

Abstract
Soluble guanylate cyclase (sGC) is a key signal-transduction enzyme activated by nitric oxide (NO). Impaired bioavailability and/or responsiveness to endogenous NO has been implicated in the pathogenesis of cardiovascular and other diseases. Current therapies that involve the use of organic nitrates and other NO donors have limitations, including non-specific interactions of NO with various biomolecules, lack of response and the development of tolerance following prolonged administration. Compounds that activate sGC in an NO-independent manner might therefore provide considerable therapeutic advantages. Here we review the discovery, biochemistry, pharmacology and clinical potential of haem-dependent sGC stimulators (including YC-1, BAY 41-2272, BAY 41-8543, CFM-1571 and A-350619) and haem-independent sGC activators (including BAY 58-2667 and HMR-1766).
Nitric oxide (NO) is a key signalling molecule that is involved in the regulation of a variety of biological and physiological processes in mammals. Vast experimental and clinical evidence indicates that reduced bioavailability and/or responsiveness to endogenously produced NO contributes to the development of cardiovascular, pulmonary, endothelial, renal and hepatic diseases, as well as erectile dysfunction. Some of these conditions are currently treated with organic nitrates (such as glyceryl trinitrate) and other NO-donor or ‘nitrovasodilator’ drugs that release NO by spontaneous decomposition1 or bioconversion2 to activate soluble guanylate cyclase (sGC). However, the use of such compounds is limited by a potential lack of response due to insufficient biometabolism3, development of tolerance following prolonged administration4 and nonspecific interactions of NO with other biological molecules, including peroxynitrite-mediated tyrosine nitration5. The latter reactions are difficult to control owing to the spontaneous release of NO from nitrovasodilators and its free diffusion in biological systems. Moreover, despite the symptomatic improvement in patients with cardiovascular disease treated with organic nitrates, there is no clear evidence that such treatment reduces mortality. Therefore compounds that activate sGC in an NO-independent manner might offer considerable advantages over current therapies (FIG. 1). In this article, we describe the NO–sGC–cGMP signalling pathway and explain the therapeutic rationale for sGC stimulators and activators. The discovery, biochemistry and pharmacology of several such compounds are reviewed, and their potential for the treatment of a range of diseases, including arterial and pulmonary hypertension, heart failure, atherosclerosis, thrombosis, erectile dysfunction, renal fibrosis and failure, and liver cirrhosis, is discussed.
Figure 1. The NO–sGC–cGMP signal transduction pathway and potential drug targets.

Nitric oxide (NO) is synthesized enzymatically from the amino acid l-arginine by three isoforms of NO synthase (NOS), including endothelial NOS (eNOS). Minute amounts of endogenously produced or exogenously administered NO activate soluble guanylate cyclase (sGC), which converts GTP to cyclic GMP, mediating various physiological and tissue protective effects. Degradation of cGMP to GMP is catalysed by several phosphodiesterase (PDE) families. Excessive amounts of NO produced under pathological conditions associated with increased inflammation and oxidative stress react avidly with superoxide anion (•O2−), to form peroxynitrite (ONOO−). Peroxynitrite, in concert with other oxidants, induces cell damage via lipid peroxidation, inactivation of enzymes and other proteins by oxidation and nitration, and activation of matrix metalloproteinases (MMP) and the nuclear enzyme poly(ADP-ribose) polymerase (PARP), which ultimately leads to cellular dysfunction and death. NO–sGC–cGMP signalling can be compromised either by reducing the bioavailability of NO (for example, via chemical interaction of NO with •O2−) or by altering the redox state of sGC itself (for example, through oxidative stress or the action of peroxynitrite), thereby making it unresponsive to endogenous NO and NO-releasing drugs. Two novel drug classes seem to be able to overcome these obstacles: sGC stimulators (stimulate sGC directly and enhance sensitivity of the reduced enzyme to low levels of bioavailable NO) and sGC activators (activate the NO-unresponsive, haem-oxidized or haem-free enzyme). Other potential therapeutic approaches that modulate this pathway are also shown (dotted lines).
The NO–sGC–cGMP signal transduction pathway
Guanine nucleotidyl (guanylyl; guanylate) cyclases (GCs) are widely distributed signal-transduction enzymes that, in response to various cellular stimuli, convert GTP into the second messenger cyclic GMP. The biological effects of cGMP are mediated by three major types of intracellular effectors: cGMP-dependent protein kinases I and II, cGMP-gated ion channels and cGMP-regulated phosphodiesterases (PDEs)6,7. Degradation of cGMP is catalysed by several differentially expressed PDE families (PDE1, 2, 3, 5, 6, 9, 10 and 11), which represent independent drug discovery targets7.
In contrast to the transmembrane particulate GC (pGC), which serves as a receptor for atrial, B-type and C-type natriuretic peptides, sGCs are receptors for gaseous ligands, namely NO and carbon monoxide (CO). They can associate with the plasma membrane through protein–protein interactions in a Ca2+-dependent manner8,9 or apparently in a constitutive manner10,11. Soluble GC is typically found as a heterodimer, consisting of a larger α-subunit and a smaller haem-binding β-subunit, although homodimers of these subunits can also form12. Four human sGC subunits exist: α1, α2, β1 and β2 of which the α1/β2 and α2/β1 heterodimers are the best characterized13,14. The 619-residue β-subunit contains an evolutionarily conserved amino-terminal haem-binding domain that has a length of about 200 residues15–17. A prosthetic haem moiety, which is crucial for the sensing of NO, is positioned in the haem-binding domain via its interaction with the axial ligand histidine-105 and the anchoring residues of the haem propionates tyrosine-135, serine-137 and arginine-139 (constituting the haem-binding motif Y-x-S-x-R; see FIG. 2)17–19. Based on sequence homology with the crystallized catalytic domains of adenylate cyclase, the carboxy-terminal catalytic domains of both sGC subunits are assumed to be orientated in a head-to-tail fashion20,21. The catalytic domains of both subunits are required for the formation of a catalytic active centre13,22,23.
Figure 2. Homology model of the haem-binding domain of the human soluble guanylate cyclase (sGC) β-subunit.
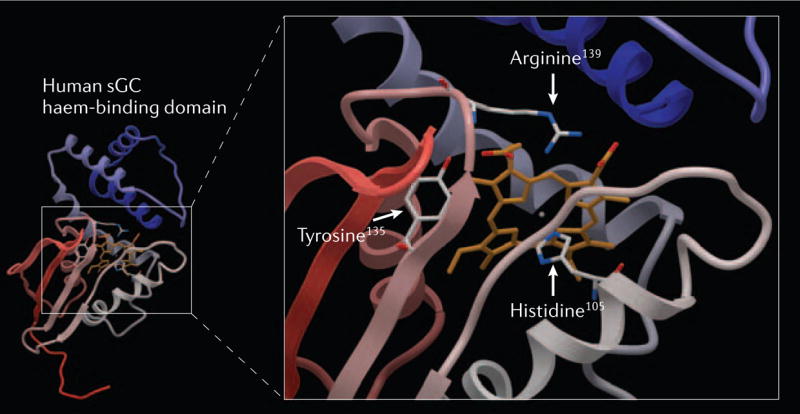
The model depicted is based on the recently resolved crystal structure of a prokaryotic haem-binding protein of Thermoanaerobacter tengcongensis with sequence homology to the sGC haem-binding domain16,17. Residues responsible for the coordination of the haem are shown in the enlargement on the right side. The axial haem ligand histidine-105 and the counterparts of the haem propionic acids tyrosine-135, serine-137 and arginine-139 comprise the unique sGC haem-binding motif Y-x-S-x-R (serine-137 was omitted for clarity)18,19,83.
The NO–sGC–cGMP pathway is crucial for the control of many physiological processes, such as host defence reactions, cell growth and proliferation, vascular homeostasis and neuronal transmission (FIG. 1)24. Although sGC is activated by nanomolar concentrations of NO, the sGC–cGMP axis might not be the only signalling pathway affected by NO. As a result of its reactivity with iron-containing catalytic sites, including the haem groups of FeS clusters, the function of various enzymes can be affected by NO, especially at higher concentrations. These alternative mechanisms could explain the distinct functional consequences of NO and cGMP signalling that have been reported, such as pro-25 and anti-aggregatory26,27 effects in platelets, and pro-28,29 and anti-atherosclerotic30,31 effects in blood vessels.
One major prerequisite for the NO-induced activation of sGC is the presence of the reduced Fe2+ haem moiety; its removal abolishes any NO-induced enzyme activation32,33. The central ferrous iron of the prosthetic group is coordinated between the four haem nitrogens and the axial ligand histidine-105, building a penta-coordinated histidyl-haem complex (FIG. 2). Binding of NO to this complex results in the formation of a hexa-coordinated histidine–haem–NO intermediate that rapidly decays into a penta-coordinated nitrosyl–haem complex. The cleavage of the haem–histidine bond is the molecular switch that leads to a ∼200-fold activation of sGC34. However, this simple model of sGC activation does not fit with some subsequently observed activation characteristics of the enzyme. For example, several research groups have demonstrated that the conversion of the hexa-coordinated intermediate state into the penta-coordinated active nitrosyl–haem complex depends on the concentration of free NO, suggesting that there is a second binding site for NO35,36. Others have confirmed this finding, and reported that spectroscopically validated, NO-bound sGC can exist in a virtually inactive form37,38. This state, which is formed in the presence of low NO concentrations, can be transformed to the fully activated enzyme by addition of NO, the sGC substrate GTP, or the reaction products cGMP and/or pyrophosphate37,38. Physiological concentrations of ATP inhibit this transformation, linking NO–sGC–cGMP signalling to cellular energy metabolism37.
In addition to removal of the haem, its oxidation39 by sGC inhibitors such as ODQ (1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one), NS2028, methylene blue or ferri-cyanide leads to the formation of an NO-insensitive form of the enzyme40–45. Endogenously, changes in the redox state of sGC can be induced by reactive oxygen and nitrogen species such as superoxide (•O2−) and peroxynitrite (ONOO−), which are generated under conditions of oxidative stress46 (FIG. 1). Indeed, the capability of the prosthetic group of sGC to exist in different redox states might allow sGC, in addition to its gas-sensing capability, to monitor intracellular redox homeostasis (FIG. 3). With respect to disease, NO–sGC–cGMP signalling can be compromised either by reducing the bioavailability of NO (for example, via the chemical interaction of NO with •O2−) or by altering the redox state of sGC itself, making it unresponsive to endogenous NO and therefore to exo genous NO from NO-releasing drugs46. Excitingly, two novel drug classes seem to be able to overcome these obstacles: sGC stimulators and sGC activators (TABLES 1,2). The former compounds stimulate sGC directly and enhance the sensitivity of the reduced enzyme to low levels of bio-available NO. Conversely, sGC activators do not modulate NO signalling at all but activate the NO-unresponsive, haem-oxidized or haem-free enzyme (FIG. 3). Clinically, the main rationale for these new therapeutic principles is based on both insufficient NO–sGC–cGMP signalling often associated with the use of conventional NO donors and the medical need to treat conditions associated with oxidative stress.
Figure 3. Soluble guanylate cyclase (sGC) redox equilibrium.

The figure illustrates the intracellular redox equilibrium of the two sGC redox states, nitric oxide (NO)-sensitive reduced (blue) and NO-insensitive oxidized sGC (pink). The equilibrium can be shifted by reactive oxygen species to the oxidized (ferric) state and by postulated but yet undiscovered reductases to the reduced (ferrous) form. Disequilibrium towards the oxidized NO-unresponsive enzyme exists under various pathophysiological conditions associated with oxidative stress. Haem-independent sGC activators such as BAY 58–2667 and HMR-1766 activate the oxidized or haem-deficient form. By contrast, haem-dependent stimulators YC-1, BAY 41-2272, BAY 41–8543, A-350619 and CFM-1571 can activate the reduced sGC and show a strong synergism when combined with NO.
Table 1. Main haem-dependent stimulators of soluble guanylate cyclase.
| Structure | Name | Comments | Refs |
|---|---|---|---|
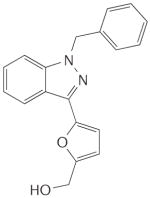
|
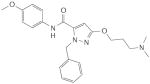
YC-1 |
|
48–50,54, 56,60,63, 154,155 |
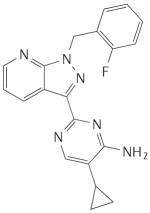
|
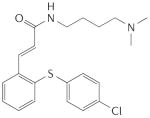
BAY 41-2272 |
|
54,57, 69,72, 73,75,142, 156–158 |
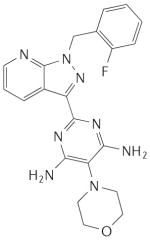
|
BAY 41-8543 |
|
67,68, 70,159 |
|
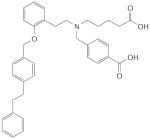
CFM-1571 |
|
53 |
|
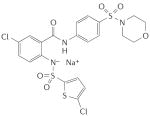
A-350619 |
|
55,78,79 |
CO, carbon monoxide; EC50, 50% effective concentration; IC50, 50% inhibitory concentration; NO, nitric oxide; PDE, phosphodiesterase; sGC, soluble guanylate cyclase.
Table 2. Main haem-independent activators of soluble guanylate cyclase.
| Structure | Name | Comments | Refs |
|---|---|---|---|
|
BAY 58–2667 |
|
18,46, 70,80 83 |
|
HMR-1766 (related derivative: S-3448) |
|
82 |
CO, carbon monoxide; EC50, 50% effective concentration; IC50, 50% inhibitory concentration; Kd, equilibrium dissociation constant; NO, nitric oxide; PDE, phosphodiesterase; sGC, soluble guanylate cyclase.
Discovery of sGC stimulators and sGC activators
Our knowledge of the function of sGC has increased tremendously during the past decade. The recent discoveries of compounds that stimulate or activate sGC independently of NO release allow this venerable pharmacological target to be approached from a completely different perspective. NO-independent but haem-dependent stimulators of sGC, as well as NO- and haem-independent sGC activators, are emerging as valuable tools that could help to elucidate the physiology and pathophysiology of the NO–sGC–cGMP pathway in more detail. Given the clinical importance of diseases that these compounds could be used to treat, and on the basis of in vitro, in vivo and initial clinical studies, expectations for these novel drug classes are high.
Haem-dependent sGC stimulators
In 1994, scientists at Bayer HealthCare AG started a search for substances that could induce an increase in NO synthesis and thereby stimulate sGC in porcine endothelial cells, in studies that involved the measurement of cGMP levels by radioimmunoassay. Nearly 20,000 compounds were tested, which led to the unexpected discovery of 5-substituted-2-furaldehyde-hydrazone derivatives as direct NO-independent sGC stimulators. Interestingly, the potency of these compounds in tests involving the stimulation of purified sGC and the relaxation of isolated blood vessels increased when they were exposed to light, which was clearly an unwanted feature for further drug development. In parallel, in December 1994, Ko and co-workers described a structurally related compound, the indazole derivative YC-1, which was subsequently characterized as an NO-independent, haem-dependent stimulator of highly purified sGC. Furthermore, its potency was unaffected by the prevailing light conditions47–50. During the following decade, various compounds that activate sGC in an NO-independent fashion were identified47,51–55. These drugs, including BAY 41–2272, BAY 41–8543, CMF-1571 and A-350619, constitute a novel class of haem-dependent sGC-stimulators, based on several shared characteristics: crucial dependency on the presence of the reduced prosthetic haem moiety and strong synergistic enzyme activation when combined with NO (TABLE 1).
Molecular mechanisms of sGC stimulators
YC-1
YC-1 activates sGC independently of NO48–50,56. However, similarly to NO, the activating effect of YC-1 crucially depends on the presence of the reduced prosthetic haem moiety of sGC. Its removal or oxidation virtually abolishes any YC-1-induced sGC activation19,32,34,42–45,57–59. In the presence of YC-1, sGC activation by NO is potentiated. This effect is based, at least partially, on the strong stabilization of the nitrosyl-haem complex60 and the postulated transformation of the NO-activated enzyme from a low- to a high-output activation state37,38. Furthermore, YC-1 was shown to potentiate the weak sGC activator CO to a level of effectiveness comparable to NO48,49,61,62, and to inhibit cGMP-metabolizing PDEs63.
The exact mechanism of direct YC-1-induced effects on sGC and its putative binding site are still a matter of debate. Guided by homology between the catalytic domains of adenylate cyclases and GCs20 and the results of mutational studies64,65, it was suggested that YC-1 might bind to the catalytic domain of sGC, activating the enzyme by a mechanism that is comparable to forskolin-induced activation of adenylate cyclases13,21. This hypothesis recently gained support from the demonstration that YC-1 and related compounds interact with the catalytic subunit of sGC66. By contrast, further spectroscopic studies demonstrated that YC-1-induced effects, such as alterations of the CO–haem bond, could still be observed in haem-containing fragments of the β-subunit in the absence of the catalytic domain62. As there is a vital crosstalk between the mechanisms of NO- and YC-1-mediated sGC activation, this hypothesis is attractive. However, at the present time, both postulated binding mechanisms could be possible and will have to be validated in the future by co-crystallization studies.
BAY 41-2272 and BAY 41-8543
BAY 41–2272 and BAY 41–8543 were synthesized based on YC-1 as a lead structure54,57,67,68. However, BAY 41–2272 and BAY 41–8543 are about two orders of magnitude more potent than YC-157. Both compounds activate purified sGC and strongly synergize with NO, reflecting the stabilization of the nitrosyl–haem complex57,67,69,70. A recent in vitro study using concentrations several orders of magnitude above those needed for sGC stimulation suggested some degree of PDE5 inhibition by BAY 41–227271. However, this effect is probably irrelevant because these high drug concentrations will not be reached therapeutically69,72,73.
Removal of the prosthetic haem moiety or its oxidation by ODQ strongly diminished BAY 41–2272/ 41–8543-induced enzyme activation. Using a photoaffinity-labelling approach based on a high-affinity analogue of BAY 41–2272, Stasch and co-workers observed labelling of cysteine-238 and -243 of the sGC α-subunit57. This result was validated with a photolabile derivative of YC-174. However, as no crystal structure of sGC is available, it is possible that these residues might be situated in the interface between the sGC subunits, thereby allowing labelling of the α-subunit even if the compound binds to the corresponding β-subunit, as discussed for YC-1. This view is supported by subsequent studies involving mutations in the α-subunit, which showed no effect on the BAY 41–2272-induced enzyme activation (P.M.S., unpublished data).
BAY 41–2272 and BAY 41–8543 produce potent relaxation of isolated systemic and coronary arteries and veins, and reduce coronary perfusion pressure in the rat heart Langendorff preparation. Both compounds also have antiproliferative properties in smooth muscles and anti-aggregatory effects in platelets57,67,68,75–77.
CFM-1571
The chemical strategy that led to the identification of CFM-1571 was based on YC-1 as a lead structure53. The enzymatic assay used to detect putative sGC-activating derivatives was performed in the presence of submaximal concentrations of NO. Although not specified by the authors, this indicates that the screening approach was designed to identify compounds that synergize with NO, a clear characteristic of haem-dependent sGC stimulators.
A-350619
The chemical structure of A-350619 (Abbott) and its analogues share no similarity with YC-155,78,79. However, like YC-1, A-350619 and its derivatives directly activate sGC and synergize with NO78. Oxidation of the sGC haem moiety by ODQ reduced the effectiveness of A-350619 in stimulating sGC. Although no competition-binding experiments have been published, Miller and co-workers proposed a common binding site for both structural classes78, based on findings showing that A-350619 and YC-1 did not have an additive effect on sGC activation when combined.
Haem-independent sGC activators
A rapid and highly sensitive cell-based assay for cGMP suitable for fully automated ultra-high-throughput screening was a key tool for further pharmacological breakthroughs. In this assay, which uses a Chinese hamster ovary cell line, intracellular cGMP production is linked to bioluminescence via an increase in Ca2+ influx through the olfactory cGMP-gated cation channel CNGA2, in the presence of aequorin, a Ca2+-sensitive luminescence indicator. This cell line was used to screen more than 900,000 compounds, leading to the identification of the primary hit BAY W 1449, an amino dicarboxylic acid80,81. A chemical derivatization programme was initiated and, in 2002, BAY 58–2667 was selected from a series of approximately 800 analogues as the first NO-independent activator of sGC that showed completely different characteristics to any of the known haem-dependent sGC stimulators. The activation of sGC by this compound was even stronger after oxidation or removal of the prosthetic haem group, indicating a previously unknown mechanism of enzyme activation80. Very recently, another compound with comparable characteristics, HMR-1766, was described, although the two chemical structures show no similarity82. These two compounds founded the novel class of NO- and haem-independent sGC activators (TABLE 2).
Molecular mechanisms of sGC activators
BAY 58-2667
BAY 58–2667 directly activates sGC with EC50 and Kd values in the low nanomolar range, making this compound the most potent NO-independent sGC activator reported to date70,80. In contrast to the haem-dependent sGC stimulators, BAY 58–2667 produces an additive, not synergistic, effect when combined with NO donors. Oxidation or removal of the prosthetic haem group potentiates BAY 58–2667-induced enzyme activation80. This unique characteristic can be explained by a mechanistic model in which the sGC prosthetic haem is replaced by BAY 58–2667, thereby resulting in activation of the enzyme. BAY 58–2667 and the haem moiety compete via their negatively charged carboxylic groups for the unique sGC haem-binding motif Y-x-S-x-R17,18,83. This hypothesis, based on activity and binding assays as well as spectroscopic studies, is further strengthened by structural alignments showing that BAY 58–2667 is able to mimic the spatial structure of the sGC porphyrin ligand18. As only the oxidized haem can be effectively replaced with BAY 58–2667 (because of the strongly reduced affinity of the oxidized haem for the sGC haem-binding pocket in comparison to the reduced moiety), this compound is able to discriminate between both redox states18. Therefore, BAY 58–2667 activates haem-deficient or oxidized sGC by binding to the unoccupied haem-binding pocket or by replacing the weakly bound oxidized haem, whereas reduced sGC is virtually unresponsive to BAY 58–2667.
BAY 58–2667 relaxes blood vessels with a potency that is several orders of magnitude greater than the NO donors sodium nitroprusside (SNP) and 3-morpho-linosydnonimine84. The compound reduces coronary perfusion pressure in the rat Langendorff heart preparation and, like its predecessors BAY 41–2272 and BAY 41–8543, remains active in tissues made tolerant to glyceryl trinitrate80.
HMR-1766 and S-3448
Submicromolar concentrations of HMR-1766 and the chemically related structure S-3448 (Sanofi-Aventis) directly activate purified sGC82. The addition of submaximal concentrations of NO results in an additive effect, whereas oxidation of the sGC haem moiety by ODQ strongly potentiates the HMR-1766 activating effect. Similarly to BAY 58–2667, competition between HMR-1766 and porphyrinic haemsite ligands was observed, indicating, at least in part, an overlapping binding site between this structural class and porphyrins. Schindler and co-workers proposed that HMR-1766 and S-3448 preferentially activate the oxidized haem-containing sGC, although activation of the haem-deficient enzyme was not ruled out82. In fact, the latter view was supported by the demonstration that a haem-free purified enzyme preparation could be activated by S-344882. Taken together these findings suggest that HMR-1766 and S-3448, similarly to BAY 58–2667, might be capable of activating both the haem-containing and the haem-deficient form of sGC. Future studies should clarify the exact mechanism of sGC activation by HMR-1766 and S-3448, and provide a direct comparison of these compounds with BAY 58–2667.
Therapeutic applications
Arterial hypertension
Arterial hypertension is one of the most important public health problems in the developed world. In 2000 in the US alone, there were at least 65 million adults with arterial hypertension, and its total prevalence rate was 31.3%85. Untreated, it leads to premature death by contributing to heart disease, stroke and renal failure. In 90–95% of cases the aetiology of arterial hypertension is unknown (essential hypertension), and therefore the therapy has to rely on symptomatic approaches rather than prevention or cure. A wide array of studies on human subjects suggests that alterations in NO–sGC–cGMP signalling that are unrelated to a decreased availability of the substrate for NO production are involved in its pathogenesis86–88. In arterial hypertension models (such as the spontaneously hypertensive rat (SHR)), endothelium-dependent vasodilation is impaired as a result, at least in part, of oxidative and nitrosative stress, and mRNA and protein levels of both the α1 and β1 subunits of sGC and sGC activity are reduced89–93. By contrast, NOS protein and activity levels seem to be upregulated to compensate for these changes30,93.
Clinically, the use of classical NO donors to treat arterial hypertension is problematic because of the development of tolerance following prolonged administration. Indeed, chronic exposure to endothelium-derived NO, as well as acute exposure to nitrovasodilator-derived NO, can lead to sGC desensitization without altering sGC expression94. Correspondingly, both acute cessation of endothelial NO formation by the pharmacological inhibition of NO synthase (NOS) in wild-type mice and chronic deficiency of NO in endothelial NOS−/− mice restore the sensitivity of sGC to NO and enhance vascular smooth muscle relaxation in response to nitrovasodilator agents94. There are therefore at least two mechanisms affecting sGC in vivo: downregulation of mRNA and protein levels, and desensitization. Non-NO-based sGC stimulators and activators potentially offer a major advantage over NO-releasing drugs by providing a means of sGC activation that is independent of NO–superoxide interactions, metabolic NO formation and NO-induced sGC desensitization (FIG. 1).
In normotensive animals, BAY 41–8543 lowers blood pressure and increases coronary blood flow67. Of note, a 500-fold higher dose of YC-1 is needed to achieve a comparable haemodynamic response50,54,95. In addition, reduction of blood pressure by YC-1 lasts only a few minutes95, whereas that of BAY 41–8543 is still present two hours after intravenous administration. In conscious SHR, orally applied YC-1 is devoid of any antihypertensive activity, unlike BAY 41–8543, which produces a dose-dependent and long-lasting (up to 24 hours) antihypertensive effect without causing tachyphylaxis following multiple administrations. In this experimental setting, BAY 41–8543 is about threefold more potent than BAY 41–227268. Furthermore, BAY 41–2272 and BAY 41–8543 prevent the increase in blood pressure induced by NOS inhibition in rats, attenuate cardiac remodelling and provide cardiorenal protection, which is associated with a reduction in mortality57,68,96. The pharmacological profiles of the sGC stimulators BAY 41–2172 and BAY 41–8543 therefore suggest that this class of compounds could have unique clinical utility in the treatment of essential hypertension and associated cardiac and renal complications.
The sGC activator BAY 58–2667 is the first NO-independent compound that has a haemodynamic profile similar to that of organic nitrates. It is about two orders of magnitude more potent as a vasodilator in isolated arterial and venous rings than BAY 41–227254,57. In anaesthetized rats, intravenous administration of BAY 58–2667 produces dose-dependent and long-lasting hypotension. ODQ does not inhibit the activity of BAY 58–2667 but, conversely, causes a significant increase in the potency and duration of the vasodilation46,97. These preliminary results suggest that, unlike glyceryl trinitrate, the efficacy of BAY 58–2667 is not reduced, and is in fact augmented, by oxidative stress, which is due to the targeting by this compound of the oxidation-impaired or haem-free sGC. This compound therefore represents an entirely new pharmacological principle, which selectively targets diseased blood vessels for antihypertensive and diagnostic purposes (FIG. 4).
Figure 4. NO–sGC–cGMP signalling in a blood vessel.

l-arginine is converted in the endothelium monolayer by the endothelial nitric oxide synthase (eNOS) to NO, which diffuses into both the vessel lumen and the vessel wall, thereby activating soluble guanylate cyclase (sGC). Haem-dependent sGC stimulators and haem-independent sGC activators increase the cellular cGMP concentration via the direct activation of sGC, which results in both vasorelaxation and inhibition of platelet aggregation. In contrast, organic nitrates require bioconversion to release NO, which is not implemented in platelets, leading to poor anti-aggregatory effect.
Pulmonary hypertension
Acute and chronic pulmonary hypertension (PH) are life-threatening conditions characterized by increased pulmonary arterial pressure that results from excessive pulmonary vasoconstriction. Chronic PH is also associated with pulmonary vascular remodelling, localized thrombosis and right heart hypertrophy. Although approaches for the diagnosis of PH have evolved dramatically in recent years, treatment of PH largely remains palliative98.
In the normal lung, the vascular endothelium and airway epithelium produce NO that regulates the processes required to match vascular perfusion with alveolar ventilation. Increased NO production causes a local increase in the synthesis of cGMP, which leads to pulmonary vasodilation. However, in PH the pulmonary production of NO and other endogenous vasodilators, such as prostacyclin, becomes markedly impaired, whereas vasoconstrictors, such as thromboxane A2 and endothelin, are released in excess98,99.
Inhalation of low concentrations of gaseous NO produces pulmonary vasodilation in well-ventilated lung regions without causing arterial hypotension. This selective pulmonary vasodilation is achieved because inhaled NO is rapidly bound by haemoglobin after entry into the intravascular space100. Limitations of NO inhalation as a chronic therapy for PH include the short duration of pulmonary vasodilation after NO is discontinued, the development of methaemoglobinaemia following inhalation of high concentrations of NO gas, and the observation that not all PH patients respond to inhaled NO100. The vasodilator effect of NO is mediated via cGMP-dependent mechanisms, and so it has been proposed that inhibition of the cGMP-metabolizing PDEs would dilate the pulmonary vasculature by increasing cGMP levels in pulmonary vascular smooth muscle cells. Subsequently, the PDE5 inhibitor sildenafil has been shown to produce pulmonary vasodilation in experimental models of PH, as well as in patients with chronic PH, and the drug has been approved for the treatment of this condition101–104. However, a significant proportion of patients with chronic PH fail to respond to sildenafil therapy, indicating that in these individuals endogenous NO production is reduced to such an extent that inhibition of cGMP degradation has no beneficial effects101. Accumulating experimental data suggest that under such conditions direct pharmacological activation of sGC could represent a promising therapeutic alternative.
In a sheep model of acute, chemically induced PH, intravenous infusion of BAY 41–2272 reduced mean pulmonary arterial pressure and pulmonary vascular resistance and increased transpulmonary cGMP release72. However, larger doses of BAY 41–2272 also elevated the cardiac index and produced systemic vaso dilation. Pharmacological inhibition of NOS abolished the systemic but not the pulmonary vasodilator effects of BAY 41–2272. The latter findings suggest that although BAY 41–2272 might act independently of endogenous NO production in the pulmonary vasculature, endogenous NO is required for the systemic vasodilator response to this compound. Furthermore, BAY 41–2272 potentiated and prolonged the pulmonary vasodilation induced by inhaled NO72. In a clinical setting, the capacity of an sGC stimulator to augment the efficacy of inhaled NO could result in an increased number of patients with PH responding to low concentrations of NO. In addition, the prolongation of the vasodilator effects of NO by an sGC stimulator might potentially facilitate chronic therapy with intermittently inhaled NO.
Similar haemodynamic effects were subsequently found following infusion of BAY 41–2272 in healthy ovine foetuses and a sheep model of persistent pulmonary hypertension of the newborn. Compared with sildenafil, the pulmonary vasodilator response to BAY 41–2272 was more prolonged105,106. In addition, BAY 41–2272 deceased mean pulmonary arterial pressure in dogs subjected to rapid ventricular pacing107. Moreover, in rodent models of chronic PH, treatment with BAY 41–2272 or BAY 58–2667 markedly attenuated an increase in right ventricular systolic pressure, right ventricular hypertrophy and structural remodelling of the lung vasculature108,109.
However, treatment of PH with oral or intravenous vasodilating agents, including NO-releasing drugs and stimulators or activators of sGC, can be associated with arterial hypotension and deterioration of arterial oxygenation due to pulmonary ventilation-perfusion mismatching72. Inhalation of vasodilating agents can provide targeted drug delivery to the lungs, thereby avoiding or reducing systemic side effects. Indeed, inhaling lipid–protein–sugar microparticles containing BAY 41–2272, BAY 41–8543 or BAY 58–2667 produced potent selective pulmonary vasodilation in lambs with acute PH without any adverse effects on pulmonary gas exchange110. Taken together, these preclinical studies provide strong evidence that direct pharmacological stimulators or activators of sGC, either alone or in combination with exogenous NO, could be an effective therapeutic intervention in PH, especially when endogenous NO–sGC–cGMP signalling is impaired. The sGC stimulator BAY 63–2521, which is structurally similar to BAY 41–2272 and BAY 41–8543, is now in clinical trials for the treatment of patients with chronic PH111.
Heart failure
Heart failure (HF), which is associated with a range of cardiovascular pathological conditions, is one of the major causes of hospitalization, morbidity and mortality worldwide, and represents a growing public-health issue. For example, in the US approximately 5 million people are affected by HF, more than 0.5 million new cases are diagnosed each year, and the total annual cost of managing patients with HF is approaching US$30 billion. Despite advances in treatment, the number of deaths from HF has increased steadily112. The progression of cardiovascular dysfunction to HF is complex and involves the activation of numerous secondary pathways113–115. In HF, sGC/pGC–cGMP signalling is disrupted either as a result of impaired production of NO or its excessive degradation, or chemical interactions with oxidants such as superoxide; in addition, inadequate release of the pGC ligands atrial and B-type natriuretic peptides, or the release of abnormal forms of these proteins, contributes to HF114,116. Conventional organic nitrates that activate sGC after bioconversion to NO have been used for more than a century to treat congestive HF; however, their therapeutic efficacy is limited because of the development of tolerance following chronic administration. The tolerance mechanism has been linked, in part, to increased vascular superoxide production, downregulation of mitochondrial aldehyde dehydrogenase, and supersensitivity to vasoconstrictors, secondary to a tonic activation of protein kinase C2,4.
In a canine model of congestive HF, intravenous administration of BAY 41–2272 increased cardiac output and renal blood flow and reduced the mean arterial, pulmonary arterial and pulmonary capillary wedge pressures, without decreasing the glomerular filtration rate or affecting the renin–angiotensin–aldosterone system, which is involved in the progression of the disease and development of complications107. Importantly, in this model BAY 41–2272 acted as a pure arterial vasodilator and, unlike glyceryl trinitrate, did not significantly decrease right atrial pressure. A possible explanation for these diverse effects is that sGC activation by glyceryl trinitrate depends on its biotransformation, which has been reported to be higher in the venous blood vessels117. Furthermore, the cGMP-independent actions of NO might also account for these differences.
Intravenous infusion of BAY 58–2667 or glyceryl trinitrate in anaesthetized dogs that were under autonomic blockade also produced a decrease in arterial blood pressure and reductions in diastolic pulmonary artery pressure and right atrial pressure. The duration of vasodilator effect was much longer for BAY 58–2667 compared with glyceryl trinitrate80. BAY 58–2667 is therefore the first new non-NO-based compound that has similar haemodynamic effects to glyceryl trinitrate, causing in vivo dilation of both arterial and venous blood vessels. In a canine model of congestive HF, intravenous administration of BAY 58–2667 resulted in dose-dependent reductions in cardiac preload and afterload, and a concomitant increase in cardiac output and renal blood flow without further neurohumoral activation118. BAY 58–2667 is currently undergoing clinical studies in patients with acute HF111. These findings suggest a novel mechanism for modulating the NO–sGC–cGMP pathway in the treatment of cardiac disease and for counteracting endothelial dysfunction without increasing vascular superoxide production and development of tolerance.
Atherosclerosis, restenosis, thrombosis and inflammation
Atherosclerosis is the underlying cause of the majority of common cardiovascular diseases, which are the leading causes of death in Western societies119. The pathophysiology of atherosclerosis is complex and involves endothelial injury, which results in the accumulation of lipids and their uptake by monocytes. This is followed by platelet and monocyte adhesion and aggregation at the site of injury, and the release of factors that promote smooth-muscle proliferation and migration, which leads to the synthesis and deposition of extracellular matrix120,121. Percutaneous transluminal coronary and peripheral artery angioplasties have become widely available and effective treatments for patients with coronary and peripheral artery diseases. Although the incidence of restenosis has been reduced to 20–30% by the use of modern stents, which have further revolutionized angioplasty procedures, it remains a significant clinical problem with a limited number of therapeutic options122.
Nitric oxide is an important anti-atherosclerotic autocoid with anti-aggregatory effects on platelets and antiproliferative and dilatory effects on vasculature (FIG. 4). Local transfer of genes encoding NOS123,124 and local or systemic administration of NO donors125,126 attenuates neointima formation after experimental vascular balloon injury (a model of restenosis). Similarly, local adenovirus-mediated gene transfer of sGC α1 and β1 subunits partially restores sGC function and NO responsiveness in balloon-injured rat carotid arteries, resulting in reduced neointima formation in the presence of low concentrations of an NO donor31. There is recent evidence demonstrating that atherosclerosis is not only associated with decreased NO bioavailability, but also with alterations in signal-transduction components downstream of NO including, among others, sGC, particularly in neointima30. sGC might therefore represent an attractive, novel pharmacological target in the treatment of atherosclerosis and restenosis.
Indeed, topical application of the sGC stimulator YC-1 inhibited vascular smooth-muscle cell proliferation through cGMP-dependent mechanisms77,127 and also reduced the expression of transforming growth factor-β (TGFβ), focal adhesion kinase and matrix metalloproteinases (MMP2 and MMP9)77,128. YC-1 also markedly inhibited neointima formation after balloon-induced carotid artery injury in rats128,129. Likewise, in the same experimental model, BAY 41–2272 reduced the neointimal response to injury through antiproliferative and antimigratory actions on vascular smooth-muscle cells130. In addition to antiproliferative effects in vascular smooth-muscle cells, BAY 41–2272, BAY 41–8543 and BAY 58–2667 also inhibited platelet aggregation and thrombosis in various experimental models57,67,68,76,80. Moreover, BAY 41–2272 inhibited P-selectin expression on platelets and endothelial cells in vitro and reduced leukocyte rolling and adhesion in vivo, indicating a previously uncharacterized role for sGC in modulating the inflammatory response131. Collectively, the studies discussed above indicate that sGC stimulators might provide considerable therapeutic benefits in atherosclerosis and restenosis, as well as other inflammatory cardiovascular disorders.
Erectile dysfunction
Erectile dysfunction (ED) is the most common sexual dysfunction in men, affecting as many as 15–30 million people in the US alone132. The physiological process of erection is governed by a complex interplay between sympathetic, parasympathetic and nitrergic nerves, neurotransmitters, blood vessels and cavernous muscles133,134. It is well established that NO, which is synthesized during sexual stimulation in the nitrergic nerve terminals of the penis and also by the endothelial cells of blood vessels in the corpora cavernosa, is pivotal in the control of erectile function through its activating effects on the sGC–cGMP axis, leading to vascular smooth-muscle relaxation and penile erection135,136. The importance of this pathway is also supported by the inhibitory effect of both sGC and NOS inhibitors on NO-mediated relaxation of the corpus cavernosum, and by the relaxant effects of NO donors and PDE5 inhibitors on the corpus cavernosum133,134. Indeed, the NO-dependent increase in intra-cellular cGMP following PDE5 inhibition by sildenafil has been shown to be an effective approach in the treatment of ED137. However, nearly 30% of patients with ED do not respond to the PDE5 inhibitor therapy, implying that endogenous NO production is impaired to such an extent that inhibition of cGMP degradation provides no significant benefit. Under such conditions the use of direct, NO-independent stimulators of sGC might represent a promising alternative approach in the treatment of ED.
Studies with YC-1 have demonstrated that this compound is effective in relaxing the rat corpus cavernosum tissue in vitro and in enhancing erection induced by stimulation of the cavernous nerve and apomorphine in vivo78,138–140. The sGC stimulator A-350619 also relaxed isolated cavernosum tissue strips and induced penile erection in a conscious rat model78. Furthermore, the relaxation effect of both YC-1 and A-350619 on isolated cavernosum tissue was potentiated by the NO donor SNP78. BAY 41–2272 has also been shown to induce rabbit and human corpus cavernosum relaxation in vitro, and is ∼30-fold more potent than YC-1141,142. Importantly, BAY 41–2272 enhanced nitrergic relaxations induced by electrical-field stimulation at concentrations known to have no PDE5 inhibitory activity69,142, further supporting the concept that BAY 41–2272 synergizes with NO. The effects of BAY 41–2272 and sildenafil on the anococcygeus muscle isolated from streptozotocin-induced diabetic rats, in which nitrergic relaxation responses are decreased, have been compared73. The residual nitrergic relaxation was enhanced by BAY 41–2272 but not by sildenafil. These results confirm the notion that endogenous NO derived from nitrergic nerves is significantly decreased in diabetes, and suggest that sGC stimulators could be more useful than PDE5 inhibitors in the treatment of diabetes-induced ED. Even when endogenous NO release was pharmacologically inhibited, BAY 41–2272 restored relaxant responses142. Moreover, BAY 41–2272 has been shown to cause penile erection in rabbits in vivo, producing a synergistic effect when combined with SNP143. The latter finding is very important because these new compounds are expected to enhance NO–sGC–cGMP signalling particularly during sexual stimulation, thereby facilitating natural penile erection. Taken together, this experimental evidence indicates that the use of sGC stimulators might represent a novel, promising strategy for the therapy of ED.
Renal fibrosis and failure
Renal fibrosis is the final common manifestation of a wide variety of chronic kidney diseases. Irrespective of the initial causes, progressive chronic kidney disease often results in widespread tissue scarring that leads to the complete destruction of kidney parenchyma and end-stage renal failure, a devastating condition that requires dialysis or kidney replacement144. There is a need for novel pharmacological approaches to delay or even prevent the onset of chronic kidney disease and fibrosis. Evidence suggests that the most relevant pathophysiological events that cause end-stage renal failure involve increased production of extracellular matrix by mesangial cells secondary to elevated expression of the profibrotic cytokine TGFβ, which is driven in a large part by increased angiotensin signalling144.
Based on the previously reported efficacy of NO donors in preventing matrix accumulation and tissue injury145, recent experimental studies evaluated whether an elevation in intracellular cGMP by direct stimulation of sGC would ameliorate renal disease. Administering BAY 41–2272 to rats with an acute form of glomerulonephritis attenuated renal dysfunction, as determined by the presence of proteinuria, an effect that correlated with decreased TGFβ production, matrix deposition and macrophage infiltration146. A subsequent study demonstrated that BAY 41–2272 elevated cGMP levels in mesangial cells, thereby reducing their proliferation and matrix production147. Interestingly, the disease process itself upregulates sGC protein expression with a concomitant increase in cGMP levels146,147, indicating that this effect could represent an endogenous protective mechanism, which adds further weight to the validity of utilizing pharmacological sGC stimulators to prevent renal disease.
Subsequent investigations in a chronic model of glomerulonephritis confirmed that BAY 41–2272 protects the kidney from progressive sclerosis and matrix deposition by limiting TGFβ expression148,149. The protective effect achieved by elevating cGMP via direct sGC stimulation with BAY 41–2272 was far superior to that produced by preventing degradation of cGMP using the PDE inhibitor pentoxifylline149. Furthermore, in rats with subtotal nephrectomy (a model of chronic renal failure), treatment with BAY 58–2667 for 18 weeks lowered blood pressure, reduced left ventricular hypertrophy and cardiac arterial wall thickness, and slowed the progression of renal disease150. In summary, these preclinical studies suggest that sGC stimulation or activation could be useful for the management of patients with chronic renal disease.
Liver cirrhosis
Cirrhosis, a pathological condition defined by deranged hepatic architecture, is the final common pathway of nearly all chronic diseases of the liver. It is now recognized that hepatic stellate cells are primarily responsible for hepatic fibrosis and subsequent progression to cirrhosis. Although substantial progress has been made in understanding the pathogenesis of hepatic fibrosis during the past 20 years, the success of current pharmacological therapies is very limited151.
The NO-dependent activation of sGC followed by an increase in intracellular cGMP has been shown to reduce hepatic stellate cell activation152. BAY 60–2770, a close chemical analogue of BAY 58–2667, has been studied in rat models of liver fibrosis (a pig serum model, a carbon tetrachloride model and an accelerated model that combines both stimuli). Oral treatment with BAY 60–2770 in doses that do not affect systemic blood pressure prevented the increase in hepatic fibrous collagen and total collagen accumulation153. These preliminary data indicate that direct activation of sGC might provide a novel approach for the treatment of liver fibrosis of necro-inflammatory and immunological origin.
Conclusions
The NO-independent stimulators and activators of sGC represent one of the major innovations in drug discovery in recent years. The first group of these compounds comprises the haem-dependent sGC stimulators (including YC-1, BAY 41–2272, BAY 41–8543, A-350619 and CFM-1571). These compounds show a strong synergy with NO and a loss of activation after oxidation or removal of the prosthetic haem moiety of sGC. The second group comprises the sGC activators (including BAY 58–2667 and HMR-1766), which have been found to require neither NO nor haem, and demonstrate even more pronounced action on the oxidized form of sGC. Emerging preclinical evidence suggests that stimulators and activators of sGC might offer a significant advantage over and/or increase the efficacy of current therapies in modulating NO–sGC–cGMP signalling in various forms of cardiovascular, pulmonary, endothelial, renal, hepatic and sexual dysfunctions. Besides their potential use as therapeutics, these compounds might be utilized as vascular functional diagnostics to elucidate vascular oxidative stress and endothelial dysfunction. Further evaluation of these drugs and their alternative routes of administration in animal models of chronic diseases, such as chronic heart failure and chronic pulmonary hypertension, will be the next step of preclinical testing. In addition, the possibility of adverse effects has to be addressed. The sGC activators and the second-generation sGC stimulators are now entering clinical development. It remains to be established whether these two novel therapeutic modalities will fulfill their initial promise in the clinical arena.
Acknowledgments
The authors thank P. Sexton for generating the sGC homology model. This publication was supported in part by the National Heart, Lung, and Blood Institute and the Intramural Research Program of National Institutes of Health (USA) and the Alexander von Humboldt Foundation (Germany).
- Haem-binding domain
A conserved domain, present in various proteins that are involved in gas and/or redox sensing, that can bind a prosthetic haem moiety, which is, in turn, responsible for binding gaseous ligands such as NO, O2 or CO.
- Prosthetic haem moiety
Haem is the prosthetic group of various gas and/or redox-sensing proteins and consists of a large heterocyclic organic ring called porphyrin and a central metal atom (for example, iron, copper or zinc).
- Redox state
A term used to reflect the oxidation state of the prosthetic haem moiety of sGC, which can exist in a reduced (ferrous; Fe2+) or an oxidized (ferric; Fe3+) state. The oxidized form of sGC is insensitive to NO.
- Reactive oxygen species
Collective term for highly reactive molecules formed by the incomplete one-electron reduction of oxygen, and the products of further potential reactions (for example with NO). They include singlet oxygen, superoxide, peroxides, hydroxyl radical, peroxynitrite and hypochlorous acid.
- EC50
The molecular concentration of an agonist that is required to produce 50% of the maximum response to that agonist.
- Kd
The equilibrium dissociation constant of a compound that reflects the concentration needed to reach half-maximal saturation of binding sites. Kd reflects the strength of binding of a compound to its specific binding site.
- Spatial structure
The occupation of three-dimensional space by a given chemical compound or protein.
- Porphyrin
A heterocyclic macrocycle made from four pyrrole rings joined by methine bridges (=CH−).
- Methaemoglobinaemia
The presence of increased concentrations of methaemoglobin (resulting from the oxidation of haemoglobin) in blood. Methaemoglobin lacks the electron that is needed to form a bond with oxygen and is therefore incapable of oxygen transport to tissues.
- Cardiac index
The volume of blood pumped by the heart every minute normalized to body surface area.
- Restenosis
A re-narrowing of an artery at the site of angioplasty or stent placement.
- Autocoid
A substance, such as a hormone, produced in one part of an organism and transported by blood or lymph to another part of the organism where it exerts a physiological effect.
- Neointima
A new layer of endothelial cells on the inner surface of a blood vessel graft or a vascular prosthesis.
- Mesangial cells
Phagocytic cells found in the mesangium of the glomerular capsule of the kidney that are thought to aid in cleaning the filtration apparatus.
- Hepatic stellate cells
Cells that reside between the parenchymal cells and sinusoidal endothelial cells of the hepatic lobule and are the major storage site of vitamin A. In chronic liver injury (for example, chronic hepatitis), these hepatic cells produce collagen and other extracellular matrix proteins that lead to liver fibrosis and cirrhosis.
Footnotes
Competing interests statement
The authors declare competing financial interests: see Web version for details.
DATABASES
The following terms in this article are linked online to:
Entrez Gene:
http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene
sGC | MMP2 | MMP9 | PDE5
FURTHER INFORMATION
International Conference on cGMP:
Access to this links box is available online.
References
- 1.Feelisch M. The use of nitric oxide donors in pharmacological studies. Naunyn Schmiedebergs Arch Pharmacol. 1998;358:113–122. doi: 10.1007/pl00005231. [DOI] [PubMed] [Google Scholar]
- 2.Chen Z, Zhang J, Stamler JS. Identification of the enzymatic mechanism of nitroglycerin bioactivation. Proc Natl Acad Sci USA. 2002;99:8306–8311. doi: 10.1073/pnas.122225199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Y, et al. Mitochondrial aldehyde dehydrogenase-2 (ALDH2) Glu504Lys polymorphism contributes to the variation in efficacy of sublingual nitroglycerin. J Clin Invest. 2006;116:506–511. doi: 10.1172/JCI26564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Munzel T, Daiber A, Mulsch A. Explaining the phenomenon of nitrate tolerance. Circ Res. 2005;97:618–628. doi: 10.1161/01.RES.0000184694.03262.6d. [DOI] [PubMed] [Google Scholar]
- 5.Dikalov S, Fink B, Skatchkov M, Stalleicken D, Bassenge E. Formation of reactive oxygen species by pentaerithrityltetranitrate and glyceryl trinitrate in vitro and development of nitrate tolerance. J Pharmacol Exp Ther. 1998;286:938–944. [PubMed] [Google Scholar]
- 6.Hofmann F, Feil R, Kleppisch T, Schlossmann J. Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol Rev. 2006;86:1–23. doi: 10.1152/physrev.00015.2005. [DOI] [PubMed] [Google Scholar]
- 7.Beavo JA. Cyclic nucleotide phosphodiesterases: functional implications of multiple isoforms. Physiol Rev. 1995;75:725–748. doi: 10.1152/physrev.1995.75.4.725. [DOI] [PubMed] [Google Scholar]
- 8.Agullo L, et al. Membrane association of nitric oxide-sensitive guanylyl cyclase in cardiomyocytes. Cardiovasc Res. 2005;68:65–74. doi: 10.1016/j.cardiores.2005.05.021. [DOI] [PubMed] [Google Scholar]
- 9.Zabel U, et al. Calcium-dependent membrane association sensitizes soluble guanylyl cyclase to nitric oxide. Nature Cell Biol. 2002;4:307–311. doi: 10.1038/ncb775. This study describes membrane association of sGC and the spatial confinement of enzymes involved in the NO–sGC–cGMP signalling pathway. [DOI] [PubMed] [Google Scholar]
- 10.Burette A, Zabel U, Weinberg RJ, Schmidt HHHW, Valtschanoff JG. Synaptic localization of nitric oxide synthase and soluble guanylyl cyclase in the hippocampus. J Neurosci. 2002;22:8961–8970. doi: 10.1523/JNEUROSCI.22-20-08961.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Russwurm M, Wittau N, Koesling D. Guanylyl cyclase/PSD-95 interaction: targeting of the nitric oxide-sensitive α2β1 guanylyl cyclase to synaptic membranes. J Biol Chem. 2001;276:44647–44652. doi: 10.1074/jbc.M105587200. [DOI] [PubMed] [Google Scholar]
- 12.Zabel U, Hausler C, Weeger M, Schmidt HHHW. Homodimerization of soluble guanylyl cyclase subunits. Dimerization analysis using a glutathione s-transferase affinity tag. J Biol Chem. 1999;274:18149–18152. doi: 10.1074/jbc.274.26.18149. [DOI] [PubMed] [Google Scholar]
- 13.Mayer B, Koesling D. cGMP signalling beyond nitric oxide. Trends Pharmacol Sci. 2001;22:546–548. doi: 10.1016/s0165-6147(00)01889-7. [DOI] [PubMed] [Google Scholar]
- 14.Zabel U, Weeger M, La M, Schmidt HHHW. Human soluble guanylate cyclase: functional expression and revised isoenzyme family. Biochem J. 1998;335:51–57. doi: 10.1042/bj3350051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iyer LM, Anantharaman V, Aravind L. Ancient conserved domains shared by animal soluble guanylyl cyclases and bacterial signaling proteins. BMC Genomics. 2003;4:5. doi: 10.1186/1471-2164-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nioche P, et al. Femtomolar sensitivity of a NO sensor from Clostridium botulinum. Science. 2004;306:1550–1553. doi: 10.1126/science.1103596. The first crystallization study of a prokaryotic homologue of the sGC haem-binding domain termed SONO. [DOI] [PubMed] [Google Scholar]
- 17.Pellicena P, Karow DS, Boon EM, Marletta MA, Kuriyan J. Crystal structure of an oxygen-binding heme domain related to soluble guanylate cyclases. Proc Natl Acad Sci USA. 2004;101:12854–12859. doi: 10.1073/pnas.0405188101. The first crystallization study of a prokaryotic homologue of the sGC haem-binding domain termed HNOX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmidt PM, Schramm M, Schroder H, Wunder F, Stasch JP. Identification of residues crucially involved in the binding of the heme moiety of soluble guanylate cyclase. J Biol Chem. 2004;279:3025–3032. doi: 10.1074/jbc.M310141200. The first description of the haem-binding motif Y-xS-x-R. This work also postulates a mechanism of sGC activation by the haem-independent activator BAY 58–2667 and explains its selective interaction with the oxidized enzyme. [DOI] [PubMed] [Google Scholar]
- 19.Wedel B, et al. Mutation of His-105 in the β1 subunit yields a nitric oxide-insensitive form of soluble guanylyl cyclase. Proc Natl Acad Sci USA. 1994;91:2592–2596. doi: 10.1073/pnas.91.7.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sunahara RK, et al. Exchange of substrate and inhibitor specificities between adenylyl and guanylyl cyclases. J Biol Chem. 1998;273:16332–16338. doi: 10.1074/jbc.273.26.16332. [DOI] [PubMed] [Google Scholar]
- 21.Tesmer JJ, Sunahara RK, Gilman AG, Sprang SR. Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gsα–GTPγS. Science. 1997;278:1907–1916. doi: 10.1126/science.278.5345.1907. [DOI] [PubMed] [Google Scholar]
- 22.Hobbs A. Soluble guanylate cyclase. Emerg Therap Targets. 2000:735–749. [Google Scholar]
- 23.Winger JA, Marletta MA. Expression and characterization of the catalytic domains of soluble guanylate cyclase: interaction with the heme domain. Biochemistry. 2005;44:4083–4090. doi: 10.1021/bi047601d. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt HHHW, Walter U. NO at work. Cell. 1994;78:919–925. doi: 10.1016/0092-8674(94)90267-4. [DOI] [PubMed] [Google Scholar]
- 25.Li Z, et al. A stimulatory role for cGMP-dependent protein kinase in platelet activation. Cell. 2003;112:77–86. doi: 10.1016/s0092-8674(02)01254-0. [DOI] [PubMed] [Google Scholar]
- 26.Gambaryan S, et al. Potent inhibition of human platelets by cGMP analogs independent of cGMP-dependent protein kinase. Blood. 2004;103:2593–2600. doi: 10.1182/blood-2003-09-3349. [DOI] [PubMed] [Google Scholar]
- 27.Schwarz UR, Walter U, Eigenthaler M. Taming platelets with cyclic nucleotides. Biochem Pharmacol. 2001;62:1153–1161. doi: 10.1016/s0006-2952(01)00760-2. [DOI] [PubMed] [Google Scholar]
- 28.Feil R, Feil S, Hofmann F. A heretical view on the role of NO and cGMP in vascular proliferative diseases. Trends Mol Med. 2005;11:71–75. doi: 10.1016/j.molmed.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 29.Wolfsgruber W, et al. A proatherogenic role for cGMP-dependent protein kinase in vascular smooth muscle cells. Proc Natl Acad Sci USA. 2003;100:13519–13524. doi: 10.1073/pnas.1936024100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Melichar VO, et al. Reduced cGMP signaling associated with neointimal proliferation and vascular dysfunction in late-stage atherosclerosis. Proc Natl Acad Sci USA. 2004;101:16671–16676. doi: 10.1073/pnas.0405509101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sinnaeve P, et al. Soluble guanylate cyclase α1 and β1 gene transfer increases NO responsiveness and reduces neointima formation after balloon injury in rats via antiproliferative and antimigratory effects. Circ Res. 2001;88:103–109. doi: 10.1161/01.res.88.1.103. [DOI] [PubMed] [Google Scholar]
- 32.Foerster J, Harteneck C, Malkewitz J, Schultz G, Koesling D. A functional heme-binding site of soluble guanylyl cyclase requires intact N-termini of α1 and β1 subunits. Eur J Biochem. 1996;240:380–386. doi: 10.1111/j.1432-1033.1996.0380h.x. [DOI] [PubMed] [Google Scholar]
- 33.Ignarro LJ, Adams JB, Horwitz PM, Wood KS. Activation of soluble guanylate cyclase by NO-hemo-proteins involves NO-heme exchange. Comparison of heme-containing and heme-deficient enzyme forms. J Biol Chem. 1986;261:4997–5002. [PubMed] [Google Scholar]
- 34.Ignarro LJ, Wood KS, Wolin MS. Activation of purified soluble guanylate cyclase by protoporphyrin IX. Proc Natl Acad Sci USA. 1982;79:2870–2873. doi: 10.1073/pnas.79.9.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ballou DP, Zhao Y, Brandish PE, Marletta MA. Revisiting the kinetics of nitric oxide (NO) binding to soluble guanylate cyclase: the simple NO-binding model is incorrect. Proc Natl Acad Sci USA. 2002;99:12097–12101. doi: 10.1073/pnas.192209799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao Y, Brandish PE, Ballou DP, Marletta MA. A molecular basis for nitric oxide sensing by soluble guanylate cyclase. Proc Natl Acad Sci USA. 1999;96:14753–14758. doi: 10.1073/pnas.96.26.14753. This paper suggests the existence of a second non-haem NO-binding site to explain the kinetics of the NO-induced sGC activation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cary SP, Winger JA, Marletta MA. Tonic and acute nitric oxide signaling through soluble guanylate cyclase is mediated by nonheme nitric oxide, ATP, and GTP. Proc Natl Acad Sci USA. 2005;102:13064–13069. doi: 10.1073/pnas.0506289102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Russwurm M, Koesling D. NO activation of guanylyl cyclase. EMBO J. 2004;23:4443–4450. doi: 10.1038/sj.emboj.7600422. The first evidence that NO-bound sGC can exist in a virtually inactive state. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feelisch M, Kotsonis P, Siebe J, Clement B, Schmidt HHHW. The soluble guanylyl cyclase inhibitor 1H-[1,2,4]oxadiazolo[4,3,-a] quinoxalin-1-one is a nonselective heme protein inhibitor of nitric oxide synthase and other cytochrome P-450 enzymes involved in nitric oxide donor bioactivation. Mol Pharmacol. 1999;56:243–253. doi: 10.1124/mol.56.2.243. [DOI] [PubMed] [Google Scholar]
- 40.Hwang TL, Wu CC, Teng CM. Comparison of two soluble guanylyl cyclase inhibitors, methylene blue and ODQ, on sodium nitroprusside-induced relaxation in guinea-pig trachea. Br J Pharmacol. 1998;125:1158–1163. doi: 10.1038/sj.bjp.0702181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kannan MS, Johnson DE. Modulation of nitric oxide-dependent relaxation of pig tracheal smooth muscle by inhibitors of guanylyl cyclase and calcium activated potassium channels. Life Sci. 1995;56:2229–2238. doi: 10.1016/0024-3205(95)00212-o. [DOI] [PubMed] [Google Scholar]
- 42.Mayer B, Brunner F, Schmidt K. Novel actions of methylene blue. Eur Heart J. 1993;14 (Suppl 1):22–26. [PubMed] [Google Scholar]
- 43.Olesen SP, et al. Characterization of NS 2028 as a specific inhibitor of soluble guanylyl cyclase. Br J Pharmacol. 1998;123:299–309. doi: 10.1038/sj.bjp.0701603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schrammel A, Behrends S, Schmidt K, Koesling D, Mayer B. Characterization of 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one as a heme-site inhibitor of nitric oxide-sensitive guanylyl cyclase. Mol Pharmacol. 1996;50:1–5. [PubMed] [Google Scholar]
- 45.Zhao Y, et al. Inhibition of soluble guanylate cyclase by ODQ. Biochemistry. 2000;39:10848–10854. doi: 10.1021/bi9929296. [DOI] [PubMed] [Google Scholar]
- 46.Stasch JP, et al. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J Clin Invest. 2006;116:2552–256. doi: 10.1172/JCI28371. This is the first demonstration that oxidized sGC exists in vivo and increases under conditions of oxidative stress associated with cardiovascular diseases. This study also shows the increased potency of BAY 58–2667 under these conditions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ko FN, Wu CC, Kuo SC, Lee FY, Teng CM. YC-1, a novel activator of platelet guanylate cyclase. Blood. 1994;84:4226–4233. The first description of a non-NO-based sGC stimulator. [PubMed] [Google Scholar]
- 48.Friebe A, Schultz G, Koesling D. Sensitizing soluble guanylyl cyclase to become a highly CO-sensitive enzyme. EMBO J. 1996;15:6863–6868. [PMC free article] [PubMed] [Google Scholar]
- 49.Hoenicka M, et al. Purified soluble guanylyl cyclase expressed in a baculovirus/Sf9 system: stimulation by YC-1, nitric oxide, and carbon monoxide. J Mol Med. 1999;77:14–23. doi: 10.1007/s001090050292. [DOI] [PubMed] [Google Scholar]
- 50.Mulsch A, et al. Effect of YC-1, an NO-independent, superoxide-sensitive stimulator of soluble guanylyl cyclase, on smooth muscle responsiveness to nitrovasodilators. Br J Pharmacol. 1997;120:681–689. doi: 10.1038/sj.bjp.0700982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fernandez PA, Bellamy T, Kling M, Madge DJ, Selwood DL. A convenient route to the solubkle guanylate cyclase activator YC-1 and its N2 regioisomer. Heterocycles. 2001;55:1813–1816. [Google Scholar]
- 52.Lee FY, et al. Synthesis of 1-benzyl-3-(5′-hydroxymethyl-2′-furyl)indazole analogues as novel antiplatelet agents. J Med Chem. 2001;44:3746–3749. doi: 10.1021/jm010001h. [DOI] [PubMed] [Google Scholar]
- 53.Selwood DL, et al. Synthesis and biological evaluation of novel pyrazoles and indazoles as activators of the nitric oxide receptor, soluble guanylate cyclase. J Med Chem. 2001;44:78–93. doi: 10.1021/jm001034k. [DOI] [PubMed] [Google Scholar]
- 54.Straub A, et al. NO-independent stimulators of soluble guanylate cyclase. Bioorg Med Chem Lett. 2001;11:781–784. doi: 10.1016/s0960-894x(01)00073-7. [DOI] [PubMed] [Google Scholar]
- 55.Zhang HQ, Zhiren X, Teodozyj K, Dinges J. A concise synthesis of ortho-substituted aryl-acrylamide- potent activators od soluble guanylyl cyclase. Tetrahedron Lett. 2003;44:8661–8663. [Google Scholar]
- 56.Wu CC, Ko FN, Kuo SC, Lee FY, Teng CM. YC-1 inhibited human platelet aggregation through NO-independent activation of soluble guanylate cyclase. Br J Pharmacol. 1995;116:1973–1978. doi: 10.1111/j.1476-5381.1995.tb16400.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stasch JP, et al. NO-independent regulatory site on soluble guanylate cyclase. Nature. 2001;410:212–215. doi: 10.1038/35065611. The first biochemical and pharmacological characterization of BAY 41–2272 as a novel NO-independent but haem-dependent stimulator of sGC. [DOI] [PubMed] [Google Scholar]
- 58.Martin E, Lee YC, Murad F. YC-1 activation of human soluble guanylyl cyclase has both heme-dependent and heme-independent components. Proc Natl Acad Sci USA. 2001;98:12938–12942. doi: 10.1073/pnas.231486198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Garthwaite J, et al. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol Pharmacol. 1995;48:184–188. [PubMed] [Google Scholar]
- 60.Russwurm M, Mergia E, Mullershausen F, Koesling D. Inhibition of deactivation of NO-sensitive guanylyl cyclase accounts for the sensitizing effect of YC-1. J Biol Chem. 2002;277:24883–24888. doi: 10.1074/jbc.M110570200. [DOI] [PubMed] [Google Scholar]
- 61.Stone JR, Marletta MA. Synergistic activation of soluble guanylate cyclase by YC-1 and carbon monoxide: implications for the role of cleavage of the iron-histidine bond during activation by nitric oxide. Chem Biol. 1998;5:255–261. doi: 10.1016/s1074-5521(98)90618-4. [DOI] [PubMed] [Google Scholar]
- 62.Denninger JW, et al. Interaction of soluble guanylate cyclase with YC-1: kinetic and resonance Raman studies. Biochemistry. 2000;39:4191–4198. doi: 10.1021/bi992332q. [DOI] [PubMed] [Google Scholar]
- 63.Galle J, et al. Effects of the soluble guanylyl cyclase activator, YC-1, on vascular tone, cyclic GMP levels and phosphodiesterase activity. Br J Pharmacol. 1999;127:195–203. doi: 10.1038/sj.bjp.0702495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Friebe A, Russwurm M, Mergia E, Koesling D. A point-mutated guanylyl cyclase with features of the YC-1-stimulated enzyme: implications for the YC-1 binding site? Biochemistry. 1999;38:15253–15257. doi: 10.1021/bi9908944. [DOI] [PubMed] [Google Scholar]
- 65.Friebe A, et al. Functions of conserved cysteines of soluble guanylyl cyclase. Biochemistry. 1997;36:1194–1198. doi: 10.1021/bi962047w. [DOI] [PubMed] [Google Scholar]
- 66.Yazawa S, Tsuchiya H, Hori H, Makino R. Functional characterization of two nucleotide binding sites in soluble guanylate cyclase. J Biol Chem. 2006;281:21763–21770. doi: 10.1074/jbc.M508983200. [DOI] [PubMed] [Google Scholar]
- 67.Stasch JP, et al. Pharmacological actions of a novel NO-independent guanylyl cyclase stimulator, BAY 41–8543: in vitro studies. Br J Pharmacol. 2002;135:333–343. doi: 10.1038/sj.bjp.0704484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stasch JP, Dembowsky K, Perzborn E, Stahl E, Schramm M. Cardiovascular actions of a novel NO-independent guanylyl cyclase stimulator, BAY 41–8543: in vivo studies. Br J Pharmacol. 2002;135:344–355. doi: 10.1038/sj.bjp.0704483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bischoff E, Stasch JP. Effects of the sGC stimulator BAY 41-2272 are not mediated by phosphodiesterase 5 inhibition. Circulation. 2004;110:e320–321. doi: 10.1161/01.CIR.0000142209.28862.12. [DOI] [PubMed] [Google Scholar]
- 70.Schmidt P, Schramm M, Schroder H, Stasch JP. Mechanisms of nitric oxide independent activation of soluble guanylyl cyclase. Eur J Pharmacol. 2003;468:167–174. doi: 10.1016/s0014-2999(03)01674-1. [DOI] [PubMed] [Google Scholar]
- 71.Mullershausen F, Russwurm M, Friebe A, Koesling D. Inhibition of phosphodiesterase type 5 by the activator of nitric oxide-sensitive guanylyl cyclase BAY 41–2272. Circulation. 2004;109:1711–1713. doi: 10.1161/01.CIR.0000126286.47618.BD. [DOI] [PubMed] [Google Scholar]
- 72.Evgenov OV, et al. Soluble guanylate cyclase activator reverses acute pulmonary hypertension and augments the pulmonary vasodilator response to inhaled nitric oxide in awake lambs. Circulation. 2004;110:2253–2259. doi: 10.1161/01.CIR.0000144469.01521.8A. The first in vivo demonstration that a sGC stimulator, BAY 41–2272, attenuates pulmonary hypertension and also enhances pulmonary vasodilatation induced by inhaling gaseous NO. [DOI] [PubMed] [Google Scholar]
- 73.Kalsi JS, Ralph DJ, Madge DJ, Kell PD, Cellek S. A comparative study of sildenafil, NCX-911 and BAY41–2272 on the anococcygeus muscle of diabetic rats. Int J Impot Res. 2004;16:479–485. doi: 10.1038/sj.ijir.3901224. [DOI] [PubMed] [Google Scholar]
- 74.Hering KW, Artz JD, Pearson WH, Marletta MA. The design and synthesis of YC-1 analogues as probes for soluble guanylate cyclase. Bioorg Med Chem Lett. 2006;16:618–621. doi: 10.1016/j.bmcl.2005.10.093. [DOI] [PubMed] [Google Scholar]
- 75.Bawankule DU, et al. BAY 41–2272 [5-cyclopropyl-2-[1-(2-fluoro-benzyl)-1H-pyrazolo[3,4-b]pyridine-3-yl]pyrimidin-4-ylamine]-induced dilation in ovine pulmonary artery: role of sodium pump. J Pharmacol Exp Ther. 2005;314:207–213. doi: 10.1124/jpet.105.083824. [DOI] [PubMed] [Google Scholar]
- 76.Hobbs AJ, Moncada S. Antiplatelet properties of a novel, non-NO-based soluble guanylate cyclase activator, BAY 41–2272. Vascul Pharmacol. 2003;40:149–154. doi: 10.1016/s1537-1891(03)00046-6. [DOI] [PubMed] [Google Scholar]
- 77.Wu CH, Chang WC, Chang GY, Kuo SC, Teng CM. The inhibitory mechanism of YC-1, a benzyl indazole, on smooth muscle cell proliferation: an in vitro and in vivo study. J Pharmacol Sci. 2004;94:252–260. doi: 10.1254/jphs.94.252. [DOI] [PubMed] [Google Scholar]
- 78.Miller LN, et al. A-350619: a novel activator of soluble guanylyl cyclase. Life Sci. 2003;72:1015–1025. doi: 10.1016/s0024-3205(02)02361-5. Biochemical and pharmacological characterization of A-350619 as a NO-independent but haem-dependent stimulator of sGC. [DOI] [PubMed] [Google Scholar]
- 79.Nakane M. Soluble guanylyl cyclase: physiological role as an NO receptor and the potential molecular target for therapeutic application. Clin Chem Lab Med. 2003;41:865–870. doi: 10.1515/CCLM.2003.131. [DOI] [PubMed] [Google Scholar]
- 80.Stasch JP, et al. NO- and haem-independent activation of soluble guanylyl cyclase: molecular basis and cardiovascular implications of a new pharmacological principle. Br J Pharmacol. 2002;136:773–783. doi: 10.1038/sj.bjp.0704778. The first biochemical and pharmacological characterization of a compound, BAY 58–2667, capable of activating the haem-free and/or oxidized form of sGC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wunder F, et al. A cell-based cGMP assay useful for ultra-high-throughput screening and identification of modulators of the nitric oxide/cGMP pathway. Anal Biochem. 2005;339:104–112. doi: 10.1016/j.ab.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 82.Schindler U, et al. Biochemistry and pharmacology of novel anthranilic acid derivatives activating heme-oxidized soluble guanylyl cyclase. Mol Pharmacol. 2006;69:1260–1268. doi: 10.1124/mol.105.018747. Biochemical and pharmacological characterization of a novel structural class (HMR-1766 and S-3448) capable of activating the oxidized and/or haem-free forms of sGC. [DOI] [PubMed] [Google Scholar]
- 83.Schmidt PM, Rothkegel C, Wunder F, Schroder H, Stasch JP. Residues stabilizing the heme moiety of the nitric oxide sensor soluble guanylate cyclase. Eur J Pharmacol. 2005;513:67–74. doi: 10.1016/j.ejphar.2005.02.046. [DOI] [PubMed] [Google Scholar]
- 84.Hobbs AJ. Soluble guanylate cyclase: an old therapeutic target re-visited. Br J Pharmacol. 2002;136:637–640. doi: 10.1038/sj.bjp.0704779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fields LE, et al. The burden of adult hypertension in the United States 1999 to 2000: a rising tide. Hypertension. 2004;44:398–404. doi: 10.1161/01.HYP.0000142248.54761.56. [DOI] [PubMed] [Google Scholar]
- 86.Panza JA, Casino PR, Badar DM, Quyyumi AA. Effect of increased availability of endothelium-derived nitric oxide precursor on endothelium-dependent vascular relaxation in normal subjects and in patients with essential hypertension. Circulation. 1993;87:1475–1481. doi: 10.1161/01.cir.87.5.1475. [DOI] [PubMed] [Google Scholar]
- 87.Schlaich MP, et al. Impaired L-arginine transport and endothelial function in hypertensive and genetically predisposed normotensive subjects. Circulation. 2004;110:3680–3686. doi: 10.1161/01.CIR.0000149748.79945.52. [DOI] [PubMed] [Google Scholar]
- 88.Taddei S, et al. Defective L-arginine-nitric oxide pathway in offspring of essential hypertensive patients. Circulation. 1996;94:1298–1303. doi: 10.1161/01.cir.94.6.1298. [DOI] [PubMed] [Google Scholar]
- 89.Kojda G, Kottenberg K, Hacker A, Noack E. Alterations of the vascular and the myocardial guanylate cyclase/cGMP-system induced by long-term hypertension in rats. Pharm Acta Helv. 1998;73:27–35. doi: 10.1016/s0031-6865(97)00044-7. [DOI] [PubMed] [Google Scholar]
- 90.Kloss S, Bouloumie A, Mulsch A. Aging and chronic hypertension decrease expression of rat aortic soluble guanylyl cyclase. Hypertension. 2000;35:43–47. [PubMed] [Google Scholar]
- 91.Ruetten H, Zabel U, Linz W, Schmidt HHHW. Downregulation of soluble guanylyl cyclase in young and aging spontaneously hypertensive rats. Circ Res. 1999;85:534–541. doi: 10.1161/01.res.85.6.534. This paper shows that, apart from NO synthesis and bioavailability, sGC can be affected in vascular disease models. [DOI] [PubMed] [Google Scholar]
- 92.Morawietz H, et al. Upregulation of vascular NAD(P)H oxidase subunit gp91phox and impairment of the nitric oxide signal transduction pathway in hypertension. Biochem Biophys Res Commun. 2001;285:1130–1135. doi: 10.1006/bbrc.2001.5312. [DOI] [PubMed] [Google Scholar]
- 93.Kagota S, et al. Disturbances in nitric oxide/cyclic guanosine monophosphate system in SHR/NDmcr-cp rats, a model of metabolic syndrome. Life Sci. 2006;78:1187–1196. doi: 10.1016/j.lfs.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 94.Brandes RP, et al. Increased nitrovasodilator sensitivity in endothelial nitric oxide synthase knockout mice: role of soluble guanylyl cyclase. Hypertension. 2000;35:231–236. doi: 10.1161/01.hyp.35.1.231. [DOI] [PubMed] [Google Scholar]
- 95.Rothermund L, Friebe A, Paul M, Koesling D, Kreutz R. Acute blood pressure effects of YC-1-induced activation of soluble guanylyl cyclase in normotensive and hypertensive rats. Br J Pharmacol. 2000;130:205–208. doi: 10.1038/sj.bjp.0703320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zanfolin M, et al. Protective effects of BAY 41–2272 (sGC stimulator) on hypertension, heart, and cardiomyocyte hypertrophy induced by chronic L-NAME treatment in rats. J Cardiovasc Pharmacol. 2006;47:391–395. [PubMed] [Google Scholar]
- 97.Rothkegel C, et al. Beyond NO and heme: biochemical and pharmacological opportunities. BMC Pharmacol. 2005;5:S18. [Google Scholar]
- 98.Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351:1655–1665. doi: 10.1056/NEJMra035488. [DOI] [PubMed] [Google Scholar]
- 99.Machado RF, et al. Nitric oxide and pulmonary arterial pressures in pulmonary hypertension. Free Radic Biol Med. 2004;37:1010–1017. doi: 10.1016/j.freeradbiomed.2004.06.039. [DOI] [PubMed] [Google Scholar]
- 100.Ichinose F, Roberts JD, Jr, Zapol WM. Inhaled nitric oxide: a selective pulmonary vasodilator: current uses and therapeutic potential. Circulation. 2004;109:3106–3111. doi: 10.1161/01.CIR.0000134595.80170.62. [DOI] [PubMed] [Google Scholar]
- 101.Chockalingam A, et al. Efficacy and optimal dose of sildenafil in primary pulmonary hypertension. Int J Cardiol. 2005;99:91–95. doi: 10.1016/j.ijcard.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 102.Michelakis E, et al. Oral sildenafil is an effective and specific pulmonary vasodilator in patients with pulmonary arterial hypertension: comparison with inhaled nitric oxide. Circulation. 2002;105:2398–2403. doi: 10.1161/01.cir.0000016641.12984.dc. [DOI] [PubMed] [Google Scholar]
- 103.Stocker C, et al. Intravenous sildenafil and inhaled nitric oxide: a randomised trial in infants after cardiac surgery. Intensive Care Med. 2003;29:1996–2003. doi: 10.1007/s00134-003-2016-4. [DOI] [PubMed] [Google Scholar]
- 104.Weimann J, et al. Sildenafil is a pulmonary vasodilator in awake lambs with acute pulmonary hypertension. Anesthesiology. 2000;92:1702–1712. doi: 10.1097/00000542-200006000-00030. [DOI] [PubMed] [Google Scholar]
- 105.Deruelle P, Grover TR, Abman SH. Pulmonary vascular effects of nitric oxide-cGMP augmentation in a model of chronic pulmonary hypertension in fetal and neonatal sheep. Am J Physiol Lung Cell Mol Physiol. 2005;289:L798–806. doi: 10.1152/ajplung.00119.2005. [DOI] [PubMed] [Google Scholar]
- 106.Deruelle P, Grover TR, Storme L, Abman SH. Effects of BAY 41–2272, a soluble guanylate cyclase activator, on pulmonary vascular reactivity in the ovine fetus. Am J Physiol Lung Cell Mol Physiol. 2005;288:L727–733. doi: 10.1152/ajplung.00409.2004. [DOI] [PubMed] [Google Scholar]
- 107.Boerrigter G, et al. Cardiorenal and humoral properties of a novel direct soluble guanylate cyclase stimulator BAY 41–2272 in experimental congestive heart failure. Circulation. 2003;107:686–689. doi: 10.1161/01.cir.0000055737.15443.f8. The first demonstration that a sGC stimulator, BAY 41–2272, attenuates experimental congestive heart failure. [DOI] [PubMed] [Google Scholar]
- 108.Deruelle P, et al. BAY 41–2272, a direct activator of soluble guanylate cyclase, reduces right ventricular hypertrophy and prevents pulmonary vascular remodeling during chronic hypoxia in neonatal rats. Biol Neonate. 2006;90:135–144. doi: 10.1159/000092518. [DOI] [PubMed] [Google Scholar]
- 109.Dumitrascu R, et al. Activation of soluble guanylate cyclase reverses experimental pulmonary hypertension and vascular remodeling. Circulation. 2006;113:286–295. doi: 10.1161/CIRCULATIONAHA.105.581405. [DOI] [PubMed] [Google Scholar]
- 110.Evgenov OV, et al. Inhalation of microparticles containing stimulators of soluble guanylate cyclase produces potent pulmonary vasodilation. Proc Am Thorac Soc. 2006;3:A686. [Google Scholar]
- 111.Austin S. Drug discovery technology Europe 2006- IBC’s Tenth Annual Conference and Exhibition. IDrugs. 2006;9:256–260. [PubMed] [Google Scholar]
- 112.Hunt S. ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure) J Am Coll Cardiol. 2005;46:e1–e82. doi: 10.1016/j.jacc.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 113.Hare JM, Stamler JS. NO/redox disequilibrium in the failing heart and cardiovascular system. J Clin Invest. 2005;115:509–517. doi: 10.1172/JCI200524459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pacher P, Schulz R, Liaudet L, Szabo C. Nitrosative stress and pharmacological modulation of heart failure. Trends Pharmacol Sci. 2005;26:302–310. doi: 10.1016/j.tips.2005.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ungvari Z, Gupte SA, Recchia FA, Batkai S, Pacher P. Role of oxidative-nitrosative stress and downstream pathways in various forms of cardiomyopathy and heart failure. Curr Vasc Pharmacol. 2005;3:221–229. doi: 10.2174/1570161054368607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Boerrigter G, Burnett JC., Jr Recent advances in natriuretic peptides in congestive heart failure. Expert Opin Investig Drugs. 2004;13:643–652. doi: 10.1517/13543784.13.6.643. [DOI] [PubMed] [Google Scholar]
- 117.Mulsch A, Bara A, Mordvintcev P, Vanin A, Busse R. Specificity of different organic nitrates to elicit NO formation in rabbit vascular tissues and organs in vivo. Br J Pharmacol. 1995;116:2743–2749. doi: 10.1111/j.1476-5381.1995.tb17236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Boerrigter G, Costello-Boerrigter LC, Lapp H, Stasch JP, Burnett JC. Co-activation of soluble and particulate guanylate cyclase by BAY 58–2667 and BNP enhances cardiorenal function in experimental heart failure. BMC Pharmacol. 2005;5:P5. [Google Scholar]
- 119.Strong JP, et al. Prevalence and extent of atherosclerosis in adolescents and young adults: implications for prevention from the Pathobiological Determinants of Atherosclerosis in Youth Study. JAMA. 1999;281:727–735. doi: 10.1001/jama.281.8.727. [DOI] [PubMed] [Google Scholar]
- 120.Ip JH, et al. Syndromes of accelerated atherosclerosis: role of vascular injury and smooth muscle cell proliferation. J Am Coll Cardiol. 1990;15:1667–1687. doi: 10.1016/0735-1097(90)92845-s. [DOI] [PubMed] [Google Scholar]
- 121.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 122.Serruys PW, Kutryk MJ, Ong AT. Coronary-artery stents. N Engl J Med. 2006;354:483–495. doi: 10.1056/NEJMra051091. [DOI] [PubMed] [Google Scholar]
- 123.Shears LL, et al. Efficient inhibition of intimal hyperplasia by adenovirus-mediated inducible nitric oxide synthase gene transfer to rats and pigs in vivo. J Am Coll Surg. 1998;187:295–306. doi: 10.1016/s1072-7515(98)00163-x. [DOI] [PubMed] [Google Scholar]
- 124.von der Leyen HE, et al. Gene therapy inhibiting neointimal vascular lesion: in vivo transfer of endothelial cell nitric oxide synthase gene. Proc Natl Acad Sci USA. 1995;92:1137–1141. doi: 10.1073/pnas.92.4.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Marks DS, et al. Inhibition of neointimal proliferation in rabbits after vascular injury by a single treatment with a protein adduct of nitric oxide. J Clin Invest. 1995;96:2630–2638. doi: 10.1172/JCI118328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Seki J, Nishio M, Kato Y, Motoyama Y, Yoshida K. FK409, a new nitric-oxide donor, suppresses smooth muscle proliferation in the rat model of balloon angioplasty. Atherosclerosis. 1995;117:97–106. doi: 10.1016/0021-9150(95)05563-c. [DOI] [PubMed] [Google Scholar]
- 127.Tulis DA, et al. YC-1, a benzyl indazole derivative, stimulates vascular cGMP and inhibits neointima formation. Biochem Biophys Res Commun. 2000;279:646–652. doi: 10.1006/bbrc.2000.3942. This study shows for the first time that a direct stimulator of sGC reduces post-angioplasty stenosis through endogenous NO-mediated, cGMP-dependent processes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Liu YN, et al. YC-1 [3-(5′-hydroxymethyl-2′-furyl)-1-benzyl indazole] inhibits neointima formation in balloon-injured rat carotid through suppression of expressions and activities of matrix metalloproteinases 2 and 9. J Pharmacol Exp Ther. 2006;316:35–41. doi: 10.1124/jpet.105.090563. [DOI] [PubMed] [Google Scholar]
- 129.Tulis DA. Salutary properties of YC-1 in the cardiovascular and hematological systems. Curr Med Chem Cardiovasc Hematol Agents. 2004;2:343–359. doi: 10.2174/1568016043356200. [DOI] [PubMed] [Google Scholar]
- 130.Tulis DA, Durante W, Schafer A. Nitric oxide-independent regulation of the vascular injury response. BMC Pharmacol. 2005;5:S26. [Google Scholar]
- 131.Ahluwalia A, et al. Antiinflammatory activity of soluble guanylate cyclase: cGMP-dependent down-regulation of P-selectin expression and leukocyte recruitment. Proc Natl Acad Sci USA. 2004;101:1386–1391. doi: 10.1073/pnas.0304264101. The first demonstration that a sGC stimulator attenuates vascular inflammation by inhibiting P-selectin expression and leukocyte recruitment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Feldman HA, Goldstein I, Hatzichristou DG, Krane RJ, McKinlay JB. Impotence and its medical and psychosocial correlates: results of the Massachusetts Male Aging Study. J Urol. 1994;151:54–61. doi: 10.1016/s0022-5347(17)34871-1. [DOI] [PubMed] [Google Scholar]
- 133.Andersson KE. Pharmacology of penile erection. Pharmacol Rev. 2001;53:417–450. [PubMed] [Google Scholar]
- 134.Andersson KE, Wagner G. Physiology of penile erection. Physiol Rev. 1995;75:191–236. doi: 10.1152/physrev.1995.75.1.191. [DOI] [PubMed] [Google Scholar]
- 135.Ignarro LJ, et al. Nitric oxide and cyclic GMP formation upon electrical field stimulation cause relaxation of corpus cavernosum smooth muscle. Biochem Biophys Res Commun. 1990;170:843–850. doi: 10.1016/0006-291x(90)92168-y. [DOI] [PubMed] [Google Scholar]
- 136.Rajfer J, Aronson WJ, Bush PA, Dorey FJ, Ignarro LJ. Nitric oxide as a mediator of relaxation of the corpus cavernosum in response to nonadrenergic, noncholinergic neurotransmission. N Engl J Med. 1992;326:90–94. doi: 10.1056/NEJM199201093260203. [DOI] [PubMed] [Google Scholar]
- 137.Boolell M, et al. Sildenafil: an orally active type 5 cyclic GMP-specific phosphodiesterase inhibitor for the treatment of penile erectile dysfunction. Int J Impot Res. 1996;8:47–52. [PubMed] [Google Scholar]
- 138.Hsieh GC, O’Neill AB, Moreland RB, Sullivan JP, Brioni JD. YC-1 potentiates the nitric oxide/ cyclic GMP pathway in corpus cavernosum and facilitates penile erection in rats. Eur J Pharmacol. 2003;458:183–189. doi: 10.1016/s0014-2999(02)02730-9. [DOI] [PubMed] [Google Scholar]
- 139.Mizusawa H, Hedlund P, Brioni JD, Sullivan JP, Andersson KE. Nitric oxide independent activation of guanylate cyclase by YC-1 causes erectile responses in the rat. J Urol. 2002;167:2276–2281. The first in vivo study showing that direct pharmacological stimulation of sGC evokes erectile response and enhances erection induced by neural stimulation. [PubMed] [Google Scholar]
- 140.Nakane M, et al. Activation of soluble guanylate cyclase causes relaxation of corpus cavernosum tissue: synergism of nitric oxide and YC-1. Int J Impot Res. 2002;14:121–127. doi: 10.1038/sj.ijir.3900843. [DOI] [PubMed] [Google Scholar]
- 141.Baracat JS, et al. Relaxing effects induced by the soluble guanylyl cyclase stimulator BAY 41–2272 in human and rabbit corpus cavernosum. Eur J Pharmacol. 2003;477:163–169. doi: 10.1016/j.ejphar.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 142.Kalsi JS, et al. BAY41–2272, a novel nitric oxide independent soluble guanylate cyclase activator, relaxes human and rabbit corpus cavernosum in vitro. J Urol. 2003;169:761–766. doi: 10.1097/01.ju.0000043880.58140.69. [DOI] [PubMed] [Google Scholar]
- 143.Bischoff E, Schramm M, Straub A, Feurer A, Stasch JP. BAY 41–2272: a stimulator of soluble guanylyl cyclase induces nitric oxide-dependent penile erection in vivo. Urology. 2003;61:464–467. doi: 10.1016/s0090-4295(02)02121-0. [DOI] [PubMed] [Google Scholar]
- 144.Gaedeke J, Neumayer HH, Peters H. Pharmacological management of renal fibrotic disease. Expert Opin Pharmacother. 2006;7:377–386. doi: 10.1517/14656566.7.4.377. [DOI] [PubMed] [Google Scholar]
- 145.Peters H, et al. NO mediates antifibrotic actions of l-arginine supplementation following induction of anti-thy1 glomerulonephritis. Kidney Int. 2003;64:509–518. doi: 10.1046/j.1523-1755.2003.00112.x. [DOI] [PubMed] [Google Scholar]
- 146.Peters H, et al. Expression and activity of soluble guanylate cyclase in injury and repair of anti-thy1 glomerulonephritis. Kidney Int. 2004;66:2224–2236. doi: 10.1111/j.1523-1755.2004.66012.x. The first in vivo evidence that pharmacological stimulation of sGC reduces fibrotic changes in glomerular disease. [DOI] [PubMed] [Google Scholar]
- 147.Hohenstein B, Daniel C, Wagner A, Stasch JP, Hugo C. Stimulation of soluble guanylyl cyclase inhibits mesangial cell proliferation and matrix accumulation in experimental glomerulonephritis. Am J Physiol Renal Physiol. 2005;288:F685–693. doi: 10.1152/ajprenal.00280.2004. [DOI] [PubMed] [Google Scholar]
- 148.Wang Y, et al. Stimulation of soluble guanylate cyclase slows progression in anti-thy1-induced chronic glomerulosclerosis. Kidney Int. 2005;68:47–61. doi: 10.1111/j.1523-1755.2005.00380.x. [DOI] [PubMed] [Google Scholar]
- 149.Wang Y, et al. Enhancing cGMP in experimental progressive renal fibrosis: soluble guanylate cyclase stimulation vs. phosphodiesterase inhibition. Am J Physiol Renal Physiol. 2006;290:F167–F176. doi: 10.1152/ajprenal.00197.2005. [DOI] [PubMed] [Google Scholar]
- 150.Kalk P, et al. NO-independent activation of soluble guanylate cyclase prevents disease progression in rats with 5/6 nephrectomy. Br J Pharmacol. 2006;148:853–859. doi: 10.1038/sj.bjp.0706792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Albanis E, Friedman SL. Antifibrotic agents for liver disease. Am J Transplant. 2006;6:12–19. doi: 10.1111/j.1600-6143.2005.01143.x. [DOI] [PubMed] [Google Scholar]
- 152.Perri RE, et al. Defects in cGMP-PKG pathway contribute to impaired NO-dependent responses in hepatic stellate cells upon activation. Am J Physiol Gastrointest Liver Physiol. 2006;290:G535–542. doi: 10.1152/ajpgi.00297.2005. [DOI] [PubMed] [Google Scholar]
- 153.Hirth-Dietrich C, et al. Antifibrotic effects of an sGC activator in rat models of liver fibrosis. BMC Pharmacol. 2005;P24 [Google Scholar]
- 154.Lee YC, Martin E, Murad F. Human recombinant soluble guanylyl cyclase: expression, purification, and regulation. Proc Natl Acad Sci USA. 2000;97:10763–10768. doi: 10.1073/pnas.190333697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Friebe A, et al. YC-1 potentiates nitric oxide- and carbon monoxide-induced cyclic GMP effects in human platelets. Mol Pharmacol. 1998;54:962–967. doi: 10.1124/mol.54.6.962. [DOI] [PubMed] [Google Scholar]
- 156.Cellek S. The Rho-kinase inhibitor Y-27632 and the soluble guanylyl cyclase activator BAY41–2272 relax rabbit vaginal wall and clitoral corpus cavernosum. Br J Pharmacol. 2003;138:287–290. doi: 10.1038/sj.bjp.0705090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Teixeira CE, Priviero FB, Todd J, Jr, Webb RC. Vasorelaxing effect of BAY 41–2272 in rat basilar artery: involvement of cGMP-dependent and independent mechanisms. Hypertension. 2006;47:596–602. doi: 10.1161/01.HYP.0000199914.36936.1b. [DOI] [PubMed] [Google Scholar]
- 158.Wharton J, et al. Antiproliferative effects of phosphodiesterase type 5 inhibition in human pulmonary artery cells. Am J Respir Crit Care Med. 2005;172:105–113. doi: 10.1164/rccm.200411-1587OC. [DOI] [PubMed] [Google Scholar]
- 159.Straub A, et al. Metabolites of orally active NO-independent pyrazolopyridine stimulators of soluble guanylate cyclase. Bioorg Med Chem. 2002;10:1711–1717. doi: 10.1016/s0968-0896(02)00034-2. [DOI] [PubMed] [Google Scholar]