

Abstract
Macro- and microvascular disease are the most common causes of morbidity and mortality in patients with diabetes mellitus. Diabetic cardiovascular dysfunction represents a problem of great clinical importance underlying the development of various severe complications including retinopathy, nephropathy, neuropathy and increase the risk of stroke, hypertension and myocardial infarction. Hyperglycemic episodes, which complicate even well-controlled cases of diabetes, are closely associated with increased oxidative and nitrosative stress, which can trigger the development of diabetic complications. Hyperglycemia stimulates the production of advanced glycosylated end products, activates protein kinase C, and enhances the polyol pathway leading to increased superoxide anion formation. Superoxide anion interacts with nitric oxide, forming the potent cytotoxin peroxynitrite, which attacks various biomolecules in the vascular endothelium, vascular smooth muscle and myocardium, leading to cardiovascular dysfunction. The pathogenetic role of nitrosative stress and peroxynitrite, and downstream mechanisms including poly(ADP-ribose) polymerase (PARP) activation, is not limited to the diabetes-induced cardiovascular dysfunction, but also contributes to the development and progression of diabetic nephropathy, retinopathy and neuropathy. Accordingly, neutralization of peroxynitrite or pharmacological inhibition of PARP is a promising new approach in the therapy and prevention of diabetic complications. This review focuses on the role of nitrosative stress and downstream mechanisms including activation of PARP in diabetic complications and on novel emerging therapeutical strategies offered by neutralization of peroxynitrite and inhibition of PARP.
Keywords: peroxynitrite, nitric oxide, superoxide, nitrotyrosine, diabetes, vascular, cardiomyopathy, nephropathy, neuropathy, retinopathy
INTRODUCTION
Diabetic state is associated with increased oxidative stress, which plays an important role in the development of diabetic complications. Hyperglycemia stimulates the production of advanced glycosylated end products, activates protein kinase C, and enhances the polyol pathway leading to increased superoxide anion formation [1–3]. Superoxide anion interacts with nitric oxide, which is produced, physiologically, by constitutive sources, such as the endothelial isoform of nitric oxide synthase (eNOS). This process leads to the formation of the strong oxidant peroxynitrite, which attacks various biomolecules leading to cellular dysfunction via multiple mechanisms (Table 1) [1,2,4–47]. One of these pathways involves DNA strand breakage and activation of the nuclear enzyme poly(ADP-ribose) polymerase, which has been covered by separate overviews [6,48,49]. The present review summarizes the accumulating experimental and clinical evidence implicating the pathogenetic role of increased nitrosative stress, peroxynitrite formation in the development of diabetic complications (Table 2). Although peroxynitrite generation also plays a role in the pathogenesis of islet-cell destruction [18,30,50], this is a separate area which is not the main focus of the present review.
Table 1.
Selected Cytotoxic Processes Initiated by Peroxynitrite, with Potential Relevance to Diabetic Complications
| Action | Mechanism(s) |
|---|---|
| Cytosolic enzyme inhibition | Oxidation, nitration |
| Membrane pump inhibition | Oxidation, nitration |
| Antioxidant enzyme inhibition | Oxidation, nitration |
| Signal transduction pathway disturbances | Oxidation, nitration |
| DNA injury | Oxidation, nitration, deamination, adduct formation |
| Surfactant protein damage | Nitration |
| Metalloproteinase activation | S-glutoxidation of prometalloproteinases |
| Antioxidant enzyme depletion | Glutathione, cysteine oxidation |
| Inhibition of BH4-dependent enzymes | Direct BH4 oxidation |
| Inhibition of NAD-dependent enzymes | NAD oxidation, NAD depletion via PARP |
| Lipid peroxidation | Peroxidation |
| Oxidative chain reactions | Lipid peroxidation, generation of reactive alpha-oxoaldehydes from glucose |
| Mitochondrial dysfunction | Inhibition of cytochromes, NADH-COQ1, etc. |
| Upregulation of adhesion receptors | NF- κB activation |
| GAPDH inhibition | Multiple, including PARP activation |
| Protein kinase C activation | Multiple, including GAPDH inhibition via PARP activation |
| Active DNA fragmentation | Caspase activation |
| Calcium dysregulation | Dysfunctional calcium pumps and cell energetics |
| Cell Necrosis | Mitochondrial injury, energetic collapse, oxidation, nitration, antioxidant depletion, calcium dysregulation |
| Apoptosis | Mitochondrial injury, DNA injury, caspase activation, signal transduction disturbances, calcium dysregulation |
(Please note that to date many of the mechanisms listed have been demonstrated in vitro but not in experimental or clinical diabetes in vivo).
Table 2.
Evidence Implicating Nitrosative Stress and Peroxynitrite Formation in Diabetes and Diabetic Complications
| Organ/Tissue, function Investigated | Species/ cells | Disease Model, trigger | Main Finding | Reference |
|---|---|---|---|---|
| Plasma | Human | T2DM | Cardiopulmonary bypass induced greater oxidative and nitrosative stress in diabetic patients. | 7 |
| Plasma | Human | T2DM | Increased plasma nitrotyrosine levels in diabetic patients | 8 |
| Plasma | Human | T2DM | Increased plasma nitrite/nitrate levels in diabetic patients. | 9 |
| Plasma | Human | T2DM | Postprandial hypertriglyceridemia and hyperglycemia induced endothelial dysfunction in diabetic patients and increased plasma nitrotyrosine levels, which was attenuated by simvastatin treatment. | 10 |
| Plasma | Human | T2DM | Increased plasma nitrotyrosine levels in diabetic patients, which correlate with postprandial hyperglycemia. | 11 |
| Plasma | Human | T1DM | Increased plasma nitrite, nitrate and nitrotyrosine , which correlate with the insulin requirements of the diabetic patients. | 12 |
| Plasma | Human | T1DM | Increased plasma nitrite/nitrate, nitrotyrosine and elevated blood pressure in diabetic patients. | 13 |
| LDL | Human | T1DM | Incubation of human aortic endothelial cells with LDL from T1DM patients increased Na+/K+-ATPase and Ca2+- ATPase activities, NOS activity and peroxynitrite production. | 14 |
| Platelets | Human | T1DM, T2DM | Increased iNOS derived peroxynitrite formation in diabetic platelets | 15 |
| Aorta, vascular function | Mouse | Mouse STZ-induced diabetes | Increased eNOS expression, nitrotyrosine formation and PARP activation in endothelium and vascular smooth muscle. | 16 |
| Skin microvasculature | Human | T2DM and prediabetic | Increased nitrotyrosine formation and PARP activation in endothelial cells of diabetic and prediabetic patients. | 17 |
| Aorta, cardiac and vascular function, pancreatic islet beta- cells | Mouse | STZ-induced diabetes. | A peroxynitrite decomposition catalyst improved vascular and cardiac function and protected against diabetes. | 18 |
| Human aortic endothelial cells | Human | High glucose | Increased ONOO- formation, tyrosine nitration and inhibition of prostacycline synthase. | 19 |
| Human umbilical vein endothelial cells | Human | Stable or intermittent high glucose | Stable or intermittent high glucose stimulated nitrotyrosine formation through PKC-dependent activation of NAD(P)H oxidase. | 20 |
| Human aortic endothelial cells | Human | High glucose | Glucose-induced activation of PKC resulted in Peroxynitrite formation and nitration of prostacyclin synthase. | 21 |
| Bovine endothelial cells | Bovine | Hyperglycemia (HG) | HG induced increased lipid peroxidation, increased superoxide and peroxynitrite formation, and PKC activity. | 22 |
| Aorta, liver, kidney | Rat | STZ-induced diabetes | Increased free radical and NO concentrations in the liver, kidney and aorta; increased ONOO− formation in aorta. | 23 |
| Aorta, vascular function | Rat | Zucker diabetic rats | Age-dependent increase of nitrotyrosine formation in the vasculature and development of endothelial dysfunction, which is attenuated by a peroxynitrite scavenger, ebselen. | 24 |
| Cardiac myocytes | Human | T2DM, hypertension | Increased apoptosis, necrosis, angiotensine II and nitrotyrosine formation in myocytes. | 25 |
| Cardiac myocytes | Mouse | STZ-induced diabetes | Increased apoptosis, H2O2, .OH, angiotensine II and nitrotyrosine formation in myocytes, which is decreased by IGF-1 overexpression. | 26 |
| Heart mitochondria | Rat | STZ-induced diabetes | Increased nitration and inactivation of succinyl-CoA:3- oxoacid CoA-transferase (SCOT). | 27 |
| Heart | Rat | High glucose | Perfusion of isolated hearts with high glucose increased superoxide generation, NO, nitrotyrosine formation and iNOS expression. | 28 |
| Heart | Mouse | Alloxan-induced diabtes | Tyrosine nitration of mitochondrial proteins. | 29 |
| Pancreatic islet beta-cells | Mouse | NOD mice | Increased nitrotyrosine formation in pancreatic islet beta-cells. | 30 |
| Placenta | Human | T1DM | Increased nitration in vascular endothelium and villous stroma. | 31 |
| Placental vasculature | Human | T1DM, preeclampsia | Increased nitrotyrosine formation, attenuated vasoconstrictor and vasodilatory responses in diabetes and preeclampsia. | 32 |
| Kidney | Human | Patients with diabetic nephropathy | Increased nitrotyrosine immunostaining in renal tubuli of diabetic patients. | 33 |
| Kidney | Rat | STZ-induced diabetes | Increased superoxide and nitrotyrosine formation in renal cortex. | 34 |
| Kidney | Mouse | STZ-induced diabetes | Increased renal nitrotyrosine and advanced glycation end product formation, which is attenuated by ramipril or aminoguanidine. | 35 |
| Kidney | Rat | STZ-induced diabetes | Increased renal expression of p47phox, hydrogen peroxide production and nitrotyrosine formation. | 36 |
| Retina | Rat | STZ-induced diabetes | Increased nitrosative stress, which is attenuated by aminoguanidine. | 37 |
| Retina | Rat | BBZ/Wor rat model of NIDDM | Increased iNOS and nitrotyrosine immunoreactivity in diabetic retinas. | 38 |
| Retina | Rat | STZ-induced diabetes | Increased retinal lipid peroxidation and nitrotyrosine formation, which was only slightly attenuated by reinstitution of good glycemic control. | 39 |
| Retina | Rat | STZ-induced diabetes | Increased tyrosine nitration and expression of vascular endothelial growth factor contribute to the breakdown of the blood-retina barrier in diabetes. | 40 |
| Retinal endothelial cells | High glucose | High glucose induced increased nitrotyrosine formation in retinal endothelial cells, which was blocked by superoxide or peroxynitrite scavengers, NOS or aldose reductase inhibitors. | 41 | |
| Peripheral nerves, epineurial arterioles, endoneurial blood flow | Rat | STZ-induced diabetes | Antioxidants reduced the production of superoxide and peroxynitrite in epineurial arterioles and improved endoneural blood flow. | 42 |
| Epineurial arterioles, endoneurial blood flow | Rat | STZ-induced diabetes | Antioxidant (M40403) reduced the production of superoxide and peroxynitrite in epineurial arterioles and improved endoneural blood flow. | 43 |
| Peripheral motor nerve function | Human | T1DM | Decreased motor nerve function in diabetic patients correlates with increased nitrosative stress. | 44 |
| Peripheral sensory neurons | Rat | STZ-induced diabetes | Rise in cytoplasmic labeling of nitrotyrosine, PARP activation. | 45 |
| Peripheral nerves, epineurial arterioles, endoneurial blood flow | Rat | STZ-induced diabetes | Antioxidants reduced the production of superoxide and peroxynitrite in epineurial arterioles and improved endoneural blood flow. | 46 |
| Bladder | Rat | STZ-induced diabetes | Increased proteasomal activation and nitrotyrosine formation during diabetic cystopathy. | 47 |
THE ROLE OF OXIDATIVE AND NITROSATIVE STRESS IN THE PATHOGENESIS OF DIABETES-INDUCED VASCULAR DYSFUNCTION
Various neurohumoral mediators and mechanical forces acting upon the innermost layer of blood vessels, the endothelium, are involved in the regulation of the vascular tone. A main pathway of vasoregulation involves the activation of the eNOS resulting in NO production [51]. Endothelium-dependent vasodilatation is frequently used as a reproducible and accessible parameter to probe endothelial function in various pathophysiological conditions. It is well established that endothelial dysfunction, in many diseases, precedes and predicts as well as predisposes for the subsequent, more severe vascular alterations. Endothelial dysfunction has been documented in various forms of diabetes, and even in pre-diabetic individuals [3,17,21,52–57]. The pathogenesis of this endothelial dysfunction involves many components including increased polyol pathway flux, altered cellular redox state, increasedformation of diacylglycerol and the subsequent activation of specific protein kinase C isoforms, and accelerated nonenzymatic formation of advanced glycation end products [58– 63]. Many of these pathways, in concert, trigger the production of oxygen- and nitrogen-derived oxidants and free radicals, such as superoxide anion and peroxynitrite, which play a significant role in the pathogenesis of diabetes-associated endothelial dysfunction [59–61,64]. The cellular sources of reactive oxygen species such as superoxide anion are multiple and include advanced glycation end products, NAD(P)H oxidases, the mitochondrial respiratory chain, xanthine oxidase, the arachidonic acid cascade (lipoxygenase and cycloxygenase), and microsomal enzymes [1,59].
Superoxide anion may quench NO, thereby reducing the efficacy of a potent endothelium-derived vasodilator system that participates in the homeostatic regulation of the vasculature, and evidence suggests that during hyperglycemia, reduced NO availability exists [65]. Hyperglycemia-induced superoxide generation contributes to the increased expression of NAD(P)H oxidase, which in turn generate more superoxide anion. Hyperglycemia also favors, through the activation of NF-κB an increased expression of iNOS, which may increase the generation of NO [56,66].
Superoxide anion interacts with nitric oxide, forming the strong cytotoxin peroxynitrite (ONOO−), which attacks various biomolecules, leading — among other processes —to the production of a modified amino acid, nitrotyrosine [67]. Although nitrotyrosine was initially considered a specific marker of peroxynitrite generation, other pathways can also induce tyrosine nitration. Thus, nitrotyrosine is now generally considered a collective index of reactive nitrogen species, rather than a specific indicator of peroxynitrite formation [68,69]. The possibility that diabetes is associated with increased nitrosative stress is supported by the recent detection of increased nitrotyrosine plasma levels in type 2 diabetic patients [8] and iNOS-dependent peroxynitrite production in diabetic platelets [15]. Nitrotyrosine formation is detected in the artery wall of monkeys during hyperglycemia [70] and in diabetic patients during an increase of postprandial hyperglycemia [10,11]. In a recent study we have demonstrated increased nitrotyrosine immunoreactivity in microvasculature of type 2 diabetic patients [17]. In the same study significant correlations were observed between nitrotyrosine immunostaining intensity and fasting blood glucose, HbA1c, intracellular adhesion molecule (ICAM), and vascular cellular adhesion molecule (VCAM).
The toxic actions of nitrotyrosine in the cardiovascular system are also highlighted by the evidence showing that there is increased apoptosis of endothelial cells, myocytes and fibroblasts in heart biopsies from diabetic patients [25], in hearts from streptozotocin-induced diabetic rats [26], and in working hearts from rats during hyperglycemia [28]. Importantly, the degree of cell death and/or dysfunction shows a correlation with levels of nitrotyrosine found in those cells. There is also evidence that nitrotyrosine can be directly harmful to endothelial cells [71]. In addition, high glucose-induced oxidative and nitrosative stress pathologically alters prostanoid profile in human endothelial cells [19,21].
Recent evidence indicates that there may be several phases to the pathogenesis of the endothelial injury induced by high glucose: the short-term effect appear to depend on a combined oxidative and nitrosative stress with peroxynitrite formation, whereas the long-term effect is related to reactive oxygen species generation; in both cases, protein kinase C ultimately mediates the vascular permeability changes [22].
Angiotensin II is a known factor in the pathogenesis of diabetic complications, perhaps most importantly, in nephropathy, cardiomyopathy and retinopathy. Recent studies indicate that the protective effects of angiotensin converting enzyme inhibitors or angiotensin receptor antagonists may go beyond the blood pressure lowering effects of these agents [72–74]. Furthermore, ACE inhibition in vivo reduces the apparent formation of peroxynitrite [35]. In this context it is noteworthy that angiotensin II can induce direct, pro-oxidative effects on the vascular endothelium. These effects are, at least in part, mediated by intraendothelial reactive species formation via a new family of NAD(P)H oxidase subunits, known as the non-phagocytic NAD(P)H oxidase proteins. Reactive oxidant species produced following angiotensin II-mediated stimulation of NAD(P)H oxidases can exert direct oxidative effects, but can also signal through pathways such as mitogen-activated protein kinases, tyrosine kinases and transcription factors, and lead to events such as inflammation, hypertrophy, remodeling and angiogenesis [54]. Recent work demonstrates that angiotensin II can also induce intraendothelial peroxynitrite formation [6,75–77], as well as PARP activation [76,77].
THE ROLE OF OXIDATIVE AND NITROSATIVE STRESS IN THE PATHOGENESIS OF DIABETIC CARDIOMYOPATHY
Diabetic cardiomyopathy is characterized by complex changes in the mechanical, biochemical, structural, and electrical properties of the heart, which may be responsible for the development of an early diastolic dysfunction and increased incidence of cardiac arrhythmias in diabetic patients. The mechanism of diastolic dysfunction remains unknown but it does not appear to be due to changes in blood pressure, microvascular complications or elevated circulating glycated hemoglobin levels [78–82]. There is circumstantial clinical and experimental evidence suggesting that increased sympathetic activity, activated cardiac renin-angiotensin system, myocardial ischemia/functional hypoxia, and elevated circulating levels of glucose result in oxidative and nitrosative stress in cardiovascular system of diabetic animals and humans. Oxidative stress associated with an impaired antioxidant defense status may play a critical role in subcellular remodeling, calcium-handling abnormalities, and subsequent diabetic cardiomyopathy [26,82,83].
Oxidative and nitrosative damage may be critical in the early onset of diabetic cardiomyopathy [18,25,26,28]. Even in simple model systems, e.g. placement of beating myocytes into culture medium containing elevated glucose, the pathophysiological alterations can be attenuated by antioxidants, NOS inhibitors, as well as by peroxynitrite neutralizing agents [84]. Consistently with the in vivo importance of this latter pathomechanism, significant nitrotyrosine formation was reported in cardiac myocytes from myocardial biopsy samples obtained from diabetic and diabetic hypertensive patients [25] and in a mouse model of STZ-induced diabetes [26]. Perfusion of isolated hearts with high glucose caused a significant upregulation of iNOS, increased the coronary perfusion pressure and both NO and superoxide generation, a condition favoring the production of peroxynitrite, accompanied by the formation of nitrotyrosine and cardiac cell apoptosis [28]. Fig. (1) shows increased NT formation in STZ-induced diabetic rat tissues.
Fig. 1. Increased nitrotyrosine (NT) formation in diabetic tissues.
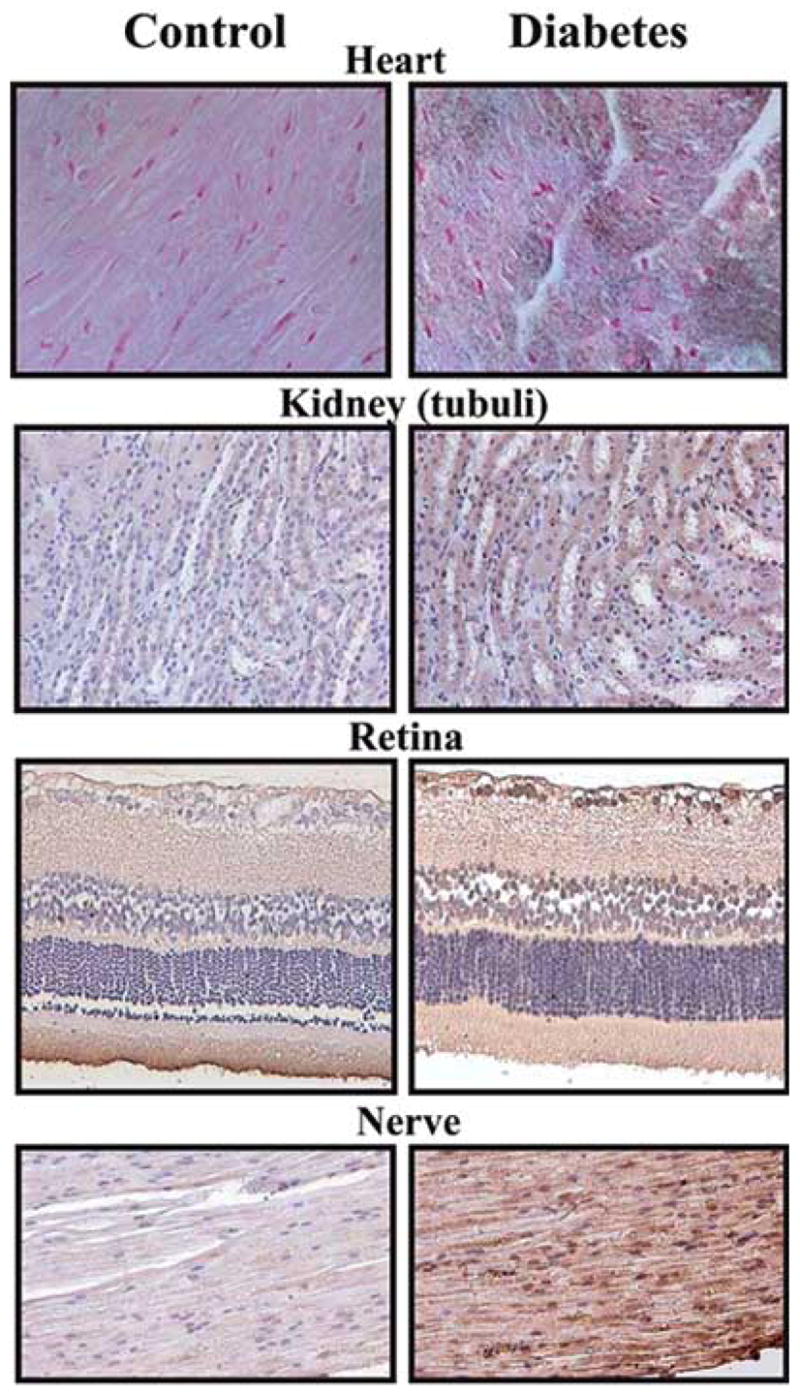
Immunohistochemical staining for NT, an indicator of peroxynitrite formation, in control (left column), and 8 weeks old STZ-induced diabetic (right column) rat heart, kidney, retina and sciatic nerve tissue samples.
PEROXYNITRITE NEUTRALIZATION IMPROVES CARDIAC AND VASCULAR DYSFUNCTION IN DIABETES
As mentioned above there is circumstantial evidence that nitrosative stress and peroxynitrite formation importantly contribute to the pathogenesis of diabetic cardiomyopathy both in animals and humans. We have tested a novel metalloporphyrin peroxynitrite decomposition catalyst, FP15, in murine models of diabetic cardiovascular complications [18]. We hypothesized that neutralization of peroxynitrite with FP15 would ameliorate the development of cardiovascular dysfunction in a streptozotocin-induced murine model of diabetes. In order to ensure that the animals received the FP15 treatment at a time when islet cell destruction was already complete and hyperglycemia has stabilized the treatment was initiated six week after the injection of streptozotocin. Although FP15 did not affect blood glucose levels, it provided a marked protection against the loss of endothelium-dependent relaxant ability of the blood vessels (Fig. 2A) and improved the depression of both diastolic (Fig. 2B) and systolic function of the heart [18]. The mechanism by which FP15 protects diabetic hearts from dysfunction may involve protection against vascular and myocardial tyrosine nitration, PARP activation, lipid peroxidation, and multiple other mechanisms, as all these mechanisms have previously been linked to diabetic cardiomyopathy as well as to peroxynitrite-induced cardiac injury. Additional mechanisms of peroxynitrite-mediated diabetic cardiac dysfunction may include inhibition of myofibrillar creatine kinase [85] and of succinyl-CoA:3-oxoacid CoAtransferase [27] or activation of metalloproteinases [45,86].
Fig. 2. Panel A. Reversal of diabetes-induced endothelial dysfunction by the porphyrinic peroxynitrite decomposition catalyst, FP15, in vascular rings from STZ-diabetic mice.
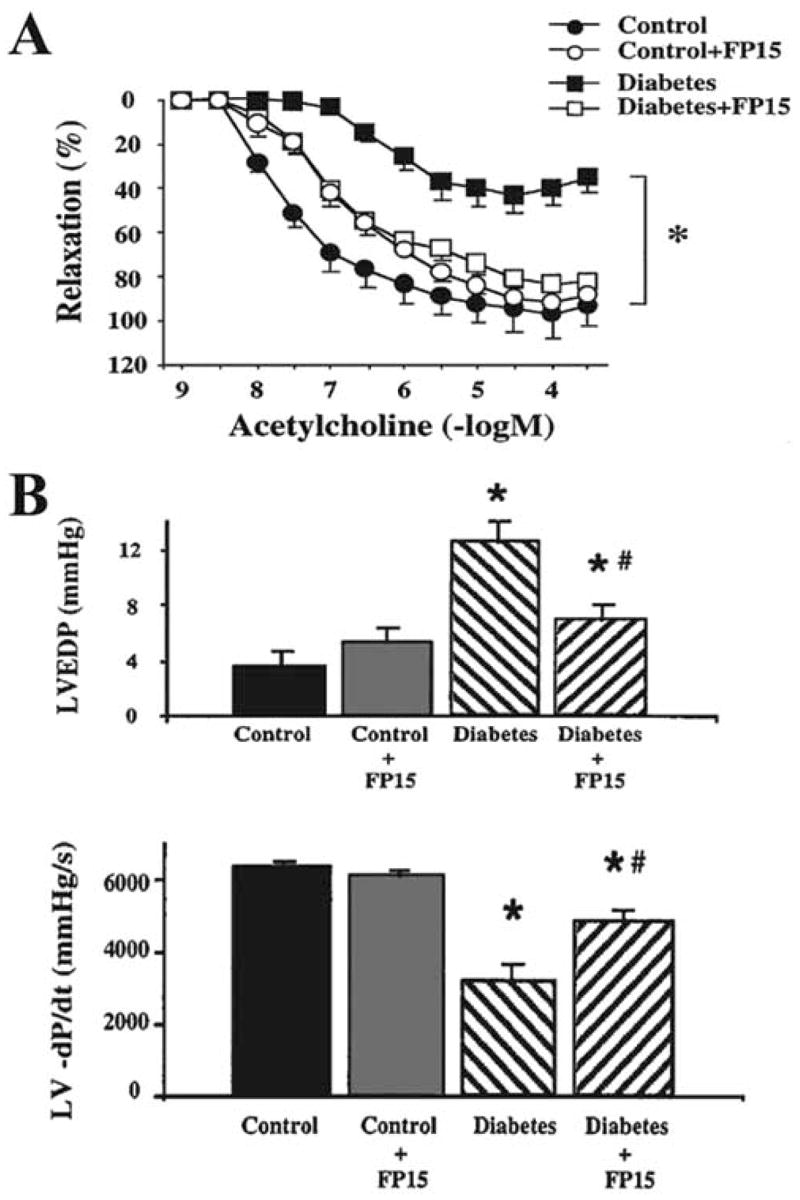
Acetylcholine (Ach) induced endothelium-dependent relaxation is impaired in rings from diabetic mice, which is markedly improved by FP15 treatment. Each point of the curve represents the mean ± SEM of 5–7 pairs of experiments in vascular rings. *p< 0.05 in FP15 treated diabetic mice versus vehicle-treated diabetic mice.
Panel B. Reversal of streptozotocin-evoked diabetes-induced diastolic cardiac dysfunction by the porphyrinic peroxynitrite decomposition catalyst, FP15, in mice.
Effect of diabetes (9–10 weeks) and FP15 treatment in diabetic mice on left ventricular end diastolic pressure (LVEDP) and left ventricular -dP/dt (LV -dP/dt). Results are mean ± SEM of seven experiments in each group. *p< 0.05 diabetic animals versus control; #p< 0.05 in FP15-treated diabetic mice versus vehicle-treated diabetic mice. Reproduced with permission from 18.
There are many pathophysiological conditions of the heart that are associated with peroxynitrite formation, including acute myocardial infarction, chronic ischemic heart failure, doxorubicin-induced and diabetic cardiomyopathy [86–91]. It appears that peroxynitrite decomposition catalysts improve cardiac function and overall outcome in these models. For instance, FP15 reduced myocardial necrosis in our current rat model of acute myocardial infarction [86] as well as in a recent porcine study [87]. Furthermore, FP15 significantly improved cardiac function in a doxorubicin-induced model of heart failure [86]. These observations-coupled with the recently reported protective effect of FP15 against diabetic cardiomyopathy-support the concept that peroxynitrite is a major mediator of myocardial injury in various pathophysiological conditions, and its effective neutralization can be of significant therapeutic benefit.
THE ROLE OF OXIDATIVE AND NITROSATIVE STRESS IN THE PATHOGENESIS OF DIABETIC RETINOPATHY, NEPHROPATHY AND EUROPATHY
Recent studies have suggested that increased oxidative and nitrosative stress is involved in the pathogenesis of diabetic microvascular injury in retinopathy nephropathy and neuropathy [33–46, 92,93] (Table 2, Fig. (1)).
Retinal endothelial cells maintained in high glucose had significant increased eNOS expression and activity as well as increased formation of superoxide anion and nitrotyrosine [40,41]. Each of these alterations was blocked by the NOS inhibitor, L-NAME, or the peroxynitrite scavenger, uric acid. Consistently with these observation there is increased oxidative and nitrosative stress in retinas of diabetic animals, which is attenuated by antioxidant treatment [39–41]. In addition increased peroxynitrite-mediated VEGF and urokinase plasminogen activator receptor expression was demonstrated and proposed to be responsible for the breakdown of the blood-retina barrier in diabetic animals [40,41].
Increased oxidative stress and nitrotyrosine formation have also been demonstrated both in kidneys of diabetic animals [34–36] and in biopsies from patients with diabetic nephropathy [33] suggesting pathogenetic role in the development of this complication.
Although hyperglycemia has been proven to cause peripheral nerve dysfunction in patients with diabetes, the biochemical mechanisms for this effect are poorly understood [94]. Recent studies in experimental animals have indicated that hyperglycemia stimulates the production of nitric oxide, which reacts with superoxide anion to form peroxynitrite, which is damaging the endothelium and perineurium [42–44,46,93]. In a recent murine study, sciatic motor nerve conduction velocity and hind-limb digital sensory conduction velocity were reduced in diabetic mice versus controls, and both indices were normalized by FP15, a peroxynitrite decomposition catalyst compound [95], which also ameliorated the accumulation of poly(ADP-ribose) accumulation in diabetic nerves [95].
It is noteworthy that in preclinical studies, administration of the aldose reductase inhibitors sorbinil or fidarestat to diabetic rats not only corrected diabetes-induced depletion of glutathione and ascorbate, downregulation of SOD activity and accumulation of lipid peroxidation products in the peripheral nerve, superoxide formation in vasa nervorum and of diabetes-associated retinal oxidative and nitrosative stress, but also inhibited poly(ADP-ribose) accumulation (a marker of PARP activation) in diabetic nerve and retina [96].
PEROXYNITRITE-POLY(ADP-RIBOSE) POLYMERASE CONNECTION IN THE PATHOGENESIS OF DIABETIC COMPLICATIONS
Peroxynitrite also damages DNA and thus triggers the activation of DNA repair systems. A DNA nick sensor enzyme, poly(ADP-ribose) polymerase-1 (PARP-1) also becomes activated upon sensing DNA breakage. Activated PARP-1 cleaves NAD+ into nicotinamide and ADP-ribose and polymerizes the latter on nuclear acceptor proteins. Peroxynitrite-induced overactivation of PARP consumes NAD+ and consequently ATP culminating in cell dysfunction, apoptosis or necrosis [6,97]. PARP-1 activation has recently been implicated in the pathogenesis of diabetes and diabetic complications [48] including cardiovascular dysfunction [16,37,98–103], nephropathy [104], neuropathy [99,105] and retinopathy [106].
CONCLUSIONS AND IMPLICATIONS
Taken together, multiple lines of evidence support the view that nitrosative stress and peroxyntrite-induced damage play a crucial role in multiple interrelated aspects of the pathogenesis of diabetes and its complications. Neutralization of reactive nitrogen species or inhibition of downstream pathways including PARP activation may emerge as a novel approach for the experimental therapy of diabetes, as well as for the prevention or reversal of its complications.
ABBREVIATIONS
- AGE
Advanced glycation end product
- AP-1
Activator protein
- DNA
Deoxyribonucleic acid
- eNOS
Endothelial nitric oxide synthase
- ET-1
Endothelin-1
- ICAM-1
Intracellular adhesion molecule
- iNOS
Inducible nitric oxide synthase
- NAD+
Nicotinamide adenine dinucleotide
- NT
Nitrotyrosine (an index of nitrosative stress and Peroxynitrite formation)
- ONOO−
Peroxynitrite
- PARP/ PARS
Poly(ADP-ribose) polymerase/synthase
- PKC
Protein kinase C
- STZ
Streptozotocin
- T2DM
Type 2 diabetes
- T1DM
Type 1 diabetes
- VCAM-1
Vascular cellular adhesion molecule
References
- 1.Brownlee M. Nature. 2001;14:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 2.Ceriello A. Diabetes Care. 2003;26:1589–1596. doi: 10.2337/diacare.26.5.1589. [DOI] [PubMed] [Google Scholar]
- 3.Ruderman NB, Williamson JR, Brownlee M. FASEB J. 1992;6:2905–2914. doi: 10.1096/fasebj.6.11.1644256. [DOI] [PubMed] [Google Scholar]
- 4.Hoeldtke RD. Clin Auton Res. 2003;13(6):406–421. doi: 10.1007/s10286-003-0139-x. [DOI] [PubMed] [Google Scholar]
- 5.Nagai R, Unno Y, Hayashi MC, Masuda S, Hayase F, Kinae N, Horiuchi S. Diabetes. 2002;51(9):2833–2839. doi: 10.2337/diabetes.51.9.2833. [DOI] [PubMed] [Google Scholar]
- 6.Virag L, Szabo E, Gergely P, Szabo C. Toxicol Lett. 2003;140–141:113–124. doi: 10.1016/s0378-4274(02)00508-8. [DOI] [PubMed] [Google Scholar]
- 7.Matata BM, Galinanes M. J Thorac Cardiovasc Surg. 2000;120(1):1–11. doi: 10.1067/mtc.2000.106835. [DOI] [PubMed] [Google Scholar]
- 8.Ceriello A, Mercuri F, Quagliaro L, Assaloni R, Motz E, Tonutti L, Taboga C. Diabetologia. 2001;44:834–838. doi: 10.1007/s001250100529. [DOI] [PubMed] [Google Scholar]
- 9.Aydin A, Orhan H, Sayal A, Ozata M, Sahin G, Isimer A. Clin Biochem. 2001;34:65–70. doi: 10.1016/s0009-9120(00)00199-5. [DOI] [PubMed] [Google Scholar]
- 10.Ceriello A, Taboga C, Tonutti L, Quagliaro L, Piconi L, Bais B, Da Ros R, Motz E. Circulation. 2002;106(10):1211–1218. doi: 10.1161/01.cir.0000027569.76671.a8. [DOI] [PubMed] [Google Scholar]
- 11.Ceriello A, Quagliaro L, Catone B, Pascon R, Piazzola M, Bais B, Marra G, Tonutti L, Taboga C, Motz E. Diabetes Care. 2002;25:1439–1443. doi: 10.2337/diacare.25.8.1439. [DOI] [PubMed] [Google Scholar]
- 12.Hoeldtke RD, Bryner KD, McNeill DR, Warehime SS, Van Dyke K, Hobbs G. J Clin Endocrinol Metab. 2003;88(4):1624–1628. doi: 10.1210/jc.2002-021525. [DOI] [PubMed] [Google Scholar]
- 13.Hoeldtke RD, Bryner KD, McNeill DR, Hobbs GR, Baylis C. Am J Hypertens. 2003;16(9 Pt 1):761–766. doi: 10.1016/s0895-7061(03)00976-2. [DOI] [PubMed] [Google Scholar]
- 14.Rabini RA, Vignini A, Salvolini E, Staffolani R, Martarelli D, Moretti N, Mazzanti L. Atherosclerosis. 2002;165(1):69–77. doi: 10.1016/s0021-9150(02)00197-1. [DOI] [PubMed] [Google Scholar]
- 15.Tannous M, Rabini RA, Vignini A, Moretti N, Fumelli P, Zielinski B, Mazzanti L, Mutus B. Diabetologia. 1999;42(5):539–544. doi: 10.1007/s001250051192. [DOI] [PubMed] [Google Scholar]
- 16.Garcia Soriano F, Virág L, Jagtap P, Szabó É, Mabley JG, Liaudet L, Marton A, Hoyt DG, Murthy KG, Salzman AL, Southan GJ, Szabo C. Nature Medicine. 2001;7:108–113. doi: 10.1038/83241. [DOI] [PubMed] [Google Scholar]
- 17.Szabo C, Zanchi A, Komjati K, Pacher P, Krolewski AS, Quist WC, LoGerfo FW, Horton ES, Veves A. Circulation. 2002;106:2680–2686. doi: 10.1161/01.cir.0000038365.78031.9c. [DOI] [PubMed] [Google Scholar]
- 18.Szabo C, Mabley JG, Moeller SM, Shimanovich R, Pacher P, Virag L, Soriano FG, Van Duzer JH, Williams W, Salzman AL, Groves JT. Mol Med. 2002;8:571–580. [PMC free article] [PubMed] [Google Scholar]
- 19.Zou MH, Shi C, Cohen RA. Diabetes. 2002;51:198–203. doi: 10.2337/diabetes.51.1.198. [DOI] [PubMed] [Google Scholar]
- 20.Quagliaro L, Piconi L, Assaloni R, Martinelli L, Motz E, Ceriello A. Diabetes. 2003;52(11):2795–2804. doi: 10.2337/diabetes.52.11.2795. [DOI] [PubMed] [Google Scholar]
- 21.Cosentino F, Eto M, De Paolis P, van der Loo B, Bachschmid M, Ullrich V, Kouroedov A, Delli Gatti C, Joch H, Volpe M, Luscher TF. Circulation. 2003;107:1017–1023. doi: 10.1161/01.cir.0000051367.92927.07. [DOI] [PubMed] [Google Scholar]
- 22.Pricci F, Leto G, Amadio L, Iacobini C, Cordone S, Catalano S, Zicari A, Sorcini M, Di Mario U, Pugliese G. Free Radic Biol Med. 2003;35(6):683–694. doi: 10.1016/s0891-5849(03)00401-5. [DOI] [PubMed] [Google Scholar]
- 23.Stadler K, Jenei V, von Bolcshazy G, Somogyi A, Jakus J. Free Radic Biol Med. 2003;35(10):1240–1251. doi: 10.1016/s0891-5849(03)00499-4. [DOI] [PubMed] [Google Scholar]
- 24.Brodsky SV, Gealekman O, Chen J, Zhang F, Togashi N, Crabtree M, Gross SS, Nasjletti A, Goligorsky MS. Circ Res. 2004;94(3):377–384. doi: 10.1161/01.RES.0000111802.09964.EF. [DOI] [PubMed] [Google Scholar]
- 25.Frustaci A, Kajstura J, Chimenti C, Jakoniuk I, Leri A, Maseri A, Nadal-Ginard B, Anversa P. Circ Res. 2000;87:1123–1132. doi: 10.1161/01.res.87.12.1123. [DOI] [PubMed] [Google Scholar]
- 26.Kajstura J, Fiordaliso F, Andreoli AM, Li B, Chimenti S, Medow MS, Limana F, Nadal-Ginard B, Leri A, Anversa P. Diabetes. 2001;50(6):1414–1424. doi: 10.2337/diabetes.50.6.1414. [DOI] [PubMed] [Google Scholar]
- 27.Turko IV, Marcondes S, Murad F. Am J Physiol. 2001;281:2289–2294. doi: 10.1152/ajpheart.2001.281.6.H2289. [DOI] [PubMed] [Google Scholar]
- 28.Ceriello A, Quagliaro L, D'Amico M, Di Filippo C, Marfella R, Nappo F, Berrino L, Rossi F, Giugliano D. Diabetes. 2002;51:1076–1082. doi: 10.2337/diabetes.51.4.1076. [DOI] [PubMed] [Google Scholar]
- 29.Turko IV, Li L, Aulak KS, Stuehr DJ, Chang JY, Murad F. J Biol Chem. 2003;278(36):33972–33977. doi: 10.1074/jbc.M303734200. [DOI] [PubMed] [Google Scholar]
- 30.Suarez-Pinzon WL, Szabo C, Rabinovitch A. Diabetes. 1997;46(5):907–911. doi: 10.2337/diab.46.5.907. [DOI] [PubMed] [Google Scholar]
- 31.Lyall F, Gibson JL, Greer IA, Brockman DE, Eis AL, Myatt L. Diabetes Care. 1998;21(10):1753–1758. doi: 10.2337/diacare.21.10.1753. [DOI] [PubMed] [Google Scholar]
- 32.Kossenjans W, Eis A, Sahay R, Brockman D, Myatt L. Am J Physiol Heart Circ Physiol. 2000;278(4):H1311–1319. doi: 10.1152/ajpheart.2000.278.4.H1311. [DOI] [PubMed] [Google Scholar]
- 33.Thuraisingham RC, Nott CA, Dodd SM, Yaqoob MM. Kidney Int. 2000;57(5):1968– 1972. doi: 10.1046/j.1523-1755.2000.00046.x. [DOI] [PubMed] [Google Scholar]
- 34.Ishii N, Patel KP, Lane PH, Taylor T, Bian K, Murad F, Pollock JS, Carmines PK. J Am Soc Nephrol. 2001;12(8):1630–1639. doi: 10.1681/ASN.V1281630. [DOI] [PubMed] [Google Scholar]
- 35.Forbes JM, Cooper ME, Thallas V, Burns WC, Thomas MC, Brammar GC, Lee F, Grant SL, Burrell LA, Jerums G, Osicka TM. Diabetes. 2002;51(11):3274–3282. doi: 10.2337/diabetes.51.11.3274. [DOI] [PubMed] [Google Scholar]
- 36.Onozato ML, Tojo A, Goto A, Fujita T, Wilcox CS. Kidney Int. 2002;61:186–194. doi: 10.1046/j.1523-1755.2002.00123.x. [DOI] [PubMed] [Google Scholar]
- 37.Du X, Matsumura T, Edelstein D, Rossetti L, Zsengeller Z, Szabo C, Brownlee M. J Clin Invest. 2003;112:1049–1057. doi: 10.1172/JCI18127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ellis EA, Guberski DL, Hutson B, Grant MB. Nitric Oxide. 2002;6:295–304. doi: 10.1006/niox.2001.0419. [DOI] [PubMed] [Google Scholar]
- 39.Kowluru RA. Diabetes. 2003;52(3):818–823. doi: 10.2337/diabetes.52.3.818. [DOI] [PubMed] [Google Scholar]
- 40.El-Remessy AB, Behzadian MA, Abou-Mohamed G, Franklin T, Caldwell RW, Caldwell RB. Am J Pathol. 2003;162:1995–2004. doi: 10.1016/S0002-9440(10)64332-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.El-Remessy AB, Abou-Mohamed G, Caldwell RW, Caldwell RB. Invest Ophthalmol Vis Sci. 2003;44(7):3135–3143. doi: 10.1167/iovs.02-1022. [DOI] [PubMed] [Google Scholar]
- 42.Coppey LJ, Gellett JS, Davidson EP, Dunlap JA, Lund DD, Yorek MA. Diabetes. 2001;50:1927–1937. doi: 10.2337/diabetes.50.8.1927. [DOI] [PubMed] [Google Scholar]
- 43.Coppey LJ, Gellett JS, Davidson EP, Dunlap JA, Lund DD, Salvemini D, Yorek MA. Br J Pharmacol. 2001;134(1):21–29. doi: 10.1038/sj.bjp.0704216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hoeldtke RD, Bryner KD, McNeill DR, Hobbs GR, Riggs JE, Warehime SS, Christie I, Ganser G, Van Dyke K. Diabetes. 2002;51:2817–2825. doi: 10.2337/diabetes.51.9.2817. [DOI] [PubMed] [Google Scholar]
- 45.Bai P, Mabley JG, Liaudet L, Virag L, Szabo C, Pacher P. Oncology Reports. 2004;11(2):505–509. [PubMed] [Google Scholar]
- 46.Coppey LJ, Gellett JS, Davidson EP, Yorek MA. Free Radic Res. 2003;37(1):33–40. doi: 10.1080/1071576021000028442. [DOI] [PubMed] [Google Scholar]
- 47.Poladia DP, Bauer JA. Diabetes Metab Res Rev. 2003;19(4):313–319. doi: 10.1002/dmrr.385. [DOI] [PubMed] [Google Scholar]
- 48.Pacher P, Szabo C. Antioxidants & Redox Signaling. 2005 doi: 10.1089/ars.2005.7.1568. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Szabo C. Toxicol Lett. 2003;140–141:105–112. doi: 10.1016/s0378-4274(02)00507-6. [DOI] [PubMed] [Google Scholar]
- 50.Suarez-Pinzon WL, Mabley JG, Strynadka K, Power RF, Szabo C, Rabinovitch A. J Autoimmun. 2001;16(4):449–455. doi: 10.1006/jaut.2001.0507. [DOI] [PubMed] [Google Scholar]
- 51.Furchgott RF. Biosci Rep. 1999;19:235–251. doi: 10.1023/a:1020537506008. [DOI] [PubMed] [Google Scholar]
- 52.Caballero AE, Arora S, Saouaf R, Lim SC, Smakowski P, Park JY, King GL, LoGerfo FW, Horton ES, Veves A. Diabetes. 1999;48:1856–1862. doi: 10.2337/diabetes.48.9.1856. [DOI] [PubMed] [Google Scholar]
- 53.Cai H, Harrison DG. Circ Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 54.Cai H, Griendling KK, Harrison DG. Trends Pharmacol Sci. 2003;24:471–478. doi: 10.1016/S0165-6147(03)00233-5. [DOI] [PubMed] [Google Scholar]
- 55.Calles–Escandon J, Cipolla M. Endocr Rev. 2001;22:36–52. doi: 10.1210/edrv.22.1.0417. [DOI] [PubMed] [Google Scholar]
- 56.Cosentino F, Hishikawa K, Katusic ZS, Luscher TF. Circulation. 1997;96:25–28. doi: 10.1161/01.cir.96.1.25. [DOI] [PubMed] [Google Scholar]
- 57.Cosentino F, Luscher TF. J Cardiovasc Pharmacol. 1998;32:S54–61. [PubMed] [Google Scholar]
- 58.Beckman JA. Circ Res. 2002;90:107–111. doi: 10.1161/hh0102.102359. [DOI] [PubMed] [Google Scholar]
- 59.De Vriese AS, Verbeuren TJ, Van de Voorde J, Lameire NH, Vanhoutte PM. Br J Pharmacol. 2000;130:963–974. doi: 10.1038/sj.bjp.0703393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guzik TJ, Mussa S, Gastaldi D, Sadowski J, Ratnatunga C, Pillai R, Channon KM. Circulation. 2002;105:1656–1662. doi: 10.1161/01.cir.0000012748.58444.08. [DOI] [PubMed] [Google Scholar]
- 61.Guzik TJ, West NE, Black E, McDonald D, Ratnatunga C, Pillai R, Channon KM. Circ Res. 2000;86:85–90. doi: 10.1161/01.res.86.9.1008. [DOI] [PubMed] [Google Scholar]
- 62.Kocsis E, Pacher P, Posa I, Nieszner E, Pogatsa G, Koltai MZ. Acta Physiol Scand. 2000;169(3):183–187. doi: 10.1046/j.1365-201x.2000.00731.x. [DOI] [PubMed] [Google Scholar]
- 63.Ungvari Z, Pacher P, Kecskemeti V, Papp G, Szollar L, Koller A. Cardiovasc Res. 1999;43(4):1018–1028. doi: 10.1016/s0008-6363(99)00106-6. [DOI] [PubMed] [Google Scholar]
- 64.Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 65.Giugliano D, Ceriello A, Paolisso G. Diabetes Care. 1996;19:257–267. doi: 10.2337/diacare.19.3.257. [DOI] [PubMed] [Google Scholar]
- 66.Spitaler MM, Graier WF. Diabetologia. 2002;45:476–494. doi: 10.1007/s00125-002-0782-0. [DOI] [PubMed] [Google Scholar]
- 67.Beckman JS, Koppenol WH. Am J Physiol. 1996;271:1424–1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 68.Eiserich JP, Hristova M, Cross CE, Jones AD, Freeman BA, Halliwell B, et al. Nature. 1998;391:393–397. doi: 10.1038/34923. [DOI] [PubMed] [Google Scholar]
- 69.Halliwell B. FEBS Lett. 1997;411:157–160. doi: 10.1016/s0014-5793(97)00469-9. [DOI] [PubMed] [Google Scholar]
- 70.Pennathur S, Wagner JD, Leeuwenburgh C, Litwak N, Heinecke JW. J Clin Invest. 2001;107:853–860. doi: 10.1172/JCI11194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mihm MJ, Jing L, Bauer JA. J Cardiovasc Pharmacol. 2000;36(2):182–187. doi: 10.1097/00005344-200008000-00007. [DOI] [PubMed] [Google Scholar]
- 72.Bell DS. Diabetes Care. 2003;26(8):2433–2441. doi: 10.2337/diacare.26.8.2433. [DOI] [PubMed] [Google Scholar]
- 73.Bui BV, Armitage JA, Tolcos M, Cooper ME, Vingrys AJ. Diabetologia. 2003;46(3):401–408. doi: 10.1007/s00125-003-1042-7. [DOI] [PubMed] [Google Scholar]
- 74.Lewis EJ, Lewis JB. Clin Exp Nephrol. 2003;7:1–8. doi: 10.1007/s101570300000. [DOI] [PubMed] [Google Scholar]
- 75.Mihm MJ, Wattanapitayakul SK, Piao SF, Hoyt DG, Bauer JA. Biochem Pharmacol. 2003;65:1189–1197. doi: 10.1016/s0006-2952(03)00012-1. [DOI] [PubMed] [Google Scholar]
- 76.Szabó C, Pacher P, Komjati K, Mabley JG, Benko R, Kollai M. FASEB J. 2003;17:A803. [Google Scholar]
- 77.Szabó C, Pacher P, Zsengellér Z, Vaslin A, Komjáti K, Benkö R, Mabley JG, Kollai M. Mol Med. 2004 doi: 10.2119/2004-00001.szabo. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bell DS. Diabetes Care. 1995;18:708–714. doi: 10.2337/diacare.18.5.708. [DOI] [PubMed] [Google Scholar]
- 79.Fein FS. Diabetes Care. 1990;13:1169–1179. doi: 10.2337/diacare.13.11.1169. [DOI] [PubMed] [Google Scholar]
- 80.Illan F, Valdes-Chavarri M, Tebar J, Garcia A, Pascual H, Soria F, Hernandez A, Vicente T. Clin Invest. 1992;70:403–410. doi: 10.1007/BF00235521. [DOI] [PubMed] [Google Scholar]
- 81.Joffe II, Travers KE, Perreault-Micale CL, Hampton T, Katz SE, Morgan JP, Douglas PS. J Am Coll Cardiol. 1999;34:2111–2119. doi: 10.1016/s0735-1097(99)00436-2. [DOI] [PubMed] [Google Scholar]
- 82.Regan TJ, Ahmed S, Haider B, Moschos C, Weisse A. N Engl J Med. 1994;91:776– 778. [PubMed] [Google Scholar]
- 83.Dhalla NS, Liu X, Panagia V, Takeda N. Cardiovasc Res. 1998;40:239–247. doi: 10.1016/s0008-6363(98)00186-2. [DOI] [PubMed] [Google Scholar]
- 84.Esberg LB, Ren J. Diabetologia. 2003;46(10):1419–1427. doi: 10.1007/s00125-003-1183-8. [DOI] [PubMed] [Google Scholar]
- 85.Mihm MJ, Coyle CM, Schanbacher BL, Weinstein DM, Bauer JA. Cardiovasc Res. 2001;49:798–807. doi: 10.1016/s0008-6363(00)00307-2. [DOI] [PubMed] [Google Scholar]
- 86.Pacher P, Liaudet L, Bai P, Mabley JG, Kaminski PM, Virag L, Deb A, Szabo E, Ungvari Z, Wolin MS, Groves JT, Szabo C. Circulation. 2003;107:896–904. doi: 10.1161/01.cir.0000048192.52098.dd. [DOI] [PubMed] [Google Scholar]
- 87.Bianchi C, Wakiyama H, Faro R, Khan T, McCully JD, Levitsky S, Szabo C, Sellke FW. Ann Thorac Surg. 2002;74:1201–1207. doi: 10.1016/s0003-4975(02)03953-x. [DOI] [PubMed] [Google Scholar]
- 88.Mihm MJ, Bauer JA. Biochimie. 2002;84:1013–1019. doi: 10.1016/s0300-9084(02)00005-6. [DOI] [PubMed] [Google Scholar]
- 89.Pacher P, Liaudet L, Bai P, Virag L, Mabley J, Hasko G, Szabo C. J Pharmacol Exp Ther. 2002;300:862–687. doi: 10.1124/jpet.300.3.862. [DOI] [PubMed] [Google Scholar]
- 90.Pacher P, Liaudet L, Mabley J, Komjati K, Szabo C. J Am Coll Cardiol. 2002;40:1006–1016. doi: 10.1016/s0735-1097(02)02062-4. [DOI] [PubMed] [Google Scholar]
- 91.Weinstein DM, Mihm MJ, Bauer JA. J Pharmacol Exp Ther. 2000;294:396–401. [PubMed] [Google Scholar]
- 92.Du Y, Smith MA, Miller CM, Kern TS. J Neurochem. 2002;80:771–779. doi: 10.1046/j.0022-3042.2001.00737.x. [DOI] [PubMed] [Google Scholar]
- 93.Cheng C, Zochodne DW. Diabetes. 2003;52:2363–2371. doi: 10.2337/diabetes.52.9.2363. [DOI] [PubMed] [Google Scholar]
- 94.Obrosova IG. Curr Diab Rep. 2003;3:439–445. doi: 10.1007/s11892-003-0005-1. [DOI] [PubMed] [Google Scholar]
- 95.Obrosova IG, Mabley JG, Zsengeller Z, Charniauskaya T, Abatan OI, Groves JT, Szabó C. FASEB J. 2005 doi: 10.1096/fj.04-1913fje. in press. [DOI] [PubMed] [Google Scholar]
- 96.Obrosova IG, Pacher P, Szabo C, Zsengeller Z, Hirooka H, Stevens MJ, Yorek MA. Diabetes. 2005 doi: 10.2337/diabetes.54.1.234. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Virag L, Szabo C. Pharmacol Rev. 2002;54:375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- 98.Ceriello A, Piconi L, Quagliaro L, Ros RD, Marini C, Giugliano D, Szabo C. FASEB J. 2003;17:A260. [Google Scholar]
- 99.Obrosova IG, Li F, Abatan OI, Komjáti K, Pacher P, Szabo C, Stevens MJ. Diabetes. 2004;53:711–720. doi: 10.2337/diabetes.53.3.711. [DOI] [PubMed] [Google Scholar]
- 100.Pacher P, Liaudet L, Soriano FG, Mabley JG, Szabó É, Szabó C. Diabetes. 2002;51:514–521. doi: 10.2337/diabetes.51.2.514. [DOI] [PubMed] [Google Scholar]
- 101.Reusch JE. J Clin Invest. 2003;112(7):986–988. doi: 10.1172/JCI19902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Soriano FG, Mabley JG, Pacher P, Liaudet L, Szabó C. Circ Res. 2001;89:684–691. doi: 10.1161/hh2001.097797. [DOI] [PubMed] [Google Scholar]
- 103.Soriano FG, Virag L, Szabo C. J Mol Med. 2001;79:437–448. doi: 10.1007/s001090100236. [DOI] [PubMed] [Google Scholar]
- 104.Minchenko AG, Stevens MJ, White L, Abatan OI, Komjáti K, Pacher P, Szabo C, Obrosova IG. FASEB J. 2003;17(11):1514–1516. doi: 10.1096/fj.03-0013fje. [DOI] [PubMed] [Google Scholar]
- 105.Li F, Szabo C, Pacher P, Southan GJ, Abatan OI, Charniauskaya T, Stevens MJ, Obrosova IG. Diabetologia. 2004;47:710–717. doi: 10.1007/s00125-004-1356-0. [DOI] [PubMed] [Google Scholar]
- 106.Obrosova IG, Minchenko AG, Frank RN, Seigel GM, Zsengeller Z, Pacher P, Stevens MJ, Szabo C. Int J Mol Med. 2004;14(1):55–64. [PubMed] [Google Scholar]