI), who were to be treated with primary percutaneous coronary intervention (PCI), the primary endpoint was to evaluate the safety, tolerability, and pharmacokinetic profile of INO-1001. The secondary objectives of the study were to characterize the pharmacodynamic profile of INO-1001 and to evaluate various biomarkers of necrosis and inflammation. The PARP inhibitor INO-1001 was found to induce a tendency to reduce the plasma levels of C-reactive protein and the inflammatory marker IL-6, without reducing plasma markers of myocardial injury. No drug-related serious adverse events were observed in the patients receiving the drug during the study period (Morrow et al. 2007).
INO-1001 is also being studied in combination therapy in metastatic melanoma and glioma and as a single agent in cancer for BRCA1- and BRCA2-deficient tumors. A preliminary analysis of a phase I trial, which evaluated INO-1001 in combination with temozolomide in unresectable stage III/IV melanoma, reported one patient with objective tumor regression (Wang et al. 2006). Recent preclinical data also indicate that INO-1001 is effective in enhancing the antitumor effects of chemotherapy agents such as doxorubicin against p53-deficient breast cancer (Mason et al. 2007).
Additional, early-stage human cancer trials include Abbott’s ABT-888 (Donawho et al. 2007), BiPAR Sciences’ (Brisbane, CA) BSI-201, and MGI Pharma’s (Bloomington, MN) GLP-21016. The latter compound is likely to derive from Guilford Pharmaceutical’s program (Lapidus et al. 2006; Tentori et al. 2003), a Baltimore-based pharmaceutical company that has merged with MGI. ABT-888 is being studied in a phase 0 clinical trial by the National Cancer Institute under an exploratory Investigational New Drug application (Kummar et al. 2007; Ratnam and Low 2007). BSI-201 is being tested as intravenous monotherapy in solid tumors with the objective to determine a maximum tolerated dose and a pharmacokinetic profile (Ratnam and Low 2007).
CONCLUSIONS
Taken together, the evidence summarized above strongly supports the crucial role of the ROS/RNS-PARP pathway in mediating cardiac and endothelial dysfunction associated with various forms of cardiovascular injury and heart failure. The information available to date supports the view that PARP activation is a pivotal feature of myocardial infarction and I/R of the heart, and that the pharmacological inhibition of PARP may provide significant benefits in these conditions by salvaging cardiomyocytes and endothelial cells, and by reducing the plasma markers of myocyte necrosis, as well as by downregulating the inflammatory responses. The first clinical data appear to be suggestive of a therapeutic benefit; additional human data are expected to become available in 2007–2008 on the latter subject. A multitude of novel pharmacological inhibitors of PARP have entered clinical testing as cytoprotective agents and as adjunct antitumor therapeutics, and several tetracycline antibiotics unexpectedly turned out to be potent PARP-1 inhibitors. Consequently, there is considerable expectation that these new drugs may become efficient therapies in the near future to combat cardiovascular diseases and cancer. While the clinical benefit of PARP inhibitors is being tested, additional new areas of research are also opening up in the preclinical front, which will certainly warrant additional studies. Some of these areas include the role of free poly(ADP-ribose) polymer in cell injury (Andrabi et al. 2006), the interactions between PARP and poly(ADP-glycohydrolase) (Meyer et al. 2007; Poitras et al. 2007), the role of PARP in the development of immunocompetence (Aldinucci et al. 2007), the regulatory role of PARP on the release of the proinflammatory nuclear protein HMGB-1 (Ditsworth et al. 2007) as well as the ever-increasing array of endogenous modulators of PARP activity, which now include, among others, sex hormones, the active form of vitamin D (Mabley et al. 2007), caffeine derivatives, as well as many other molecules (Szabo et al. 2006a).
ADDENDUM
The full chemical names of some of the compounds mentioned in the text are:
PJ-34: [11C]2-(dimethylamino)-N-(5,6-dihydro-6-oxophenanthridin-2-yl)acetamide
L-2286: 2-[(2-piperidin-1-yletil)thio]quinazolin-4(3H)-one
BGP 15: O-(3-piperidino-2-hydroxy-1-propyl) nicotinic acid-amidoxime
GPI 6150: 1,11b-dihydro-[2H]benzopyrano [4,3,2-de]isoquinolin-3-one
The chemical names of INO-1001, AG-014699, KU 0059436, ABT 888, BSI-201 or GLO-21016 were not found in the published literature or available databases.
Acknowledgments
This publication was supported by the Intramural Research Program of NIH/NIAAA (to PP) and by a grant from the NIH to CS (R01 GM060915).
Footnotes
Conflict of interest: CS is one of the founders and has been from 1998–2005 Chief Scientific Officer of Inotek, Inc., the developer of PARP inhibitors; PP owns stock in the company. The trust, established for the members of CS family, owns stock in Inotek, Inc. [Correction added after online publication Oct 3, 2007: PP replaced by CS]
References
- Aldinucci A, Gerlini G, Fossati S, Cipriani G, Ballerini C, Biagioli T, Pimpinelli N, Borgognoni L, Massacesi L, Moroni F, et al. A key role for poly(ADP-ribose) polymerase-1 activity during human dendritic cell maturation. J Immunol. 2007;179:305–312. doi: 10.4049/jimmunol.179.1.305. [DOI] [PubMed] [Google Scholar]
- Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M, Klaus JA, Otsuka T, Zhang Z, Koehler RC, et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Nat Acad Sci USA. 2006;103:18308–18313. doi: 10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azevedo LC, Pedro MA, Souza LC, de Souza HP, Janiszewski M, da Luz PL, Laurindo FR. Oxidative stress as a signaling mechanism of the vascular response to injury: The redox hypothesis of restenosis. Cardiovasc Res. 2000;47:436–445. doi: 10.1016/s0008-6363(00)00091-2. [DOI] [PubMed] [Google Scholar]
- Bai P, Mabley JG, Liaudet L, Virag L, Szabo C, Pacher P. Matrix metalloproteinase activation is an early event in doxorubicin-induced cardiotoxicity. Onco Rep. 2004;11:505–508. [PubMed] [Google Scholar]
- Baker CS, Dutka DP, Pagano D, Rimoldi O, Pitt M, Hall RJ, Polak JM, Bonser RS, Camici PG. Immunocytochemical evidence for inducible nitric oxide synthase and cyclooxygenase-2 expression with nitrotyrosine formation in human hibernating myocardium. Basic Res Cardiol. 2002;97:409–415. doi: 10.1007/s003950200050. [DOI] [PubMed] [Google Scholar]
- Balakumar P, Singh M. Possible role of poly(ADP-ribose) polymerase in pathological and physiological cardiac hypertrophy. Methods Findings Expl Clin Pharmacol. 2006;28:683–689. doi: 10.1358/mf.2006.28.10.1037495. [DOI] [PubMed] [Google Scholar]
- Beller CJ, Radovits T, Kosse J, Gero D, Szabo C, Szabo G. Activation of the peroxynitrite-poly(adenosine diphosphate-ribose) polymerase pathway during neointima proliferation: A new target to prevent restenosis after endarterectomy. J Vasc Surg. 2006;43:824–830. doi: 10.1016/j.jvs.2005.11.021. [DOI] [PubMed] [Google Scholar]
- Benko R, Pacher P, Vaslin A, Kollai M, Szabo C. Restoration of the endothelial function in the aortic rings of apolipoprotein E deficient mice by pharmacological inhibition of the nuclear enzyme poly(ADP-ribose) polymerase. Life Sci. 2004;75:1255–1261. doi: 10.1016/j.lfs.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Bhatnagar A. Contribution of ATP to oxidative stress-induced changes in action potential of isolated cardiac myocytes. Am J Physiol. 1997;272:H1598–1608. doi: 10.1152/ajpheart.1997.272.4.H1598. [DOI] [PubMed] [Google Scholar]
- Booz GW. PARP inhibitors and heart failure—Translational medicine caught in the act. Congest Heart Fail. 2007;13:105–112. doi: 10.1111/j.1527-5299.2007.06595.x. [DOI] [PubMed] [Google Scholar]
- Boulares HA, Giardina C, Navarro CL, Khairallah EA, Cohen SD. Modulation of serum growth factor signal transduction in Hepa 1–6 cells by acetaminophen: An inhibition of c-myc expression, NF-kappaB activation, and Raf-1 kinase activity. Toxicol Sci. 1999;48:264–274. doi: 10.1093/toxsci/48.2.264. [DOI] [PubMed] [Google Scholar]
- Bowes J, McDonald MC, Piper J, Thiemermann C. Inhibitors of poly (ADP-ribose) synthetase protect rat cardiomyocytes against oxidant stress. Cardiovasc Res. 1999;41:126–134. doi: 10.1016/s0008-6363(98)00221-1. [DOI] [PubMed] [Google Scholar]
- Bowes J, Piper J, Thiemermann C. Inhibitors of the activity of poly (ADP-ribose) synthetase reduce the cell death caused by hydrogen peroxide in human cardiac myoblasts. Br J Pharmacol. 1998a;124:1760–1766. doi: 10.1038/sj.bjp.0702009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowes J, Ruetten H, Martorana PA, Stockhausen H, Thiemermann C. Reduction of myocardial reperfusion injury by an inhibitor of poly (ADP-ribose) synthetase in the pig. Eur J Pharmacol. 1998b;359:143–150. doi: 10.1016/s0014-2999(98)00638-4. [DOI] [PubMed] [Google Scholar]
- Cesselli D, Jakoniuk I, Barlucchi L, Beltrami AP, Hintze TH, Nadal-Ginard B, Kajstura J, Leri A, Anversa P. Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ Res. 2001;89:279–286. doi: 10.1161/hh1501.094115. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Lin SH, Li F, Maiese K. The sirtuin inhibitor nicotinamide enhances neuronal cell survival during acute anoxic injury through AKT, BAD, PARP, and mitochondrial associated “antiapoptotic” pathways. Curr Neurovasc Res. 2005;2:271–285. doi: 10.2174/156720205774322584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csiszar A, Pacher P, Kaley G, Ungvari Z. Role of oxidative and nitrosative stress, longevity genes and poly(ADP-ribose) polymerase in cardiovascular dysfunction associated with aging. Curr Vasc Pharmacol. 2005;3:285–291. doi: 10.2174/1570161054368616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM. Deadly conversations: Nuclear-mitochondrial cross-talk. J Bioenerg Biomembr. 2004;36:287–294. doi: 10.1023/B:JOBB.0000041755.22613.8d. [DOI] [PubMed] [Google Scholar]
- Ditsworth D, Zong WX, Thompson CB. Activation of poly(ADP)-ribose polymerase (PARP-1) induces release of the pro-inflammatory mediator HMGB1 from the nucleus. J Biol Chem. 2007;282:17845–17854. doi: 10.1074/jbc.M701465200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docherty JC, Kuzio B, Silvester JA, Bowes J, Thiemermann C. An inhibitor of poly (ADP-ribose) synthetase activity reduces contractile dysfunction and preserves high energy phosphate levels during reperfusion of the ischaemic rat heart. Br J Pharmacol. 1999;127:1518–1524. doi: 10.1038/sj.bjp.0702705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donawho CK, Luo Y, Luo Y, Penning TD, Bauch JL, Bouska JJ, Bontcheva-Diaz VD, Cox BF, DeWeese TL, Dillehay LE, et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. 2007;13:2728–2737. doi: 10.1158/1078-0432.CCR-06-3039. [DOI] [PubMed] [Google Scholar]
- Escobales N, Crespo MJ. Oxidative-nitrosative stress in hypertension. Curr Vasc Pharmacol. 2005;3:231–246. doi: 10.2174/1570161054368643. [DOI] [PubMed] [Google Scholar]
- Evgenov OV, Liaudet L. Role of nitrosative stress and activation of poly(ADP-ribose) polymerase-1 in cardiovascular failure associated with septic and hemorrhagic shock. Curr Vasc Pharmacol. 2005;3:293–299. doi: 10.2174/1570161054368580. [DOI] [PubMed] [Google Scholar]
- Evgenov OV, Pacher P, Schmidt PM, Hasko G, Schmidt HH, Stasch JP. NO-independent stimulators and activators of soluble guanylate cyclase: Discovery and therapeutic potential. Nat Rev Drug Discov. 2006;5:755–768. doi: 10.1038/nrd2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farivar AS, McCourtie AS, MacKinnon-Patterson BC, Woolley SM, Barnes AD, Chen M, Jagtap P, Szabo C, Salerno CT, Mulligan MS. Poly (ADP) ribose polymerase inhibition improves rat cardiac allograft survival. Ann Thorac Surg. 2005;80:950–956. doi: 10.1016/j.athoracsur.2005.02.035. [DOI] [PubMed] [Google Scholar]
- Faro R, Toyoda Y, McCully JD, Jagtap P, Szabo E, Virag L, Bianchi C, Levitsky S, Szabo C, Sellke FW. Myocardial protection by PJ34, a novel potent poly (ADP-ribose) synthetase inhibitor. Ann Thorac Surg. 2002;73:575–581. doi: 10.1016/s0003-4975(01)03329-x. [DOI] [PubMed] [Google Scholar]
- Feng Q, Lu X, Jones DL, Shen J, Arnold JM. Increased inducible nitric oxide synthase expression contributes to myocardial dysfunction and higher mortality after myocardial infarction in mice. Circulation. 2001;104:700–704. doi: 10.1161/hc3201.092284. [DOI] [PubMed] [Google Scholar]
- Ferdinandy P, Schulz R. Nitric oxide, superoxide, and peroxynitrite in myocardial ischaemia-reperfusion injury and preconditioning. Br J Pharmacol. 2003;138:532–543. doi: 10.1038/sj.bjp.0705080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorillo C, Pace S, Ponziani V, Nediani C, Perna AM, Liguori P, Cecchi C, Nassi N, Donzelli GP, Formigli L, et al. Poly(ADP-ribose) polymerase activation and cell injury in the course of rat heart heterotopic transplantation. Free Radic Res. 2002;36:79–87. doi: 10.1080/10715760210168. [DOI] [PubMed] [Google Scholar]
- Fiorillo C, Ponziani V, Giannini L, Cecchi C, Celli A, Nassi N, Lanzilao L, Caporale R, Nassi P. Protective effects of the PARP-1 inhibitor PJ34 in hypoxic-reoxygenated cardiomyoblasts. Cell Mol Life Sci. 2006;63:3061–3071. doi: 10.1007/s00018-006-6345-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorillo C, Ponziani V, Giannini L, Cecchi C, Celli A, Nediani C, Perna AM, Liguori P, Nassi N, Formigli L, et al. Beneficial effects of poly (ADP-ribose) polymerase inhibition against the reperfusion injury in heart transplantation. Free Radic Res. 2003;37:331–339. doi: 10.1080/1071576021000055262. [DOI] [PubMed] [Google Scholar]
- Fong PC, Spicer J, Reade S, Reid A, Vidal L, Schellens JH, Tutt A, Harris PA, Kaye S, De Bono JS. Phase I pharmacokinetic (PK) and pharmacodynamic (PD) evaluation of a small molecule inhibitor of poly ADP-ribose polymerase (PARP), KU-0059436 (Ku) in patients (p) with advanced tumours. J Clin Oncol. 2006;24:A3022. [Google Scholar]
- Frye RA. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem Biophys Res Commun. 1999;260:273–279. doi: 10.1006/bbrc.1999.0897. [DOI] [PubMed] [Google Scholar]
- Garcia Soriano F, Virag L, Jagtap P, Szabo E, Mabley JG, Liaudet L, Marton A, Hoyt DG, Murthy KG, Salzman AL, et al. Diabetic endothelial dysfunction: the role of poly(ADP-ribose) polymerase activation. Nat Med. 2001;7:108–113. doi: 10.1038/83241. [DOI] [PubMed] [Google Scholar]
- Gilad E, Zingarelli B, Salzman AL, Szabo C. Protection by inhibition of poly (ADP-ribose) synthetase against oxidant injury in cardiac myoblasts In vitro. J Mol Cell Cardiol. 1997;29:2585–2597. doi: 10.1006/jmcc.1997.0496. [DOI] [PubMed] [Google Scholar]
- Graziani G, Szabo C. Clinical perspectives of PARP inhibitors. Pharmacol Res. 2005;52:109–118. doi: 10.1016/j.phrs.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- Grupp IL, Jackson TM, Hake P, Grupp G, Szabo C. Protection against hypoxia-reoxygenation in the absence of poly (ADP-ribose) synthetase in isolated working hearts. J Mol Cell Cardiol. 1999;31:297–303. doi: 10.1006/jmcc.1998.0864. [DOI] [PubMed] [Google Scholar]
- Ha HC, Hester LD, Snyder SH. Poly(ADP-ribose) polymerase-1 dependence of stress-induced transcription factors and associated gene expression in glia. Proc Natl Acad Sci USA. 2002;99:3270–3275. doi: 10.1073/pnas.052712399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haince JF, Rouleau M, Hendzel MJ, Masson JY, Poirier GG. Targeting poly(ADP-ribosyl)ation: A promising approach in cancer therapy. Trends Mol Med. 2005;11:456–463. doi: 10.1016/j.molmed.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Halmosi R, Berente Z, Osz E, Toth K, Literati-Nagy P, Sumegi B. Effect of poly(ADP-ribose) polymerase inhibitors on the ischemia-reperfusion-induced oxidative cell damage and mitochondrial metabolism in Langendorff heart perfusion system. Molec Pharmacol. 2001;59:1497–1505. doi: 10.1124/mol.59.6.1497. [DOI] [PubMed] [Google Scholar]
- Harrison DG, Cai H, Landmesser U, Griendling KK. Interactions of angiotensin II with NAD(P)H oxidase, oxidant stress and cardiovascular disease. J Renin Angiotensin Aldosterone Syst. 2003;4:51–61. doi: 10.3317/jraas.2003.014. [DOI] [PubMed] [Google Scholar]
- Hassa PO, Haenni SS, Elser M, Hottiger MO. Nuclear ADP-ribosylation reactions in mammalian cells: Where are we today and where are we going? Microbiol Mol Biol Rev. 2006;70:789–829. doi: 10.1128/MMBR.00040-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser B, Groger M, Ehrmann U, Albicini M, Bruckner UB, Schelzig H, Venkatesh B, Li H, Szabo C, Speit G, et al. The PARP-1 inhibitor INO-1001 facilitates hemodynamic stabilization without affecting DNA repair in porcine thoracic aortic cross-clamping-induced ischemia/reperfusion. Shock. 2006;25:633–640. doi: 10.1097/01.shk.0000209561.61951.2e. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Sawa Y, Fukuyama N, Miyamoto Y, Takahashi T, Nakazawa H, Matsuda H. Leukocyte-depleted terminal blood cardioplegia provides superior myocardial protective effects in association with myocardium-derived nitric oxide and peroxynitrite production for patients undergoing prolonged aortic crossclamping for more than 120 minutes. J Thorac Cardiovasc Surg. 2003;126:1813–1821. doi: 10.1016/s0022-5223(03)01282-0. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Sawa Y, Ohtake S, Fukuyama N, Nakazawa H, Matsuda H. Peroxynitrite formation from human myocardium after ischemia-reperfusion during open heart operation. Ann Thor Surg. 2001;72:571–576. doi: 10.1016/s0003-4975(01)02668-6. [DOI] [PubMed] [Google Scholar]
- Inoue T, Kato T, Hikichi Y, Hashimoto S, Hirase T, Morooka T, Imoto Y, Takeda Y, Sendo F, Node K. Stent-induced neutrophil activation is associated with an oxidative burst in the inflammatory process, leading to neointimal thickening. Thromb Haemost. 2006;95:43–48. [PubMed] [Google Scholar]
- Jacobson GM, Dourron HM, Liu J, Carretero OA, Reddy DJ, Andrzejewski T, Pagano PJ. Novel NAD(P)H oxidase inhibitor suppresses angioplasty-induced superoxide and neointimal hyperplasia of rat carotid artery. Circ Res. 2003;92:637–643. doi: 10.1161/01.RES.0000063423.94645.8A. [DOI] [PubMed] [Google Scholar]
- Jagtap P, Soriano FG, Virag L, Liaudet L, Mabley J, Szabo E, Hasko G, Marton A, Lorigados CB, Gallyas F, et al. Novel phenanthridinone inhibitors of poly (adenosine 5′-diphosphate-ribose) synthetase: Potent cytoprotective and antishock agents. Crit Care Med. 2002;30:1071–1082. doi: 10.1097/00003246-200205000-00019. [DOI] [PubMed] [Google Scholar]
- Jagtap P, Szabo C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov. 2005;4:421–440. doi: 10.1038/nrd1718. [DOI] [PubMed] [Google Scholar]
- Jagtap PG, Baloglu E, Southan GJ, Mabley JG, Li H, Zhou J, van Duzer J, Salzman AL, Szabo C. Discovery of potent poly(ADP-ribose) polymerase-1 inhibitors from the modification of indeno[1,2-c]isoquinolinone. J Med Chem. 2005;48:5100–5103. doi: 10.1021/jm0502891. [DOI] [PubMed] [Google Scholar]
- Jagtap PG, Southan GJ, Baloglu E, Ram S, Mabley JG, Marton A, Salzman A, Szabo C. The discovery and synthesis of novel adenosine substituted 2,3-dihydro-1H-isoindol-1-ones: potent inhibitors of poly(ADP-ribose) polymerase-1 (PARP-1) Bioorg Med Chem Lett. 2004;14:81–85. doi: 10.1016/j.bmcl.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Kooy NW, Lewis SJ, Royall JA, Ye YZ, Kelly DR, Beckman JS. Extensive tyrosine nitration in human myocardial inflammation: Evidence for the presence of peroxynitrite. Crit Care Med. 1997;25:812–819. doi: 10.1097/00003246-199705000-00017. [DOI] [PubMed] [Google Scholar]
- Kummar S, Kinders R, Rubinstein L, Parchment RE, Murgo AJ, Collins J, Pickeral O, Low J, Steinberg SM, Gutierrez M, et al. Compressing drug development timelines in oncology using phase ‘0’ trials. Nat Rev Cancer. 2007;7:131–139. doi: 10.1038/nrc2066. [DOI] [PubMed] [Google Scholar]
- Lapidus RG, Xu W, Spicer E, Hoover R, Zhang J. PARP inhibitors enhance the effect of cisplatin against tumors and ameliorate cisplatin-induced neuropathy. Am Assoc Cancer Res. 2006;47:A2141. [Google Scholar]
- Levrand S, Vannay-Bouchiche C, Pesse B, Pacher P, Feihl F, Waeber B, Liaudet L. Peroxynitrite is a major trigger of cardiomyocyte apoptosis in vitro and in vivo. Free Rad Biol Med. 2006;41:886–895. doi: 10.1016/j.freeradbiomed.2006.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaudet L, Soriano FG, Szabo E, Virag L, Mabley JG, Salzman AL, Szabo C. Protection against hemorrhagic shock in mice genetically deficient in poly(ADP-ribose)polymerase. Proc Nat Acad Sci USA. 2000;97:10203–10208. doi: 10.1073/pnas.170226797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaudet L, Szabo E, Timashpolsky L, Virag L, Cziraki A, Szabo C. Suppression of poly (ADP-ribose) polymerase activation by 3-aminobenzamide in a rat model of myocardial infarction: Long-term morphological and functional consequences. Br J Pharmacol. 2001a;133:1424–1430. doi: 10.1038/sj.bjp.0704185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaudet L, Yang Z, Al-Affar EB, Szabo C. Myocardial ischemic preconditioning in rodents is dependent on poly (ADP-ribose) synthetase. Mol Med. 2001b;7:406–417. [PMC free article] [PubMed] [Google Scholar]
- Lucchesi BR. Modulation of leukocyte-mediated myocardial reperfusion injury. Ann Rev Pysiology. 1990;52:561–576. doi: 10.1146/annurev.ph.52.030190.003021. [DOI] [PubMed] [Google Scholar]
- Mabley JG, Soriano FG. Role of nitrosative stress and poly(ADP-ribose) polymerase activation in diabetic vascular dysfunction. Curr Vasc Pharmacol. 2005;3:247–252. doi: 10.2174/1570161054368571. [DOI] [PubMed] [Google Scholar]
- Mabley JG, Wallace R, Pacher P, Murphy K, Szabo C. Inhibition of poly(adenosine diphosphate-ribose) polymerase by the active form of vitamin D. Int J Mol Med. 2007;19:947–952. [PMC free article] [PubMed] [Google Scholar]
- Martinet W, Knaapen MW, De Meyer GR, Herman AG, Kockx MM. Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques. Circulation. 2002;106:927–932. doi: 10.1161/01.cir.0000026393.47805.21. [DOI] [PubMed] [Google Scholar]
- Mason KA, Valdecanas D, Hunter NR, Milas L. INO-1001, a novel inhibitor of poly(ADP-ribose) polymerase, enhances tumor response to doxorubicin. Invest New Drugs. 2007 doi: 10.1007/s10637-007-9072-5. in press. [DOI] [PubMed] [Google Scholar]
- Mehlhorn U, Krahwinkel A, Geissler HJ, LaRosee K, Fischer UM, Klass O, Suedkamp M, Hekmat K, Tossios P, Bloch W. Nitrotyrosine and 8-isoprostane formation indicate free radical-mediated injury in hearts of patients subjected to cardioplegia. J Thorac Cardiovasc Surg. 2003;125:178–183. doi: 10.1067/mtc.2003.97. [DOI] [PubMed] [Google Scholar]
- Meyer RG, Meyer-Ficca ML, Whatcott CJ, Jacobson EL, Jacobson MK. Two small enzyme isoforms mediate mammalian mitochondrial poly(ADP-ribose) glycohydrolase (PARG) activity. Exp Cell Res. 2007;313:2920–2936. doi: 10.1016/j.yexcr.2007.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihm MJ, Coyle CM, Schanbacher BL, Weinstein DM, Bauer JA. Peroxynitrite induced nitration and inactivation of myofibrillar creatine kinase in experimental heart failure. Cardiovasc Res. 2001;49:798–807. doi: 10.1016/s0008-6363(00)00307-2. [DOI] [PubMed] [Google Scholar]
- Mihm MJ, Yu F, Weinstein DM, Reiser PJ, Bauer JA. Intracellular distribution of peroxynitrite during doxorubicin cardiomyopathy: Evidence for selective impairment of myofibrillar creatine kinase. Br J Pharmacol. 2002;135:581–588. doi: 10.1038/sj.bjp.0704495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnar A, Toth A, Bagi Z, Papp Z, Edes I, Vaszily M, Galajda Z, Papp JG, Varro A, Szuts V, et al. Activation of the poly(ADP-ribose) polymerase pathway in human heart failure. Mol Med. 2006;12:143–152. doi: 10.2119/2006-00043.Molnar. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow DA, Baran K, Krakover R, Dauerman H, Murphy SA, Kumar S, McCabe CH, Brickman CM, Salzman AL. Safety, pharmacokinetics, and pharmacodynamics of a single intravenous administration of INO-1001 in subjects with ST-elevated myocardial infarction (STEMI) undergoing primary percutaneous coronary intervention: results of the TIMI 37A trial. J Am Coll Cardiol. 2007:2002A. doi: 10.1007/s11239-008-0230-1. [DOI] [PubMed] [Google Scholar]
- Muscoli C, Sacco I, Alecce W, Palma E, Nistico R, Costa N, Clementi F, Rotiroti D, Romeo F, Salvemini D, et al. The protective effect of superoxide dismutase mimetic M40401 on balloon injury-related neointima formation: Role of the lectin-like oxidized low-density lipoprotein receptor-1. J Pharmacol Exp Ther. 2004;311:44–50. doi: 10.1124/jpet.104.068205. [DOI] [PubMed] [Google Scholar]
- Obrosova IG, Minchenko AG, Frank RN, Seigel GM, Zsengeller Z, Pacher P, Stevens MJ, Szabo C. Poly(ADP-ribose) polymerase inhibitors counteract diabetes- and hypoxia-induced retinal vascular endothelial growth factor overexpression. Int J Mol Med. 2004;14:55–64. [PubMed] [Google Scholar]
- Oliver FJ, Menissier-de Murcia J, Nacci C, Decker P, Andriantsitohaina R, Muller S, de la Rubia G, Stoclet JC, de Murcia G. Resistance to endotoxic shock as a consequence of defective NF-kappaB activation in poly (ADP-ribose) polymerase-1 deficient mice. EMBO J. 1999;18:4446–4454. doi: 10.1093/emboj/18.16.4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oumouna-Benachour K, Hans CP, Suzuki Y, Naura A, Datta R, Belmadani S, Fallon K, Woods C, Boulares AH. Poly(ADP-Ribose) polymerase inhibition reduces atherosclerotic plaque size and promotes factors of plaque stability in apolipoprotein e-deficient mice. Effects on macrophage recruitment, nuclear factor-{kappa}b nuclear translocation, and foam cell death. Circulation. 2007;115(18):2442–2450. doi: 10.1161/CIRCULATIONAHA.106.668756. [DOI] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Cziraki A, Mabley JG, Liaudet L, Papp L, Szabo C. Role of poly(ADP-ribose) polymerase activation in endotoxin-induced cardiac collapse in rodents. Biochem Pharmacol. 2002a;64:1785–1791. doi: 10.1016/s0006-2952(02)01421-1. [DOI] [PubMed] [Google Scholar]
- Pacher P, Liaudet L, Bai P, Mabley JG, Kaminski PM, Virag L, Deb A, Szabo E, Ungvari Z, Wolin MS, et al. Potent metalloporphyrin peroxynitrite decomposition catalyst protects against the development of doxorubicin-induced cardiac dysfunction. Circulation. 2003;107:896–904. doi: 10.1161/01.cir.0000048192.52098.dd. [DOI] [PubMed] [Google Scholar]
- Pacher P, Liaudet L, Bai P, Virag L, Mabley JG, Hasko G, Szabo C. Activation of poly(ADP-ribose) polymerase contributes to development of doxorubicin-induced heart failure. J Pharmacol Exp Ther. 2002b;300:862–867. doi: 10.1124/jpet.300.3.862. [DOI] [PubMed] [Google Scholar]
- Pacher P, Liaudet L, Mabley JG, Cziraki A, Hasko G, Szabo C. Beneficial effects of a novel ultrapotent poly(ADP-ribose) polymerase inhibitor in murine models of heart failure. Int J Mol Med. 2006a;17:369–375. [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Liaudet L, Mabley J, Komjati K, Szabo C. Pharmacologic inhibition of poly(adenosine diphosphate-ribose) polymerase may represent a novel therapeutic approach in chronic heart failure. J Am Coll Cardiol. 2002c;40:1006–1016. doi: 10.1016/s0735-1097(02)02062-4. [DOI] [PubMed] [Google Scholar]
- Pacher P, Liaudet L, Soriano FG, Mabley JG, Szabo E, Szabo C. The role of poly(ADP-ribose) polymerase activation in the development of myocardial and endothelial dysfunction in diabetes. Diabetes. 2002d;51:514–521. doi: 10.2337/diabetes.51.2.514. [DOI] [PubMed] [Google Scholar]
- Pacher P, Mabley JG, Liaudet L, Evgenov OV, Marton A, Hasko G, Kollai M, Szabo C. Left ventricular pressure-volume relationship in a rat model of advanced aging-associated heart failure. Am J Physiol Heart Circ Physiol. 2004a;287:H2132–2137. doi: 10.1152/ajpheart.00405.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Mabley JG, Soriano FG, Liaudet L, Komjati K, Szabo C. Endothelial dysfunction in aging animals: The role of poly(ADP-ribose) polymerase activation. Br J Pharmacol. 2002e;135:1347–1350. doi: 10.1038/sj.bjp.0704627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Mabley JG, Soriano FG, Liaudet L, Szabo C. Activation of poly(ADP-ribose) polymerase contributes to the endothelial dysfunction associated with hypertension and aging. Int J Mol Med. 2002f;9:659–664. [PubMed] [Google Scholar]
- Pacher P, Nivorozhkin A, Szabo C. Therapeutic effects of xanthine oxidase inhibitors: Renaissance half a century after the discovery of allopurinol. Pharmacol Rev. 2006b;58:87–114. doi: 10.1124/pr.58.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Obrosova IG, Mabley JG, Szabo C. Role of nitrosative stress and peroxynitrite in the pathogenesis of diabetic complications. Emerging new therapeutical strategies. Curr Med Chem. 2005a;12:267–275. doi: 10.2174/0929867053363207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Schulz R, Liaudet L, Szabo C. Nitrosative stress and pharmacological modulation of heart failure. Trends Pharmacol Sci. 2005b;26:302–310. doi: 10.1016/j.tips.2005.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Szabo C. Role of poly(ADP-ribose) polymerase-1 activation in the pathogenesis of diabetic complications: Endothelial dysfunction, as a common underlying theme. Antioxid Redox Signal. 2005;7:1568–1580. doi: 10.1089/ars.2005.7.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Szabo C. Role of peroxynitrite in the pathogenesis of cardiovascular complications of diabetes. Curr Opin Pharmacol. 2006;6:136–141. doi: 10.1016/j.coph.2006.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Vaslin A, Benko R, Mabley JG, Liaudet L, Hasko G, Marton A, Batkai S, Kollai M, Szabo C. A new, potent poly(ADP-ribose) polymerase inhibitor improves cardiac and vascular dysfunction associated with advanced aging. J Pharmacol Exp Ther. 2004b;311:485–491. doi: 10.1124/jpet.104.069658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palfi A, Toth A, Hanto K, Deres P, Szabados E, Szereday Z, Kulcsar G, Kalai T, Hideg K, Gallyas F, et al. PARP inhibition prevents postinfarction myocardial remodeling and heart failure via the protein kinase C/glycogen synthase kinase-3beta pathway. J Mol Cell Cardiol. 2006;41:149–159. doi: 10.1016/j.yjmcc.2006.03.427. [DOI] [PubMed] [Google Scholar]
- Pieper AA, Walles T, Wei G, Clements EE, Verma A, Snyder SH, Zweier JL. Myocardial postischemic injury is reduced by polyADPripose polymerase-1 gene disruption. Mol Med. 2000;6:271–282. [PMC free article] [PubMed] [Google Scholar]
- Pillai JB, Gupta M, Rajamohan SB, Lang R, Raman J, Gupta MP. Poly(ADP-ribose) polymerase-1-deficient mice are protected from angiotensin II-induced cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2006;291:H1545–1553. doi: 10.1152/ajpheart.01124.2005. [DOI] [PubMed] [Google Scholar]
- Pillai JB, Isbatan A, Imai S, Gupta MP. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. J Biol Chem. 2005a;280:43121–43130. doi: 10.1074/jbc.M506162200. [DOI] [PubMed] [Google Scholar]
- Pillai JB, Russell HM, Raman J, Jeevanandam V, Gupta MP. Increased expression of poly(ADP-ribose) polymerase-1 contributes to caspase-independent myocyte cell death during heart failure. Am J Physiol Heart Circ Physiol. 2005b;288:H486–496. doi: 10.1152/ajpheart.00437.2004. [DOI] [PubMed] [Google Scholar]
- Plummer ER. Inhibition of poly(ADP-ribose) polymerase in cancer. Curr Opin Pharmacol. 2006;6:364–368. doi: 10.1016/j.coph.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Plummer R, Lorigan P, Evans J, Steven N, Middleton M, Wilson R, Snow K, Dewji R, Calvert H. First and final report of a phase II study of the poly(ADP-ribose) polymerase (PARP) inhibitor, AG014699, in combination with temozolomide (TMZ) in patients with metastatic malignant melanoma (MM) J Clin Oncol. 2006;24:A8013. [Google Scholar]
- Poitras MF, Koh DW, Yu SW, Andrabi SA, Mandir AS, Poirier GG, Dawson VL, Dawson TM. Spatial and functional relationship between poly(ADP-ribose) polymerase-1 and poly(ADP-ribose) glycohydrolase in the brain. Neuroscience. 2007;148:198–211. doi: 10.1016/j.neuroscience.2007.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radovits T, Seres L, Gero D, Berger I, Szabo C, Karck M, Szabo G. Single dose treatment with PARP-inhibitor INO-1001 improves aging-associated cardiac and vascular dysfunction. Exp Gerontol. 2007;42:676–685. doi: 10.1016/j.exger.2007.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajesh M, Mukhopadhyay P, Batkai S, Godlewski G, Hasko G, Liaudet L, Pacher P. Pharmacological inhibition of poly(ADP-ribose) polymerase inhibits angiogenesis. Biochem Biophys Res Commun. 2006a;350:352–357. doi: 10.1016/j.bbrc.2006.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajesh M, Mukhopadhyay P, Godlewski G, Batkai S, Hasko G, Liaudet L, Pacher P. Poly(ADP-ribose)polymerase inhibition decreases angiogenesis. Biochem Biophys Res Commun. 2006b;350:1056–1062. doi: 10.1016/j.bbrc.2006.09.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnam K, Low JA. Current development of clinical inhibitors of poly(ADP-ribose) polymerase in oncology. Clin Cancer Res. 2007;13:1383–1388. doi: 10.1158/1078-0432.CCR-06-2260. [DOI] [PubMed] [Google Scholar]
- Ravalli S, Albala A, Ming M, Szabolcs M, Barbone A, Michler RE, Cannon PJ. Inducible nitric oxide synthase expression in smooth muscle cells and macrophages of human transplant coronary artery disease. Circulation. 1998;97:2338–2345. doi: 10.1161/01.cir.97.23.2338. [DOI] [PubMed] [Google Scholar]
- Rubbo H, O’Donnell V. Nitric oxide, peroxynitrite and lipoxygenase in atherogenesis: Mechanistic insights. Toxicology. 2005;208:305–317. doi: 10.1016/j.tox.2004.11.019. [DOI] [PubMed] [Google Scholar]
- Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): Novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- Schulz R. Intracellular targets of matrix metalloproteinase-2 in cardiac disease: Rationale and therapeutic approaches. Ann Rev Pharmacol Toxicol. 2007;47:211–242. doi: 10.1146/annurev.pharmtox.47.120505.105230. [DOI] [PubMed] [Google Scholar]
- Soriano FG, Liaudet L, Szabo E, Virag L, Mabley JG, Pacher P, Szabo C. Resistance to acute septic peritonitis in poly(ADP-ribose) polymerase-1-deficient mice. Shock. 2002;17:286–292. doi: 10.1097/00024382-200204000-00008. [DOI] [PubMed] [Google Scholar]
- Soriano FG, Nogueira AC, Caldini EG, Lins MH, Teixeira AC, Cappi SB, Lotufo PA, Bernik MM, Zsengeller Z, Chen M, et al. Potential role of poly(adenosine 5-diphosphate-ribose) polymerase activation in the pathogenesis of myocardial contractile dysfunction associated with human septic shock. Crit Care Med. 2006;34:1073–1079. doi: 10.1097/01.CCM.0000206470.47721.8D. [DOI] [PubMed] [Google Scholar]
- Soriano FG, Pacher P, Mabley J, Liaudet L, Szabo C. Rapid reversal of the diabetic endothelial dysfunction by pharmacological inhibition of poly(ADP-ribose) polymerase. Circ Res. 2001;89:684–691. doi: 10.1161/hh2001.097797. [DOI] [PubMed] [Google Scholar]
- Southan GJ, Szabo C. Poly(ADP-ribose) polymerase inhibitors. Curr Med Chem. 2003;10:321–340. doi: 10.2174/0929867033368376. [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Szabados E, Literati-Nagy P, Farkas B, Sumegi B. BGP-15, a nicotinic amidoxime derivate protecting heart from ischemia reperfusion injury through modulation of poly(ADP-ribose) polymerase. Biochem Pharmacol. 2000;59:937–945. doi: 10.1016/s0006-2952(99)00418-9. [DOI] [PubMed] [Google Scholar]
- Szabo A, Hake P, Salzman AL, Szabo C. 3-Aminobenzamide, an inhibitor of poly (ADP-ribose) synthetase, improves hemodynamics and prolongs survival in a porcine model of hemorrhagic shock. Shock. 1998;10:347–353. doi: 10.1097/00024382-199811000-00007. [DOI] [PubMed] [Google Scholar]
- Szabo C. Potential role of the peroxynitrate-poly(ADP-ribose) synthetase pathway in a rat model of severe hemorrhagic shock. Shock. 1998;9:341–344. doi: 10.1097/00024382-199805000-00005. [DOI] [PubMed] [Google Scholar]
- Szabo C. Multiple pathways of peroxynitrite cytotoxicity. Toxicol Lett 140–. 2003;141:105–112. doi: 10.1016/s0378-4274(02)00507-6. [DOI] [PubMed] [Google Scholar]
- Szabo C. Cardioprotective effects of poly(ADP-ribose) polymerase inhibition. Pharmacol Res. 2005a;52:34–43. doi: 10.1016/j.phrs.2005.02.017. [DOI] [PubMed] [Google Scholar]
- Szabo C. Pharmacological inhibition of poly(ADP-ribose) polymerase in cardiovascular disorders: Future directions. Curr Vasc Pharmacol. 2005b;3:301–303. doi: 10.2174/1570161054368553. [DOI] [PubMed] [Google Scholar]
- Szabo C. Roles of poly(ADP-ribose) polymerase activation in the pathogenesis of diabetes mellitus and its complications. Pharmacol Res. 2005c;52:60–71. doi: 10.1016/j.phrs.2005.02.015. [DOI] [PubMed] [Google Scholar]
- Szabo C. Poly(ADP-ribose) polymerase activation by reactive nitrogen species–relevance for the pathogenesis of inflammation. Nitric Oxide. 2006;14:169–179. doi: 10.1016/j.niox.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Szabo C. Poly (ADP-ribose) polymerase activation and circulatory shock. Novartis Found Symp. 2007;280:92–103. doi: 10.1007/0-387-36005-0_16. discussion 103–107, 160–164. [DOI] [PubMed] [Google Scholar]
- Szabo C, Billiar TR. Novel roles of nitric oxide in hemorrhagic shock. Shock. 1999;12:1–9. doi: 10.1097/00024382-199907000-00001. [DOI] [PubMed] [Google Scholar]
- Szabo C, Cuzzocrea S, Zingarelli B, O’Connor M, Salzman AL. Endothelial dysfunction in a rat model of endotoxic shock. Importance of the activation of poly (ADP-ribose) synthetase by peroxynitrite. J Clin Invest. 1997;100:723–735. doi: 10.1172/JCI119585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C, Pacher P, Swanson RA. Novel modulators of poly(ADP-ribose) polymerase. Trends Pharmacol Sci. 2006a;27:626–630. doi: 10.1016/j.tips.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C, Pacher P, Zsengeller Z, Vaslin A, Komjati K, Benko R, Chen M, Mabley JG, Kollai M. Angiotensin II-mediated endothelial dysfunction: Role of poly(ADP-ribose) polymerase activation. Mol Med. 2004a;10:28–35. doi: 10.2119/2004-00001.szabo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C, Zanchi A, Komjati K, Pacher P, Krolewski AS, Quist WC, LoGerfo FW, Horton ES, Veves A. Poly(ADP-ribose) polymerase is activated in subjects at risk of developing type 2 diabetes and is associated with impaired vascular reactivity. Circulation. 2002a;106:2680–2686. doi: 10.1161/01.cir.0000038365.78031.9c. [DOI] [PubMed] [Google Scholar]
- Szabo G, Bahrle S. Role of nitrosative stress and poly(ADP-ribose) polymerase activation in myocardial reperfusion injury. Curr Vasc Pharmacol. 2005;3:215–220. doi: 10.2174/1570161054368599. [DOI] [PubMed] [Google Scholar]
- Szabo G, Bahrle S, Sivanandam V, Stumpf N, Gero D, Berger I, Beller C, Hagl S, Szabo C, Dengler TJ. Immunomodulatory effects of poly(ADP-ribose) polymerase inhibition contribute to improved cardiac function and survival during acute cardiac rejection. J Heart Lung Transplant. 2006b;25:794–804. doi: 10.1016/j.healun.2006.03.017. [DOI] [PubMed] [Google Scholar]
- Szabo G, Bahrle S, Stumpf N, Sonnenberg K, Szabo EE, Pacher P, Csont T, Schulz R, Dengler TJ, Liaudet L, et al. Poly(ADP-ribose) polymerase inhibition reduces reperfusion injury after heart transplantation. Circ Res. 2002b;90:100–106. doi: 10.1161/hh0102.102657. [DOI] [PubMed] [Google Scholar]
- Szabo G, Bahrle S, Stumpf N, Szabo C, Hagl S. Contractile dysfunction in experimental cardiac allograft rejection: role of the poly (ADP-ribose) polymerase pathway. Transpl Int. 2006c;19:506–513. doi: 10.1111/j.1432-2277.2005.00262.x. [DOI] [PubMed] [Google Scholar]
- Szabo G, Soos P, Bahrle S, Zsengeller Z, Flechtenmacher C, Hagl S, Szabo C. Role of poly(ADP-ribose) polymerase activation in the pathogenesis of cardiopulmonary dysfunction in a canine model of cardiopulmonary bypass. Eur J Cardiothorac Surg. 2004b;25:825–832. doi: 10.1016/j.ejcts.2004.01.031. [DOI] [PubMed] [Google Scholar]
- Szabo G, Soos P, Heger U, Flechtenmacher C, Bahrle S, Zsengeller Z, Szabo C, Hagl S. Poly(ADP-ribose) polymerase inhibition attenuates biventricular reperfusion injury after orthotopic heart transplantation. Eur J Cardiothorac Surg. 2005;27:226–234. doi: 10.1016/j.ejcts.2004.10.055. [DOI] [PubMed] [Google Scholar]
- Szabo G, Soos P, Mandera S, Heger U, Flechtenmacher C, Bahrle S, Seres L, Cziraki A, Gries A, Zsengeller Z, et al. INO-1001 a novel poly(ADP-ribose) polymerase (PARP) inhibitor improves cardiac and pulmonary function after crystalloid cardioplegia and extracorporal circulation. Shock. 2004c;21:426–432. doi: 10.1097/00024382-200405000-00005. [DOI] [PubMed] [Google Scholar]
- Szabolcs MJ, Ravalli S, Minanov O, Sciacca RR, Michler RE, Cannon PJ. Apoptosis and increased expression of inducible nitric oxide synthase in human allograft rejection. Transplantation. 1998;65:804–812. doi: 10.1097/00007890-199803270-00007. [DOI] [PubMed] [Google Scholar]
- Szenczi O, Kemecsei P, Holthuijsen MF, van Riel NA, van der Vusse GJ, Pacher P, Szabo C, Kollai M, Ligeti L, Ivanics T. Poly(ADP-ribose) polymerase regulates myocardial calcium handling in doxorubicin-induced heart failure. Biochem Pharmacol. 2005;69:725–732. doi: 10.1016/j.bcp.2004.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasatargil A, Dalaklioglu S, Sadan G. Inhibition of poly(ADP-ribose) polymerase prevents vascular hyporesponsiveness induced by lipopolysaccharide in isolated rat aorta. Pharmacol Res. 2005;51:581–586. doi: 10.1016/j.phrs.2005.02.020. [DOI] [PubMed] [Google Scholar]
- Tentori L, Graziani G. Chemopotentiation by PARP inhibitors in cancer therapy. Pharmacol Res. 2005;52:25–33. doi: 10.1016/j.phrs.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Tentori L, Leonetti C, Scarsella M, D’Amati G, Vergati M, Portarena I, Xu W, Kalish V, Zupi G, Zhang J, et al. Systemic administration of GPI 15427, a novel poly(ADP-ribose) polymerase-1 inhibitor, increases the antitumor activity of temozolomide against intracranial melanoma, glioma, lymphoma. Clin Cancer Res. 2003;9:5370–5379. [PubMed] [Google Scholar]
- Thiemermann C, Bowes J, Myint FP, Vane JR. Inhibition of the activity of poly(ADP ribose) synthetase reduces ischemia-reperfusion injury in the heart and skeletal muscle. Proc Nat Acad Sci USA. 1997;94:679–683. doi: 10.1073/pnas.94.2.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas HD, Calabrese CR, Batey MA, Canan S, Hostomsky Z, Kyle S, Maegley KA, Newell DR, Skalitzky D, Wang LZ, et al. Preclinical selection of a novel poly(ADP-ribose) polymerase inhibitor for clinical trial. Mol Cancer Ther. 2007;6:945–956. doi: 10.1158/1535-7163.MCT-06-0552. [DOI] [PubMed] [Google Scholar]
- Toth-Zsamboki E, Horvath E, Vargova K, Pankotai E, Murthy K, Zsengeller Z, Barany T, Pek T, Fekete K, Kiss RG, et al. Activation of poly(ADP-ribose) polymerase by myocardial ischemia and coronary reperfusion in human circulating leukocytes. Mol Med. 2006;12:221–228. doi: 10.2119/2006-00055.Toth-Zsamboki. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turko IV, Murad F. Protein nitration in cardiovascular diseases. Pharmacol Rev. 2002;54:619–634. doi: 10.1124/pr.54.4.619. [DOI] [PubMed] [Google Scholar]
- Ungvari Z, Gupte SA, Recchia FA, Batkai S, Pacher P. Role of oxidative-nitrosative stress and downstream pathways in various forms of cardiomyopathy and heart failure. Curr Vasc Pharmacol. 2005;3:221–229. doi: 10.2174/1570161054368607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veres B, Gallyas F, Jr, Varbiro G, Berente Z, Osz E, Szekeres G, Szabo C, Sumegi B. Decrease of the inflammatory response and induction of the Akt/protein kinase B pathway by poly-(ADP-ribose) polymerase 1 inhibitor in endotoxin-induced septic shock. Biochem Pharmacol. 2003;65:1373–1382. doi: 10.1016/s0006-2952(03)00077-7. [DOI] [PubMed] [Google Scholar]
- Virag L. Structure and function of poly(ADP-ribose) polymerase-1: Role in oxidative stress-related pathologies. Curr Vasc Pharmacol. 2005;3:209–214. doi: 10.2174/1570161054368625. [DOI] [PubMed] [Google Scholar]
- Virag L, Szabo C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54:375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- Virag L, Szabo E, Gergely P, Szabo C. Peroxynitrite-induced cytotoxicity: Mechanism and opportunities for intervention. Toxicol Lett 140–. 2003;141:113–124. doi: 10.1016/s0378-4274(02)00508-8. [DOI] [PubMed] [Google Scholar]
- Wang C, Bedikian AY, Kim K, Papadopoulos NE, Hwu W, Hwu P. Evaluation of tolerability, safety, and pharmacokinetics of INO-1001 plus temozolomide (TMZ) in patients with unresectable stage III/IV melanoma. J Clin Oncol. 2006;24:A12015. [Google Scholar]
- Wayman N, McDonald MC, Thompson AS, Threadgill MD, Thiemermann C. 5-aminoisoquinolinone, a potent inhibitor of poly (adenosine 5′-diphosphate ribose) polymerase, reduces myocardial infarct size. Eur J Pharmacol. 2001;430:93–100. doi: 10.1016/s0014-2999(01)01359-0. [DOI] [PubMed] [Google Scholar]
- Weinstein DM, Mihm MJ, Bauer JA. Cardiac peroxynitrite formation and left ventricular dysfunction following doxorubicin treatment in mice. J Pharmacol Exper Ther. 2000;294:396–401. [PubMed] [Google Scholar]
- Woon EC, Threadgill MD. Poly(ADP-ribose)polymerase inhibition—where now? Curr Med Chem. 2005;12:2373–2392. doi: 10.2174/0929867054864778. [DOI] [PubMed] [Google Scholar]
- Xiao CY, Chen M, Zsengeller Z, Li H, Kiss L, Kollai M, Szabo C. Poly(ADP-Ribose) polymerase promotes cardiac remodeling, contractile failure, and translocation of apoptosis-inducing factor in a murine experimental model of aortic banding and heart failure. J Pharmacol Exper Ther. 2005;312:891–898. doi: 10.1124/jpet.104.077164. [DOI] [PubMed] [Google Scholar]
- Xiao CY, Chen M, Zsengeller Z, Szabo C. Poly(ADP-ribose) polymerase contributes to the development of myocardial infarction in diabetic rats and regulates the nuclear translocation of apoptosis-inducing factor. J Pharmacol Exper Ther. 2004;310:498–504. doi: 10.1124/jpet.104.066803. [DOI] [PubMed] [Google Scholar]
- Xu Y, Huang S, Liu ZG, Han J. Poly(ADP-ribose) polymerase-1 signaling to mitochondria in necrotic cell death requires RIP1/TRAF2-mediated JNK1 activation. J Biol Chem. 2006;281:8788–8795. doi: 10.1074/jbc.M508135200. [DOI] [PubMed] [Google Scholar]
- Yang Z, Zingarelli B, Szabo C. Effect of genetic disruption of poly (ADP-ribose) synthetase on delayed production of inflammatory mediators and delayed necrosis during myocardial ischemia-reperfusion injury. Shock. 2000;13:60–66. doi: 10.1097/00024382-200013010-00011. [DOI] [PubMed] [Google Scholar]
- Yu SW, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, Dawson VL. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Nat Acad Sci USA. 2006;103:18314–18319. doi: 10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- Zhou HZ, Swanson RA, Simonis U, Ma X, Cecchini G, Gray MO. Poly(ADP-ribose) polymerase-1 hyperactivation and impairment of mitochondrial respiratory chain complex I function in reperfused mouse hearts. Am J Physiol Heart Circ Physiol. 2006;291:H714–723. doi: 10.1152/ajpheart.00823.2005. [DOI] [PubMed] [Google Scholar]
- Zingarelli B, Cuzzocrea S, Zsengeller Z, Salzman AL, Szabo C. Protection against myocardial ischemia and reperfusion injury by 3-aminobenzamide, an inhibitor of poly (ADP-ribose) synthetase. Cardiovasc Res. 1997;36:205–215. doi: 10.1016/s0008-6363(97)00137-5. [DOI] [PubMed] [Google Scholar]
- Zingarelli B, Hake PW, Burroughs TJ, Piraino G, O’Connor M, Denenberg A. Activator protein-1 signalling pathway and apoptosis are modulated by poly(ADP-ribose) polymerase-1 in experimental colitis. Immunology. 2004a;113:509–517. doi: 10.1111/j.1365-2567.2004.01991.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingarelli B, Hake PW, O’Connor M, Denenberg A, Kong S, Aronow BJ. Absence of poly(ADP-ribose)polymerase-1 alters nuclear factor-kappa B activation and gene expression of apoptosis regulators after reperfusion injury. Mol Med. 2003;9:143–153. doi: 10.2119/2003-00011.zingarelli. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingarelli B, Hake PW, O’Connor M, Denenberg A, Wong HR, Kong S, Aronow BJ. Differential regulation of activator protein-1 and heat shock factor-1 in myocardial ischemia and reperfusion injury: Role of poly(ADP-ribose) polymerase-1. Am J Physiol Heart Circ Physiol. 2004b;286:H1408–1415. doi: 10.1152/ajpheart.00953.2003. [DOI] [PubMed] [Google Scholar]
- Zingarelli B, Salzman AL, Szabo C. Genetic disruption of poly (ADP-ribose) synthetase inhibits the expression of P-selectin and intercellular adhesion molecule-1 in myocardial ischemia/reperfusion injury. Circ Res. 1998;83:85–94. doi: 10.1161/01.res.83.1.85. [DOI] [PubMed] [Google Scholar]