

Abstract
Peroxynitrite is a potent oxidant and nitrating species proposed as a direct effector of myocardial damage in numerous cardiac pathologies. Whether peroxynitrite also acts indirectly, by modulating cell signal transduction in the myocardium, has not been investigated. We therefore examined a possible role of peroxynitrite on the activation of NF-κ B, a crucial pro-inflammatory transcription factor, in cultured H9C2 cardiomyocytes. H9C2 cells were stimulated with TNF α or LPS following a brief (20 min) exposure to peroxynitrite. NF-κB activation (phosphorylation and degradation of its inhibitor I κ B α , nuclear translocation of NF-κ B p65 and NF-κ B DNA binding) triggered by LPS or TNF α was abrogated by peroxynitrite. Peroxynitrite also inhibited NF-κ B in 2 human endothelial cell lines activated with TNF α or IL-1 β . These effects were related to oxidative, but not nitrative chemistry, being still observed while nitration was suppressed by epicatechin. The mechanism of NF-κ B inhibition by peroxynitrite was a complete blockade of phosphorylation and activation of the upstream kinase IKK β , required for canonical, pro-inflammatory NF-κ B activation. At the same time, peroxynitrite activated phosphorylation of NIK and IKK α , considered as part of an alternative, non canonical NF-κ B activation pathway. Suppression of IKK β-dependent NF-κ B activation translated into a marked inhibition of the transcription of NF-κ B dependent genes by peroxynitrite. Thus, peroxynitrite has a dual effect on NF-κ B, inhibiting canonical IKK β-dependent NF-κ B activation while activating NIK and IKK α phosphorylation, which suggests its involvement in an alternative pathway of NF-κ B activation. These findings offer new perspectives in the understanding of the relationships between redox stress and inflammation.
NF-κB is a crucial transcription factor activating inflammatory and anti-apoptotic genes in response to immunostimulation. NF-κB is a family of dimeric proteins which include NF-κB1 (p50 and its precursor p105), NF-κB2 (p52 and its precursor p100), p65 (RelA), RelB and c-Rel, the commonest dimer being formed from a p50 subunit and a p65 subunit. NF-κB is normally held in the cytoplasm in an inactive form, bound to inhibitory proteins, the IκBs. The critical step in NF-κB activation relies in its dissociation from the IκB protein, which results from stimulus-induced phosphorylation of IκB, followed by its polyubiquitination and degradation by the proteasome. IκBs are phosphorylated by a protein kinase complex, IκB kinase (IKK), composed of a heterodimer of two catalytic subunits, IKKα and IKKβ, associated with a dimer of a regulatory subunit, IKKγ or NEMO, NF-κB Essential Modulator (for review, see (1,2)). Two distinct pathways of NF-κB activation have been described. In the first one, termed the canonical pathway, IKKβ is activated by an unknown upstream activator via regulatory IKKγ, and then phosphorylates IκB. This pathway is responsible for activation of inflammatory responses and the transcription of genes which protect against apoptosis (3–6). In addition to inflammatory signals, a number of reports have indicated that canonical NF-κB activation is also redox sensitive, as it can be activated in a number of different cell types by oxidants and free radicals (7,8). The second pathway of NF-κB activation, termed the non canonical pathway, is independent from IKKγ and IKKβ, but is based on NF-κB inducing kinase (NIK) and IKKα-dependent processing of NF-κB p100 to p52, a pathway crucial for B cell maturation and lymphoid organ development (9–12). In contrast to the canonical pathway, it is currently unknown whether this alternate pathway is responsive to redox stress.
Peroxynitrite (PN) is a highly reactive oxidant and nitrating species formed by the reaction of nitric oxide (NO) with superoxide (O2−) (13). PN generation is favored when the production of both NO and O2− is enhanced, as may occur during inflammation, circulatory shock and reperfusion injury (14). In the heart, PN generation occurs during myocardial infarction (15), heart failure (16) and cardiomyopathy due to anthracyclines (17). In such conditions, it is unknown whether PN only acts as a direct cytotoxic effector, or may have further, indirect effects related to the modulation of redox sensitive cell signal transduction pathways such as NF-κB. It is also noteworthy that cardiomyocytes are not only exposed to PN in these circumstances, but also to additional inflammatory stimuli such as pro-inflammatory cytokines. Whether PN may alter the cell response to these stimuli has not been evaluated. The present study was therefore designed to explore a possible crosstalk between PN and inflammatory stimuli (TNFα and LPS) at the level of NF-κB signaling in cultured cardiomyocytes and endothelial cells. Our findings demonstrate that PN blocks IKKβ phosphorylation and activation, thereby preventing nuclear translocation of NF-κB and inflammatory gene transcription in response to immune stimulation. In parallel, PN strongly activates the phosphorylation of NIK and IKKα, whether or not the cells are exposed to immune stimuli. Thus, PN has a dual effect on NF-κB, being an inhibitor of canonical IKKβ-dependent NF-κB activation pathway while simultaneously activating the phosphorylation of NIK and IKKα, suggesting its involvement in an alternative pathway of NF-κB activation.
MATERIAL AND METHODS
Cell culture conditions and reagents
H9C2 cells, a clonal line derived from rat heart (ATCC, Manassas, VA) and human macrovascular endothelial cells (EAhy 926, a generous donation of Prof. D. Hayoz, Division of Hypertension, Lausanne University Hospital) were grown (5% CO2, 37 °C) in DMEM (Gibco BRL, Invitrogen, Basel, Switzerland ) supplemented with 10% FBS, 100 units/ml penicillin and 0.1 mg/ml streptomycin. The human microvascular endothelial cell line HMEC-1, a generous gift from Dr F.J. Candal, Center of Disease Control and Prevention, Atlanta, GA, were cultured (5% CO2, 37 °C) in MCDB 131 medium (GibcoBRL) enriched with 10% FBS, 50 units/ml penicillin, 50 μg/ml streptomycin, and 0.01 mg/ml epidermal growth factor (EGF) (Roche; Basel, Switzerland). Cells were cultured to 80 percent confluence and were serum-starved 24h before the experiments. Unless otherwise specified, all the reagents used in this study were obtained from Sigma-Aldrich (Fluka, Buchs, Switzerland).
Stimulation with PN, LPS, TNFα and IL-1β
PN (Calbiochem), was synthesized as described previously (18). Stocks of PN were stored in 0.4 M NaOH at −80°C. Prior to experimentation, the concentration of PN was determined from its absorption at 302 nm (extinction coefficient = 1670 M-1 cm-1) (13). For stimulation with PN, cells were washed once and then covered with PBS-glucose buffer (50 mM Na2HPO4, 90 mM NaCl , 5 mM KCl, 0.8 mM MgCl2, 0.2 mM CaCl2, 5 mM glucose, pH 7.4). PN was delivered as a single bolus at a 1:100 dilution. The final concentration of PN was 10–250 μM depending on the experimental conditions, in agreement with the concentrations of PN used in previous studies from our laboratory (19), and from other investigators (20–23). Such concentrations of PN are physiologically relevant, as it has been estimated that the rate of PN generation may reach up to 1mM/min in an inflamed organ (lung) in vivo (24). Cells were exposed to PN for 20 min, after which they were washed and replaced in culture medium for stimulation with either 1μg/ml LPS (E. Coli O127:B8, Sigma Chemicals), 20 ng/ml TNFα (Socochim SA, Lausanne, Switzerland) or 1 ng/ml IL-1β (PreproTech, London, UK) for the indicated times. Control experiments were performed using decomposed PN (DP) in NaOH, obtained by leaving the PN solution overnight at room temperature, after which PN is completely d e c o m p o s e d a s d e t e r m i n e d spectrophotometrically (19,25). In some experiments, hydrogen peroxide instead of PN was used to pretreat the cells before immunostimulation.
Preparation of cytoplasmic and nuclear extracts
Cells were scraped into hypotonic buffer (HEPES 10 mM, pH 7.9, EDTA 0.1 mM, EGTA 0.1 mM, KCl 10 mM, DTT 1 mM, PMSF 1 mM, Na3VO4 1 mM). After addition of NP-40 (0,625 %), the tubes were centrifuged and supernatants containing the cytoplasmic proteins were stored at −70°C. Pellets were resuspended in hypertonic buffer (HEPES 20 mM, pH 7.9; NaCl 0.4 M, EDTA 1 mM, EGTA 1 mM, DTT 1 mM, PMSF 1 mM). After centrifugation, supernatants (nuclear extract) were collected and stored at −70°C. Amount of proteins was quantified by BCA assay.
SDS Page and Western immunoblotting
30 μg of cytoplasmic or 10 μg of nuclear proteins were separated by SDS-Page, transferred to nitrocellulose membranes and blocked for 1 h at room temperature in PBS/0.1% Tween/3% nonfat dry milk. For nuclear extracts, the membrane was incubated with a rabbit anti-NF-κB RelA/p65 Ab (Santa Cruz Biotechnology, Santa Cruz, CA) and for cytoplasmic extracts, the membrane was incubated with appropriate dilutions of primary anti-IκBα, anti-phospho-IκBα (both from Santa Cruz), anti-IKKα, anti-IKKβ, anti-phospho-IKKα/β, anti-phospho-IKKα, anti-NIK, anti-phospho NIK, anti-NF-κ B RelA/p65, anti-α-tubulin (all from Cell Signaling, Beverly, MA) or anti-nitrotyrosine antibody (Upstate biotechnology, Lake Placid, NY), followed by incubation with a 1:5000 dilution of the appropriate horseradish peroxidase-conjugated secondary antibody (Bio-Rad, Hercules, CA). The immunoblot signal was visualized using enhanced chemiluminescence (ECL, Amersham Biosciences).
Electromobility shift assay (EMSA)
An NF-κ B oligonucleotide probe (5′-AGTTGAGGGGACTTTCCCAGG-3′) was labeled with α-32PdCTP using the Klenow enzyme (Roche). 10 μg of nuclear proteins were incubated with EMSA buffer (20 mM Hepes pH 7.9, 50 mM KCl, 0.5 mM EDTA, NP40 0.1 %, BSA 1 mg/ml, Glycerol 5%), 170 μg/ml poly dIdC and the probe for 20 min at room temperature. Samples were resolved on a non-denaturing polyacrylamide gel. Gels were transferred to Whatman 3M paper, dried under vacuum, and exposed to photographic film at −70°C with intensifying screens. Densitometric analysis of autoradiographs were performed using a Personal Densitometer and TotalLab Software.
NF-κB gene reporter gene assay
Cells were transiently transfected with 1 μg of a multimeric NF-κB pGL2 luciferase vector and 0.1 μg of the Renilla pRL-TK vector (kind gifts from Prof. T. Calandra, Department of Infectious Diseases, Lausanne University Hospital) using Lipofectamine 2000 (Invitrogen, Basel, Switzerland). The media was changed 6 h after transfection and the cells were grown overnignt in fresh complete media. Luciferase activity was determined using a standard kit (Dual-Luciferase Reporter assay system, Promega Biosciences Inc, San Luis Obispo, CA). The activity of the NF-κB reporter luciferase was standardized to that of Renilla luciferase.
Immunoprecipitation and IKK kinase assay
Cells were lysed in LB (50 mM HEPES, 150 mM NaCl, 1 mM EDTA, 2 mM MgCl2, 1 mM Na3VO4, 1 mM PMSF, 0.1% NP-40, 10 μg/ml Leupeptin, 10 μg/ml Aprotinin,1μg/ml Pepstatin, 0.5 mM DTT, 100 μM NaF). IKK was immunoprecipitated by incubation of 200 μg of protein with an anti-IKKβ antibody overnight at 4°C, followed by addition of Protein A Sepharose beads CL-4B (Amersham) for 3 h. Precipitates were washed 5x with LB and 3x with Kinase Buffer (20 mM HEPES pH 7.9, 20 mM β-glycerophosphate, 1 mM MnCl2, 5 mM MgCl2, 2 mM NaF, 1 mM DTT). The kinase reaction was performed by adding 1 μg of recombinant GST-IκBα (aminoacids1–54, MTR Scientific, Ijamsville, MD) and 5 μCi γ-32PATP and incubating 30′ à 30°C. Reactions were stopped by addition of 2xLaemmli sample buffer. Proteins were separated on a 12% polyacrylamide gel, and gels were fixed, dried and examined by autoradiography.
RNA isolation and polymerase chain reaction
Cells were stimulated with LPS or TNFα for 2 h and total RNA was isolated by TRIZOL (Invitrogen). RNA was reverse-transcribed to cDNA and amplified by PCR using the One Step RT-PCR kit from Qiagen (Qiagen AG, Hombrechtikon, Switzerland) with a Tpersonal Thermocycler (Biometra, Goettingen, Germany). The sequences of the primer pairs were as follows: rat iNOS (sense, 5′-CTGCATGGAACAGTATAAGGCAAAC-3′; antisense, 5′-AGACAGTTTCTGGTCGATGTCATGA-3′); rat TNF a (sense, 5′-TCTGTCTACTGAACTTCGGGGTGAT-3′; antisense, 5′-CAGCCTTGTCCCTTGAAGAGAACC-3′); rat GAPDH, used as an internal control (sense, 5′-ACCACAGTCCATGCCATCAC-3′; antisense : 5′-TCCACCACCCTGTTGCTGTA-3′). PCR products were run by electrophoresis on a 2% agarose gel and stained with ethidium bromide.
Measurement of cell viability by MTT assay
Cell viability was assessed by the mitochondrial-dependent reduction of 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) to formazan. Cells were stimulated for the indicated periods, and then incubated for 60 minutes with MTT (0.5 mg/ml), followed by aspiration and solubilisation of the cells in 100μl DMSO, and absorbance was measured at 540 nm.
Immunocytochemistry
Cells plated on glass coverslips were treated with PN (20 min), and then fixed with 10% paraformaldehyde. Cells were then incubated with polyclonal rabbit anti-nitrotyrosine (Upstate) at room temperature for 2h. A standard immunostaining protocol was then followed using Vectastain ABC kit from Vecor Laboratories (Burlinghame, CA).
Statistical analysis
Densitometric analyses of blots are shown as mean±sem of n observations. Statistical analysis was performed using analysis of variance followed by Bonferroni adjustment, and p<0.05 was considered statistically significant.
RESULTS
Peroxynitrite inhibits IκBα phosphorylation and degradation, and NF-κB p65 nuclear translocation elicited by LPS or TNFa
Phosphorylation of IκBα, which targets IκBα for degradation in the proteasome, allows inactive cytoplasmic NF-κ B dimers to translocate into the nucleus (1,2). Stimulation of H9C2 cells with LPS or TNFα (Fig. 1A and B) triggered IκBα phosphorylation that was maximal at 10 min and rapidly declined thereafter, most likely due to its known proteasome-dependent degradation. Phosphorylation of IκBα was associated with IκB degradation, as evidenced by the marked reduction of IκB signal 30 min after LPS or TNFα (Fig. 1C and D). At 60 min post-stimulation, the IκB α signal returned to baseline levels, consistent with Iκ B α resynthesis triggered by the activation of NF-κB (26). Pretreatment of cells with 250 μM PN for 20 min before LPS or TNFα prevented Iκ B α to be phosphorylated, an effect associated to a complete inhibition of IκBα degradation. In contrast to the effects of PN, decomposed peroxynitrite (DP) did not exert any influence on IκBα (Fig. 1A–D). There were no significant changes in the expression levels of α-tubulin between the different conditions, indicating equal loading of proteins (Fig. 1E and F).
Fig. 1.
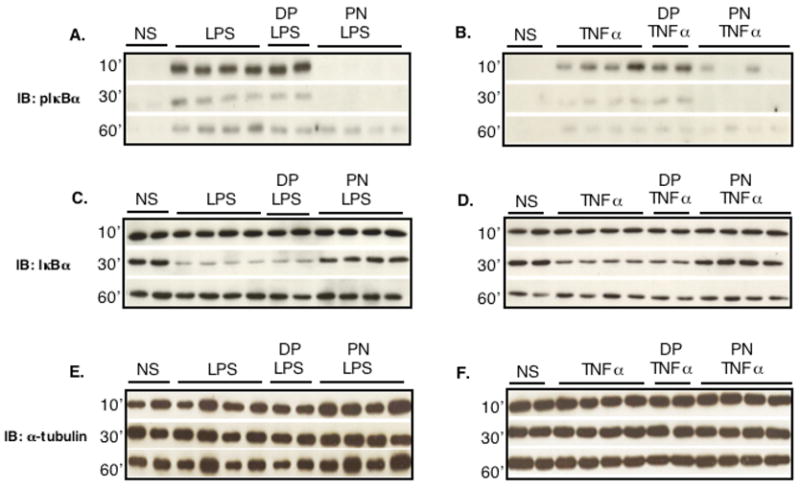
Peroxynitrite inhibits IκBα phosphorylation and degradation triggered by inflammatory stimuli in H9C2 cells. H9C2 cells were either not stimulated (NS) or stimulated with LPS (1μg/ml, A, C, E) or TNFα (20 ng/ml, B, D, F) after pretreatment with either decomposed peroxynitrite (DP) or authentic peroxynitrite (PN, 250 μM) for 20 min. Both LPS and TNFα induced IκBα phosphorylation (A, B) and degradation (C, D), which were prevented by PN but not DP. Expression levels of α-tubulin (E, F) are shown as loading controls.
Nuclear translocation of NF-κB p65 was evaluated by monitoring the levels of p65 (RelA) protein in nuclear extracts. Both LPS and TNFα induced RelA/p65 nuclear translocation, that occurred earlier with TNFα (10 min) than with LPS (30 min) and was maintained up to 1 hour after stimulation (Fig. 2A and B). In keeping with its effects on IκBα phosphorylation and degradation, PN (but not DP) abrogated the nuclear translocation of RelA/p65 elicited by LPS or TNF. The changes in nuclear RelA/p65 were clearly due to its nuclear translocation, but not to changes in the expression levels of RelA/p65, as shown by RelA immunoblots in cytoplasmic extracts (Fig. 2C and D).
Fig. 2.
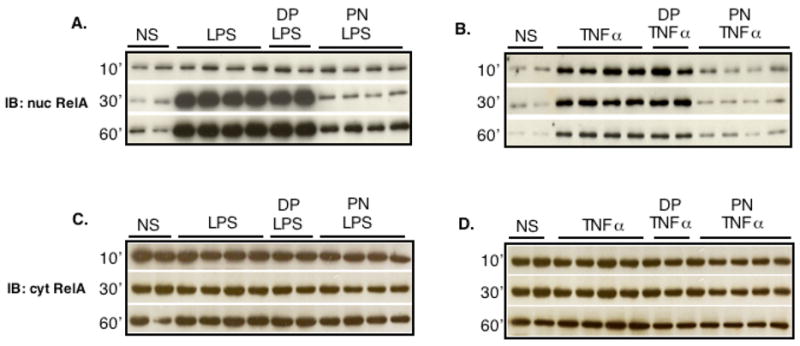
Peroxynitrite prevents the nuclear translocation of the p65 (RelA) subunit of NF-κB induced by LPS or TNFα. H9C2 cells were left unstimulated (NS) or were stimulated with LPS (1μg/ml, A, C) or TNFα (20 ng/ml, B, D) after a 20 min pretreatment with 250 μM peroxynitrite (PN) or decomposed peroxynitrite (DP) for 20 min. Nuclear RelA accumulation was markedly increased following LPS or TNFα (A, B), with no changes in the levels of cytoplasmic RelA (C, D). PN, but not DP, prevented RelA nuclear translocation.
Peroxynitrite prevents NF-κB DNA binding in H9C2, HMEC-1 and EAhy 926 cells
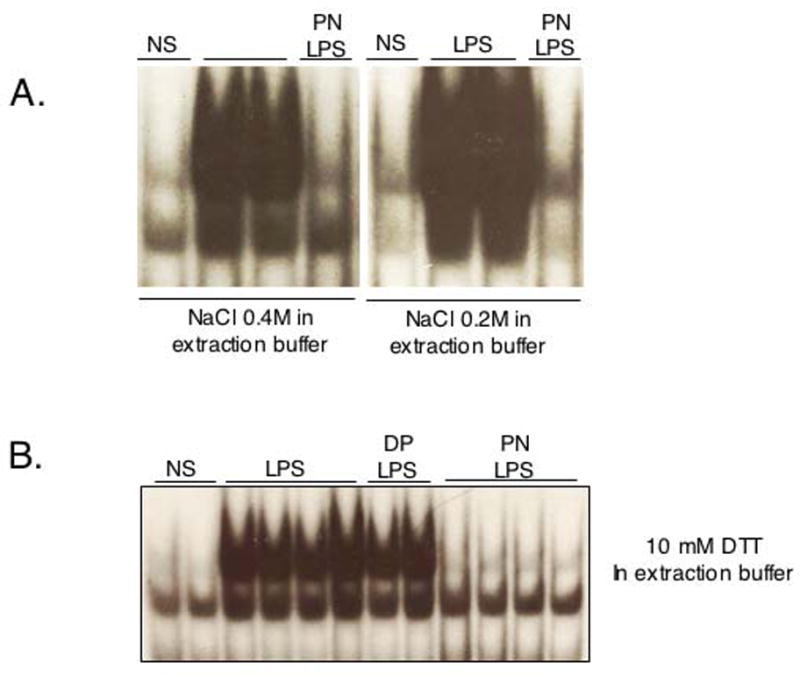
There was no nuclear NF-κB DNA binding in quiescent H9C2 cells, as shown by bandshift experiments (Fig. 3). After 30 minutes of stimulation, LPS or TNFα strongly induced NF-κB DNA binding activity, which was abrogated by PN, but not DP (Fig. 3A and B). Concentration-response experiments indicated that PN already significantly inhibited DNA binding activity at a concentration of 10 μM, with almost complete inhibition obtained at 100 μM (Fig. 3C and D). To evaluate possible cell specificity of this effect of PN, NF-κB DNA binding was determined in two human endothelial cell lines, HMEC-1 and Eahy 926 stimulated with TNFα, IL-1β, or both. In HMEC-1 cells, NF-κB was activated by TNFα and IL-1β, and this was eliminated by PN (250μM), but not DP (Fig. 3E). Eahy 926 cells were found to be responsive to TNFα, but not IL-1β. NF-κB DNA binding elicited by TNFα was again markedly inhibited by PN pretreatment (Fig. 3F). Thus, the inhibitory effects of PN on NF-κB activation is observed independently from the stimulus (LPS, TNFα, IL-1β) or the cell type studied.
Fig. 3.
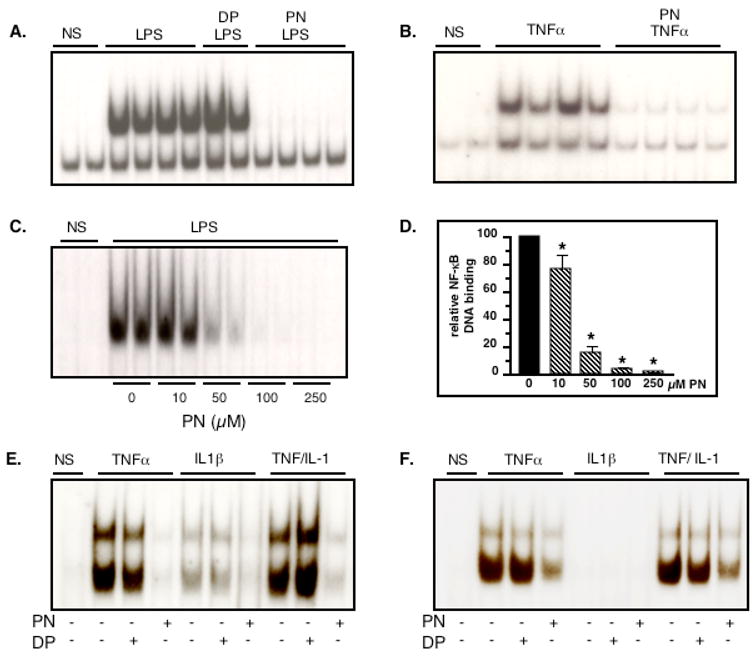
Peroxynitrite inhibits NF-κB DNA binding activity triggered by inflammatory stimuli in H9C2, HMEC-1 and EAhy 926 cells. H9C2 cells were not stimulated (NS) or treated with LPS (1μg/ml, A) or TNFα (20 ng/ml, B). Both LPS and TNF triggered NF-κB DNA binding, an effect suppressed by pretreatment with peroxynitrite (PN, 250 μM, 20 min), but not decomposed PN (DP). The inhibitory effect of PN on LPS-mediated NF-κB DNA binding was concentration-dependent (C, D). PN, but not DP, also inhibited NF-κB DNA binding in HMEC-1 cells (E) and EAhy 926 cells (F) stimulated with TNFα (20 ng/ml), IL1β (1 ng/ml) or both. Bar graph shows mean±sem of at least 4 observations. * p<0.05 (ANOVA followed by Bonferroni adjustments)
Peroxynitrite-dependent NF-κB inhibition depends on oxidation, but not nitration
The chemistry of PN encompasses both oxidation reactions and nitration of protein tyrosine residues (27). Distinction between these two kinds of reactions can be obtained with epicatechin, which acts as a potent inhibitor of tyrosine nitration elicited by PN (21). Stimulation with 250 μM PN for 15 minutes induced a widespread nitration of cellular proteins in H9C2 cells, as evidenced both by western blotting and by immunocytochemical detection (Fig. 4A and B). Nitration was concentration-dependently inhibited by epicatechin, in accordance with previous data by other investigators (21), with complete inhibition obtained at 100 μM and above. We thus used a concentration of 100 μM epicatechin to evaluate the role of nitration on NF-κB inhibition by PN. As shown in Fig. 4C, the PN-dependent prevention of IκBα phosphorylation in response to LPS was not affected by epicatechin, indicating that PN does not block NF-κB activation via nitration reactions and supporting that an oxidative type of chemistry is responsible for this effect. To further confirm that the inhibition of NF-κB by PN was due to oxidation, we assessed the possible effect of another oxidant species, hydrogen peroxide (H2O2), on the process of NF-κB activation by LPS in H9C2 cells. As shown in Fig. 4D and E, H2O2 prevented the DNA binding activity of NF-κB as did PN. However, in contrast to PN, the concentrations of H2O2 required for this inhibition were higher (100 μM and above). Also, the effect of H2O2 could be fully reversed in the presence of catalase, whereas the latter did not affect the inhibition of NF-κB activation by PN (Fig. 4F and G). Finally, the PN-mediated inhibition of NF-κB-DNA binding triggered by LPS was completely prevented when cells were pretreated for 2h with 250μM of the superoxide dismutase (SOD) mimetics and antioxidant agent Mn(III)-tetrakis-(4-benzoic acid) porphyrin (MnTBAP) (Fig. 4H). Overall, these data indicate that PN-dependent inhibition of NF-κB occurs as a result of oxidative, but not nitrative processes.
Fig. 4.
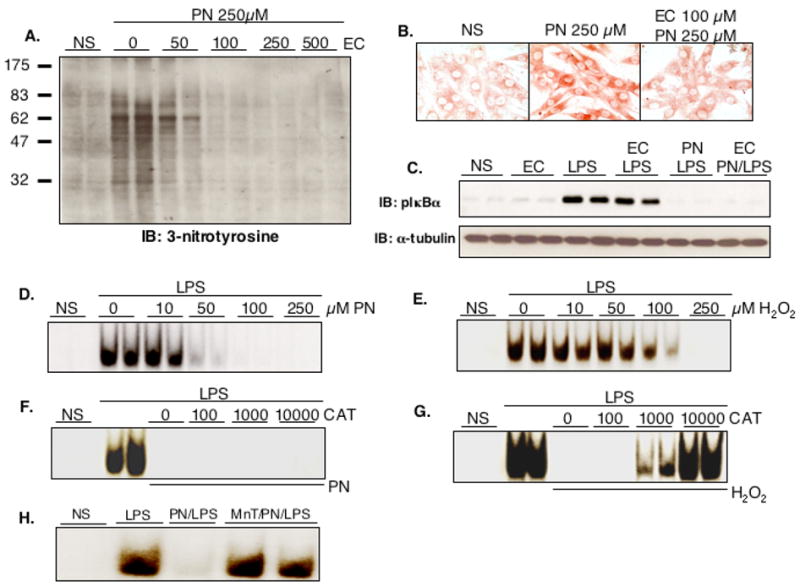
Peroxynitrite inhibits NF-κB independently from nitration reactions. Treatment of H9C2 cells for 20 min with 250 μM peroxynitrite (PN) induced nitration of cellular proteins, as indicated by the formation of 3-nitrotyrosine, which was abrogated by pretreatment with epicatechin (EC) at concentrations of 100 μM and above (A, western, B, immunochemistry). Although 100 μM EC efficiently prevented tyrosine nitration, it did not influence the PN dependent inhibition of NF-κB activation in cells stimulated with LPS, as assessed by IκBα phosphorylation (C). Expression levels of α-tubulin are shown as a loading control. Hydrogen peroxide (H2O2), another oxidant species, was also able to inhibit NF-κB, as shown by band shift experiments, although this effect required higher concentrations than PN (D, E). The inhibitory effect of H2O2 on NF-κB DNA binding was reversed in the presence of 100–10000 Units catalase (CAT), whereas the effect of PN was not influenced by catalase (F, G). Finally, the prevention of NF-κB-DNA binding activation produced by PN was reversed after 2h pretreatment with 250 μM of the SOD mimetics and antioxidant MnTBAP (H). NS: Not Stimulated. MnT: MnTBAP.
Peroxynitrite inhibits IKKβ phosphorylation and IKK kinase activity, but activates the phosphorylation of NIK and IKKα
Activation of IKK represents the crucial step in the process of NF-κB activation (1). As shown in Fig. 5A and B, both LPS and TNFα enhanced IKK activity, as demonstrated by kinase assay experiments. Following LPS, IKK activation started at 10 min, reached a peak at 15–20 min and then steadily declined until 60 min. Upon stimulation with TNFα, the pattern was similar, although IKK activity was induced earlier than with LPS, starting as early as 5 min after TNFα, peaking at 5 to10 min and disappearing thereafter. IKK activation was associated with an enhanced phosphorylation of both IKKα and IKKβ by LPS and TNFα, which was maximal at 20 min after LPS and 10 min after TNFα (Fig. 5A an B).
Fig. 5.
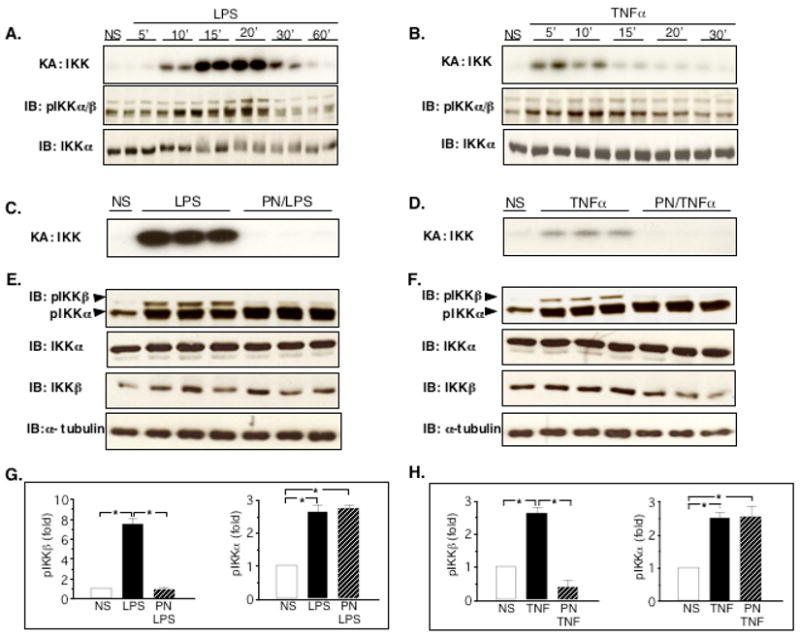
Peroxynitrite inhibits IKK kinase activity by preventing specifically the phosphorylation of IKKβ in H9C2 cells. A and B: IKKkinase assay (KA) in cells treated with LPS or TNFα. LPS strongly activated IKK kinase from 10 to 30 minutes (A), whereas the effects of TNFα were most pronounced at 5 and 10 minutes (B). The activation of IKK was associated with an increased phosphorylation of IKKα and IKKβ by LPS and TNFα. IKKα levels are shown as an internal control. C and D: IKK kinase activity in cells pretreated with peroxynitrite (PN, 250 μM, 20 min) and stimulated with LPS (C, 20 min) or TNFα (D, 10 min). PN abrogated IKK activity triggered by LPS or TNFα. E and F: Phosphorylation of IKKα and β was induced both by LPS (20 min, E) and TNFα (10 min, F). PN completely prevented the phosphorylation of IKKβ triggered by LPS and TNFα, whereas it did not reduce IKKα phosphorylation in response to LPS and TNFα. No significant changes in the levels of IKKα, IKKβ and α-tubulin were found between the different conditions. G and H: densitometric analysis of phospho-IKK western blots showing mean±sem of 3 observations. NS: Not Stimulated. * p<0.05, ANOVA followed by Bonferroni adjustment.
Due to the inhibition of NF-κB activation by PN, we assessed whether this effect could be related to the inhibition of IKK activation. The possible influence of PN was explored in cells stimulated with LPS for 20 min, or with TNFα for 10 min, as these time points corresponded to the peak activation of IKK by these stimuli. Fig. 5C (LPS) and 5 D (TNFα), unequivocally demonstrate that PN completely prevented the activation of IKK triggered by the two inflammatory stimuli.
Since IKK activation relies on the phosphorylation of two serine residues within the activation loops of IKKα and IKKβ(1), we examined the effects of PN on IKK phosphorylation. Both LPS (Fig. 5E) and TNFα (Fig. 5F) triggered the phosphorylation of IKKα and IKKβ after 20, respectively 10 min. When cells were pretreated with PN for 20 min, the phosphorylation of IKKβ was totally prevented, while PN did not exert any inhibition on the phosphorylation of IKKα (Fig. 5E and F, densitometric analysis, Fig. 5G and H). The effects of PN were not associated with significant changes in the expression levels of IKKα, IKKβ and α-tubulin. Thus, PN prevents IKK activation by impairing specifically the phosphorylation of IKKβ.
We then sought to determine whether PN on its own has the ability to phoshorylate IKKα. As shown in figure 6A and B, The phosphorylation of IKKα in cells treated with LPS or TNFα was maintained for up to 2h only if they were pretreated with PN. Furthermore, PN alone, independently from any stimulation with LPS or TNFα, activated the phosphorylation of IKKα, an effect evident already after 10 min and further increasing at 15 and 20 min (Fig. 6C). Previous studies have shown that IKKα phosphorylation relies on the upstream activation of NIK (12,28), and we thus determined whether PN might activate this particular signaling cascade. As indicated in Fig. 6C, PN induced the phosphorylation of NIK, with a time-course parallel to that of IKKα phosphorylation.
Fig. 6.
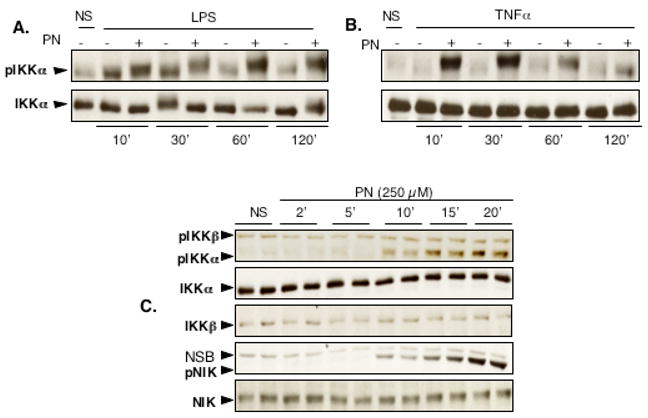
PN activates the phosphorylation of IKKα and NIK. IKKα phosphorylation was monitored in cells stimulated with LPS (A) or TNFα (B) for 10 to 120 min, in the absence or presence of peroxynitrite (PN). In the presence of PN, there was a strong phosphorylation of IKKα, maintained for up to 120 min after LPS or TNFα, with no change in total IKKα. C: PN alone strongly activated the phosphorylation of IKKα and NIK, an effect starting after 10 min. NS: Not Stimulated. NSB: non specific band
Peroxynitrite inhibits the transcription of NF-κB-dependent genes
In order to evaluate whether PN-dependent NF-κB inhibition might influence NF-κB-dependent gene transcription, we first addressed the influence of PN on NF-κB-luciferase reporter gene expression determined 4 hours after TNFα stimulation. While PN alone had no effect, TNFα induced more than a 20-fold increase in the expression of the transfected NF-κB-dependent luciferase gene (Fig. 7A). This increase was not modified by pretreatment of cells with DP, whereas PN induced an almost complete inhibition of luciferase expression. We then evaluated the concentration-dependence of this effect by pretreating cells with increasing concentrations (10–250 μM) of PN and then TNFα or LPS for 4h. Fig. 7B and C show that PN at all concentrations significantly inhibited the TNFα and LPS-dependent luciferase expression. To make sure that these effects were not related to nonspecific cytotoxicity by PN, we assessed cell viability (MTT assay) in H9C2 cells 4 hours after a 20 min exposure to 250 μM PN. As shown in Fig. 7D, PN only marginally affected cell viability, which indicates that the decrease in luciferase expression was indeed consecutive to NF-κB inhibition but not to cell death.
Fig. 7.
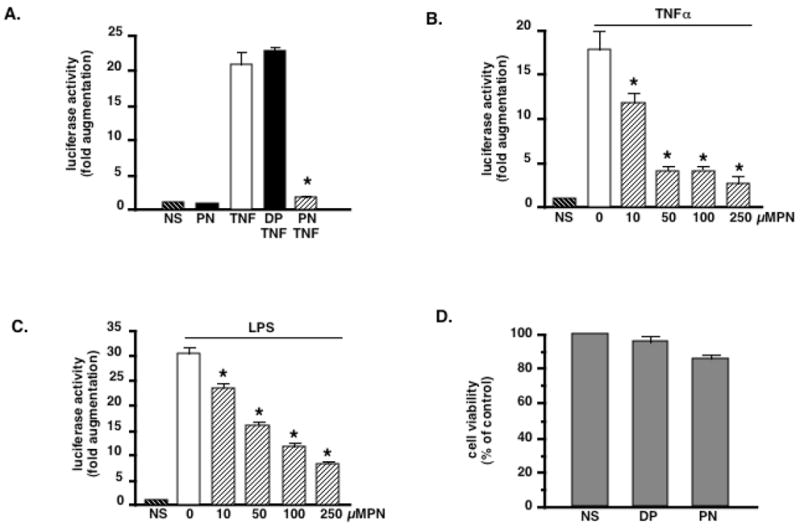
PN inhibits the activity of a NF-κB-luciferase gene reporter activated by LPS or TNFα. Cells were either not stimulated (NS), or stimulated with peroxynitrite alone (PN, 250μM, 20 min), or TNFα (20 ng/ml) following pretreatment for 20 min with decomposed peroxynitrite (DP) or PN. Luciferase activity was measured after 4 h (A). PN alone had no effect, while TNFα induced a 20-fold increase in luciferase activity, an effect unchanged by DP, but significantly prevented by PN. B and C: PN-dependent inhibition of luciferase activity triggered by TNFα (B) or LPS (C) is concentration–dependent. D. The effects of PN are not related to non specific cytotoxicity. Cells were treated with PN (250 μM, 20 min) or DP (20 min). Cell viability was assessed 4 h later by the MTT assay. PN only marginally affected cell survival (86% viability at 4h). Bar graphs are means ± sem of at least 4 measurements. * p< 0.05 vs TNF or LPS alone (ANOVA followed by Bonferroni adjustments)
We also explored whether endogenous genes, which are known to be transcriptionnally regulated by NF-κB, would be affected by PN. Both LPS and TNFα induced the transcription of the genes encoding inducible NO synthase (iNOS) (Fig. 8A and B) and TNFα (Fig. 8C and D), and this was not influenced when the cells were pretreated with DP. In contrast, PN completely abrogated the transcription of iNOS and TNFα in cells stimulated either with LPS or TNFα.
Fig. 8.
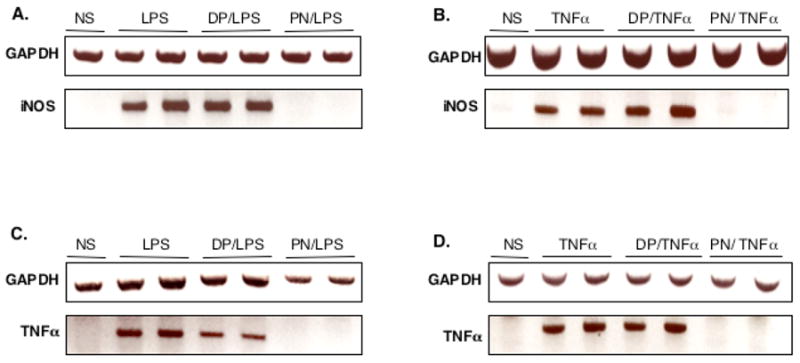
PN inhibits the transcription of endogenous NF-κB-dependent genes. The transcription of inducible nitric oxide synthase (iNOS) and TNFα mRNA was evaluated in response to LPS (A, C) or TNFα (B, D) stimulation for 2h. LPS and TNF induced iNOS and TNFα gene transcription, an effect abrogated by pretreatment with peroxynitrite (PN, 250 μM, 20 min), but not decomposed PN (DP, 20 min). NS: Not Stimulated.
DISCUSSION
Recent studies have indicated that PN is generated in large amounts in the myocardium during myocardial reperfusion injury (15), chronic heart failure (16), circulatory shock (29) and anthracycline cardiomyopathy (17). While PN has generally been considered as directly biotoxic towards cardiomyocytes, we now provide the novel evidence that PN also behaves as an important modulator of cell signal transduction at the level of NF-κB. A short exposure to PN exerted a marked inhibition towards IκBα phosphorylation and degradation, p65 nuclear accumulation and NF-κB DNA binding triggered by LPS or TNFα in H9C2 cardiomyocytes (Fig.1–3). This inhibition was already significant at low concentrations of PN (10 μM), with more than 80% inhibition obtained at 50 μM of the oxidant. Similarly, PN inhibited NF-κB activation in 2 additional cell lines, HMEC-1 and EAhy 926 endothelial cells, stimulated with TNFα and IL-1β (Fig. 3E and F), which indicates that the inhibitory effect of PN on NF-κB is independent of type of cell and stimulus.
PN has been associated with two different types of chemistry, namely oxidation reactions and nitration of tyrosine residues. Our finding that epicatechin, which fully reversed the nitration induced by PN, did not affect the inhibition of NF-κB activation (Fig. 4A–C), indicates that oxidation rather than nitration was the relevant chemical reaction underlying this effect of PN. This conclusion is further supported by the findings that (a) another strong oxidant species, hydrogen peroxide, also inhibited NF-κB activation in H9C2 cells stimulated with LPS, although this effect was observed at concentrations higher than those of PN (Fig. 4D and E), and (b) the inhibition of NF-κB-DNA binding activity by PN was totally prevented by the SOD mimetics and antioxidant compound MnTBAP (Fig. 4H). These observations argue against the concept that NF-κB is a general sensor of cellular redox stress. Such redox sensitivity of NF-κB has been proposed on the basis that anti-oxidants can prevent NF-κB activation in response to cytokines and that NF-κB can be activated upon the direct exposure of cells to reactive oxygen species (7,30), but this issue is currently debated for a number of reasons. First, NF-κB can be activated by oxidant stress only in some, but not all cell types (8). Second, there are multiple mechanisms accounting for NF-κB activation in response to redox stress (8). Oxidants have been shown to activate NF-κB either via IKK-dependent IκBα serine phosphorylation (31), or via tyrosine phosphorylation of IκBα at Tyr 42 (32). It has been claimed that this variability in the modes of action of oxidants does not support their role as universal NF-κ B activators (8). Third, it has been demonstrated that endogenously produced reactive oxygen species do not mediate NF-κB activation in Hela, HT-29, L-929 and Jurkat T-cells (33). Fourth, a recent report indicated that oxidant stress inhibited NF-κB activation by TNFα in alveolar RLE and C10 cells (34). Our data further challenge the issue of the redox responsiveness of NF-κB, by providing strong experimental evidence that a short episode of redox stress with PN potently inactivates consecutive activation of NF-κB by pro-inflammatory signals. Since PN is generated in many different pathological conditions, these findings may have important consequences to understand the ramification between PN formation and the regulation of inflammatory processes in vivo.
The key regulatory step in NF-κB activation is the activation of the upstream kinase IKK (1). The activation of IKK subunits relies in the phosphorylation of serine residues within their activation loops (Ser176 and 180 for IKKα and Ser177 and 181 for IKKβ) (35). Here, we provide the first experimental evidence that PN prevents LPS and TNFα-dependent NF-κB activation through a complete blockade of IKK activity, due to inhibition of IKKβ phosphorylation (Fig. 5). These findings are consistent with previous data from the literature indicating that IKKβ represents a key target of various NF-κB inhibitors, including cyclopentone prostaglandins (36), arsenite (37) and NO (whose chemical reactivity is distinct from that of PN (14)).
Another striking observation of the present study was the opposite influence of PN towards the α and β subunits of IKK. Whereas PN blocked LPS and TNFα-dependent IKKβ phosphorylation, it did not reduce, but instead promoted IKKα phosphorylation (Fig. 5 and 6). Such a contrasted influence of a single compound on IKKα and β has not been previously reported, but is consistent with the fact that phosphorylation of IKKα and IKKβ proceeds through distinct mechanisms. Whereas IKKβ phosphorylation critically depends on the regulatory subunit IKKγ, IKKα can be directly phosphorylated by the upstream kinase NIK (1). Recent reports indicate that NIK/IKKα mediate an alternative, non canonical NF-κB pathway, in lymphoid cells in response to certain stimuli including LTβR, BAFF and CD40, resulting into the processing of the NF-κB subunit p100 into p52 (9–11,38). Our finding that phosphorylation of IKKα by PN was associated with the simultaneous phosphorylation of NIK (Fig. 6C), suggests that redox stress mediated by PN may activate a similar pathway in cardiomyocytes. However, we did not find evidence of p100 processing into p52 in cells stimulated with PN (data not shown), implying that the downstream events triggered by PN-mediated phosphorylation of NIK and IKKα remain yet to be established.
Beside its function in non canonical NF-κB activation, a novel role of IKKα in the canonical pathway of NF-κB activation has been recently identified by Lawrence and coworkers (39). In this study, mice expressing an inactivatable variant IKKα (ikkαAA/AA) showed a markedly increased inflammatory response following LPS administration, characterized by an increased expression of NF-κB-target genes in tissues (lung and liver). Furthermore, LPS-stimulated ikkα AA/AA macrophages disclosed a markedly prolonged NF-κB-DNA binding activity relative to wild-type cells, related to a reduced degradation of the NF-κB subunits RelA (p65) and c-Rel in ikkαAA/AA macrophages (39). Overall, the results of this study clearly identified a previously unrecognized function of IKKα as a negative regulator of NF-κB activation, indispensable for the resolution of inflammation. Thus, IKKα and IKKβ play opposite (down-versus upregulation), yet complimentary roles in inflammation. Our results that PN-mediated inhibition of NF-κB activation is associated with an opposite regulation of IKKα (activation) and IKKβ (inhibition) is therefore consistent with the distinct functions of these subunits reported in the study by Lawrence et al.
In keeping with the findings that PN blocked IKKβ-dependent NF-κB activation, we found that it also markedly reduced the transcription of NF-κB dependent genes in cardiomyocytes. Using a luciferase reporter assay, we found a marked reduction of luciferase activity in cells stimulated with TNFα or LPS after PN pretreatment (Fig. 7A–C). Also, PN completely blocked the transcription of the endogenous inflammatory genes encoding iNOS and TNFα (Fig. 8). Importantly, both TNFα and NO generated from iNOS have been proposed as potential myocardial depressant effectors in pathologies such as septic shock (40) and chronic heart failure (41), where enhanced myocardial formation of PN has also been documented. Our data therefore suggest that PN generation in vivo might unexpectedly down-regulate the expression of pro-inflammatory mediators and cardiodepressant molecules, providing a counterregulatory mechanism to prevent overt inflammation and myocardial failure in various pathological conditions.
In summary, our data show that PN, acting through oxidative but not nitrative chemistry, is a potent inhibitor of canonical NF-κB activation triggered by inflammatory cytokines or LPS, as proposed in the diagrammatic scheme presented in figure 9. This effect is related to the blockade of IKKβ phosphorylation, resulting in the inactivation of the kinase and downstream signaling. The inhibition of IKKβ dependent NF-κ B activation by PN translates into a complete inhibition of the transcription of NF-κB dependent genes. Furthermore, PN simultaneously activates NIK and IKKα phosphorylation, suggesting its potential role in an alternative pathway of NF-κB activation, whose cellular consequences remain to be clarified. These findings offer new perspectives in understanding the relationships between oxidant stress and inflammation.
Fig. 9.
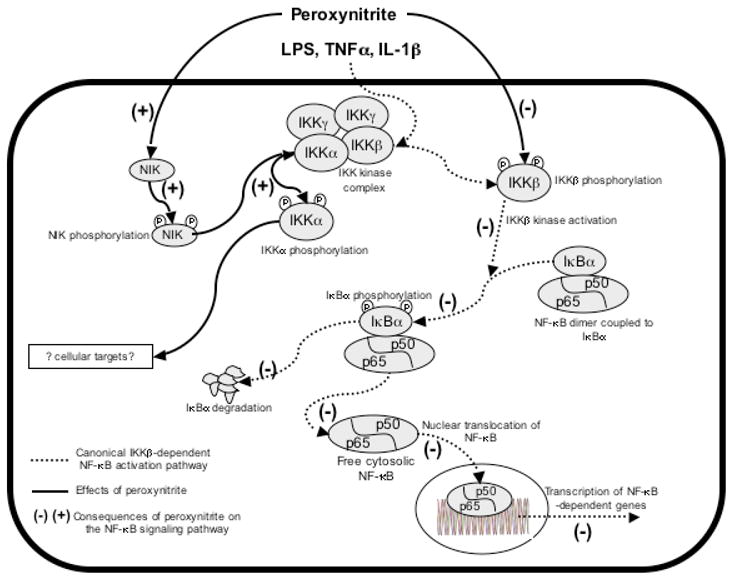
A model for peroxynitrite-mediated interference with the NF-κB pathway. Immune stimuli activate the canonical NF-κB pathway, by inducing phosphorylation and activation of IKKβ, in a process regulated by IKKγ. In turn, IKKβ phosphorylates IκBα, leading to its proteasomal degradation. NF-κB dimers (mostly p50/p65) then translocate into the nucleus, bind to κB consensus sequences, and activate the transcription of target genes. In the presence of peroxynitrite, IKKβ phosphorylation is impaired, thereby blocking IKKβ activation and downstream signaling (as indicated by the (−) signs). At the same time, peroxynitrite activates the phosphorylation of IKKα, most probably via the phosphorylation and activation of the upstream kinase NIK. The cellular targets of IKKα in this model remain to be defined.
Fig. 10.
Acknowledgments
Supported by grants from the Swiss National Fund for Scientific Research (3100-65005.01 and PP00B-68882/1) to LL.
References
- 1.Bonizzi G, Karin M. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 2.Ghosh S, Karin M. Cell. 2002;109(Suppl):S81–96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 3.Egan LJ, Eckmann L, Greten FR, Chae S, Li ZW, Myhre GM, Robine S, Karin M, Kagnoff MF. Proc Natl Acad Sci U S A. 2004;101:2452–2457. doi: 10.1073/pnas.0306734101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 5.Chen LW, Egan L, Li ZW, Greten FR, Kagnoff MF, Karin M. Nat Med. 2003;9:575–581. doi: 10.1038/nm849. [DOI] [PubMed] [Google Scholar]
- 6.Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, Johnson R, Karin M. J Exp Med. 1999;189:1839–1845. doi: 10.1084/jem.189.11.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Janssen-Heininger YM, Poynter ME, Baeuerle PA. Free Radic Biol Med. 2000;28:1317–1327. doi: 10.1016/s0891-5849(00)00218-5. [DOI] [PubMed] [Google Scholar]
- 8.Li N, Karin M. Faseb J. 1999;13:1137–1143. [PubMed] [Google Scholar]
- 9.Drayton DL, Bonizzi G, Ying X, Liao S, Karin M, Ruddle NH. J Immunol. 2004;173:6161–6168. doi: 10.4049/jimmunol.173.10.6161. [DOI] [PubMed] [Google Scholar]
- 10.Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C, Li ZW, Karin M, Ware CF, Green DR. Immunity. 2002;17:525–535. doi: 10.1016/s1074-7613(02)00423-5. [DOI] [PubMed] [Google Scholar]
- 11.Bonizzi G, Bebien M, Otero DC, Johnson-Vroom KE, Cao Y, Vu D, Jegga AG, Aronow BJ, Ghosh G, Rickert RC, Karin M. Embo J. 2004;23:4202–4210. doi: 10.1038/sj.emboj.7600391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC, Karin M. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 13.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Proc Natl Acad Sci U S A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liaudet L, Soriano FG, Szabo C. Crit Care Med. 2000;28:N37–52. doi: 10.1097/00003246-200004001-00005. [DOI] [PubMed] [Google Scholar]
- 15.Liaudet L, Yang Z, Al-Affar EB, Szabo C. Mol Med. 2001;7:406–417. [PMC free article] [PubMed] [Google Scholar]
- 16.Pacher P, Liaudet L, Mabley J, Komjati K, Szabo C. J Am Coll Cardiol. 2002;40:1006–1016. doi: 10.1016/s0735-1097(02)02062-4. [DOI] [PubMed] [Google Scholar]
- 17.Pacher P, Liaudet L, Bai P, Mabley JG, Kaminski PM, Virag L, Deb A, Szabo E, Ungvari Z, Wolin MS, Groves JT, Szabo C. Circulation. 2003;107:896–904. doi: 10.1161/01.cir.0000048192.52098.dd. [DOI] [PubMed] [Google Scholar]
- 18.Uppu RM, Pryor WA. Methods Enzymol. 1996;269:322–329. doi: 10.1016/s0076-6879(96)69033-8. [DOI] [PubMed] [Google Scholar]
- 19.Pesse B, Levrand S, Feihl F, Waeber B, Gavillet B, Pacher P, Liaudet L. J Mol Cell Cardiol. 2005;38:765–775. doi: 10.1016/j.yjmcc.2005.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klotz LO, Schieke SM, Sies H, Holbrook NJ. Biochem J. 2000;352(Pt 1):219–225. [PMC free article] [PubMed] [Google Scholar]
- 21.Schroeder P, Klotz LO, Buchczyk DP, Sadik CD, Schewe T, Sies H. Biochem Biophys Res Commun. 2001;285:782–787. doi: 10.1006/bbrc.2001.5210. [DOI] [PubMed] [Google Scholar]
- 22.Zhang P, Wang YZ, Kagan E, Bonner JC. J Biol Chem. 2000;275:22479–22486. doi: 10.1074/jbc.M910425199. [DOI] [PubMed] [Google Scholar]
- 23.van der Vliet A, Hristova M, Cross CE, Eiserich JP, Goldkorn T. J Biol Chem. 1998;273:31860–31866. doi: 10.1074/jbc.273.48.31860. [DOI] [PubMed] [Google Scholar]
- 24.Ischiropoulos H, Zhu L, Beckman JS. Arch Biochem Biophys. 1992;298:446–451. doi: 10.1016/0003-9861(92)90433-w. [DOI] [PubMed] [Google Scholar]
- 25.Upmacis RK, Deeb RS, Resnick MJ, Lindenbaum R, Gamss C, Mittar D, Hajjar DP. Am J Physiol Cell Physiol. 2004;286:C1271–1280. doi: 10.1152/ajpcell.00143.2003. [DOI] [PubMed] [Google Scholar]
- 26.Eaves-Pyles T, Murthy K, Liaudet L, Virag L, Ross G, Soriano FG, Szabo C, Salzman AL. J Immunol. 2001;166:1248–1260. doi: 10.4049/jimmunol.166.2.1248. [DOI] [PubMed] [Google Scholar]
- 27.Klotz LO, Schroeder P, Sies H. Free Radic Biol Med. 2002;33:737–743. doi: 10.1016/s0891-5849(02)00892-4. [DOI] [PubMed] [Google Scholar]
- 28.Matsushima A, Kaisho T, Rennert PD, Nakano H, Kurosawa K, Uchida D, Takeda K, Akira S, Matsumoto M. J Exp Med. 2001;193:631–636. doi: 10.1084/jem.193.5.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lancel S, Tissier S, Mordon S, Marechal X, Depontieu F, Scherpereel A, Chopin C, Neviere R. J Am Coll Cardiol. 2004;43:2348–2358. doi: 10.1016/j.jacc.2004.01.047. [DOI] [PubMed] [Google Scholar]
- 30.Schreck R, Rieber P, Baeuerle PA. Embo J. 1991;10:2247–2258. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kamata H, Manabe T, Oka S, Kamata K, Hirata H. FEBS Lett. 2002;519:231–237. doi: 10.1016/s0014-5793(02)02712-6. [DOI] [PubMed] [Google Scholar]
- 32.Imbert V, Rupec RA, Livolsi A, Pahl HL, Traenckner EB, Mueller-Dieckmann C, Farahifar D, Rossi B, Auberger P, Baeuerle PA, Peyron JF. Cell. 1996;86:787–798. doi: 10.1016/s0092-8674(00)80153-1. [DOI] [PubMed] [Google Scholar]
- 33.Hayakawa M, Miyashita H, Sakamoto I, Kitagawa M, Tanaka H, Yasuda H, Karin M, Kikugawa K. Embo J. 2003;22:3356–3366. doi: 10.1093/emboj/cdg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Korn SH, Wouters EF, Vos N, Janssen-Heininger YM. J Biol Chem. 2001;276:35693–35700. doi: 10.1074/jbc.M104321200. [DOI] [PubMed] [Google Scholar]
- 35.Delhase M, Hayakawa M, Chen Y, Karin M. Science. 1999;284:309–313. doi: 10.1126/science.284.5412.309. [DOI] [PubMed] [Google Scholar]
- 36.Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, Santoro MG. Nature. 2000;403:103–108. doi: 10.1038/47520. [DOI] [PubMed] [Google Scholar]
- 37.Kapahi P, Takahashi T, Natoli G, Adams SR, Chen Y, Tsien RY, Karin M. J Biol Chem. 2000;275:36062–36066. doi: 10.1074/jbc.M007204200. [DOI] [PubMed] [Google Scholar]
- 38.Viatour P, Merville MP, Bours V, Chariot A. Trends Biochem Sci. 2005;30:43–52. doi: 10.1016/j.tibs.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 39.Lawrence T, Bebien M, Liu GY, Nizet V, Karin M. Nature. 2005;434:1138–1143. doi: 10.1038/nature03491. [DOI] [PubMed] [Google Scholar]
- 40.Kumar A, Haery C, Parrillo JE. Crit Care Clin. 2000;16:251–287. doi: 10.1016/s0749-0704(05)70110-x. [DOI] [PubMed] [Google Scholar]
- 41.Torre-Amione G, Kapadia S, Lee J, Durand JB, Bies RD, Young JB, Mann DL. Circulation. 1996;93:704–711. doi: 10.1161/01.cir.93.4.704. [DOI] [PubMed] [Google Scholar]