

Abstract
Both increased aldose reductase (AR) activity and oxidative/nitrosative stress have been implicated in the pathogenesis of diabetic nephropathy, but the relation between the two factors remains a subject of debate. This study evaluated the effects of AR inhibition on nitrosative stress and poly(ADP-ribose) polymerase (PARP) activation in diabetic rat kidney and high-glucose-exposed human mesangial cells. In animal experiments, control (C) and streptozotocin-diabetic (D) rats were treated with/without the AR inhibitor fidarestat (F, 16 mg kg−1 day−1) for 6 weeks starting from induction of diabetes. Glucose, sorbitol, and fructose concentrations were significantly increased in the renal cortex of D vs C (p < 0.01 for all three comparisons), and sorbitol pathway intermediate, but not glucose, accumulation, was completely prevented in D + F. F at least partially prevented diabetes-induced increase in kidney weight as well as nitrotyrosine (NT, a marker of peroxynitrite-induced injury and nitrosative stress), and poly(ADP-ribose) (a marker of PARP activation) accumulation, assessed by both immunohistochemistry and Western blot analysis, in glomerular and tubular compartments of the renal cortex. In vitro studies revealed the presence of both AR and PARP-1 in human mesangial cells, and none of these two variables were affected by high glucose or F treatment. Nitrosylated and poly(ADP-ribosyl)ated proteins (Western blot analysis) accumulated in cells cultured in 30 mM D-glucose (vs 5.55 mM glucose, p < 0.01), but not in cells cultured in 30 mM L-glucose or 30 mM D-glucose plus 10 μM F. AR inhibition counteracts nitrosative stress and PARP activation in the diabetic renal cortex and high-glucose-exposed human mesangial cells. These findings reveal new beneficial properties of the AR inhibitor F and provide the rationale for detailed studies of F on diabetic nephropathy.
Keywords: Aldose reductase, Fidarestat, Oxidative-nitrosative stress, Poly(ADP-ribose) polymerase, Streptozotocin-diabetic rats, Superoxide, Free radicals
Introduction
Diabetes accounts for at least ~35% of all new cases of end-stage renal disease in the United States [1], and diabetic patients make up the fastest growing group of renal dialysis and transplant recipients. The Diabetes Control and Complications Trial [2] and the U.K. Prospective Diabetes Study [3] strongly suggest the importance of hyperglycemia in the pathogenesis of chronic complications of diabetes mellitus including diabetic renal disease. Hyperglycemia leads to diabetic nephropathy (DN) via multiple mechanisms, and among them increased aldose reductase (AR) activity [4,5], nonenzymatic glycation and glycooxidation [6,7], activation of protein kinase C (PKC) and hexosamine pathway [8–10], arachidonic acid metabolism via 12/15-lipoxygenase pathway [11,12], and triose phosphate accumulation [13] are the best studied. Growing evidence obtained in diabetic animals (primarily, STZ (streptozotocin)-diabetic rats and mice) [14–17] as well as cell culture models [18–20] implicates free radicals and the potent oxidant peroxynitrite (a product of superoxide anion radical reaction with nitric oxide) in both hemodynamic and metabolic abnormalities leading to DN.
Oxidative stress affects all three compartments of the renal cortex, i.e., glomeruli [21], tubulo-interstitium [22], and vasculature [23]. Renal hydrogen peroxide overproduction and lipid peroxide accumulation occur at a very early stage of STZ-diabetes [24] and are associated with clearly manifest impairment of antioxidative defense and, in particular, GSH and ascorbate (AA) depletion, changes in glutathione and ascorbate redox states reflecting in increased oxidized glutathione(GSSG)/GSH and dehydroAA/AA ratios, and upregulated superoxide dismutase, GSH peroxidase, GSH transferase, and GSSG reductase activities [14,25]. Enhanced lipid peroxidation and GSH depletion have also been documented in the model of advanced DN [15]. Evidence for the presence of peroxynitrite-induced injury in the diabetic kidney is emerging [17,26].
Oxidative-nitrosative stress triggers several important downstream mechanisms, i.e., activation of mitogen-activated protein kinases [20,27], the nuclear transcription factor NF-κB [28,29], and upregulation of growth factors such as transforming growth factor-β (TGF-β) [14,15], cytokines [30,31], and vascular endothelial growth factor [32] implicated in diabetic renal disease [12,27–29,33,34]. In vitro and in vivo studies in nondiabetic models of oxidative injury as well as animal and cell culture models of diabetic complications revealed that free radical and peroxynitrite-induced DNA single-strand breakage is also responsible for activation of the nuclear enzyme poly (ADP-ribose) polymerase (PARP) and resultant energy failure, profound metabolic imbalances, and changes in transcriptional regulation and gene expression [35–37]. Recent studies including those from our group generated evidence of an important role of PARP activation in diabetic endothelial [38] and myocardial [39] dysfunction, peripheral neuropathy [40,41], and retinopathy [42,43]. We have also demonstrated (1) tubular PARP activation manifested by increased poly(ADP-ribose) immunoreactivity, and (2) the key role of PARP in upregulation of endothelin-1 and endothelin (A) and (B) receptors, known to play an important role in DN, in the renal cortex of STZ-diabetic rats with 4-week duration of diabetes [44].
The interactions among various hyperglycemia-initiated mechanisms are not completely understood, and the relation between increased AR activity and oxidative-nitrosative stress/ PARP activation in the renal cortex has never been explored. The present study was designed to evaluate the effect of pharmacological AR inhibition with the potent and highly specific AR inhibitor (ARI) fidarestat [45–47] on nitrosative stress and PARP activation in diabetic rat kidney and high-glucose-exposed human mesangial cells. Our animal studies performed in the STZ-diabetic rat model as well as in vitro studies in high-glucose-exposed human mesangial cells provide evidence of the major contribution of increased AR activity to diabetes- and hyperglycemia-associated nitrosative stress and PARP activation in the renal cortex, and, specifically, the key cell target in DN, i.e., glomerular mesangial cells. Furthermore, they identify abundant AR protein expression and early high-glucose-induced peroxynitrite formation and PARP activation in human mesangial cells, thus suggesting the importance of these mechanisms in human DN.
Methods
Reagents
Unless otherwise stated, all chemicals were of reagent-grade quality, and were purchased from Sigma Chemical Co. (St. Louis, MO). Methanol (HPLC grade), perchloric acid, hydrochloric acid, and sodium hydroxide were obtained from Fisher Scientific (Pittsburgh, PA). Reagents for immunohistochemistry were purchased from Vector Laboratories, Inc., Burlingdale, CA and Dako Laboratories, Inc. (Santa Barbara, CA) as specified in the procedures. Monoclonal anti-nitrotyrosine (NT) antibody, clone 1A6 was purchased from Upstate Biotechnology Inc. (Lake Placid, NY). Monoclonal anti-PARP antibody, clone C-2-10, and monoclonal anti-poly(ADP-ribose) antibody were obtained from Biomol (Plymouth Meeting, PA). Polyclonal anti-AR 2 antibody and AR 2 protein were obtained from Santa Cruz (Santa Cruz, CA). Three other AR antibodies were obtained from Drs. D. Carper, R.L. Sorenson, and J.M.Petrash, and human AR protein was from Dr. J.M. Petrash. Human mesangial cells and mesangial cell medium were purchased from ScienCell Research Laboratories (San Diego, CA).
Animals
The experiments were performed in accordance with regulations specified by the National Institutes of Health Principles of Laboratory Animal Care, 1985 Revised Version, and University of Michigan Protocol for Animal Studies. Male Wistar rats (Charles River, Wilmington, MA), body weight 250–300 g, were fed a standard rat chow (PMI Nutrition Int., Brentwood, MO) and had access to water ad libitum. STZ-diabetes was induced as we described previously [25,32,40,41,43]. Blood samples for glucose measurements were taken from the tail vein ~48 h after the STZ injection and the day before the animals were killed. The rats with blood glucose ~13.8 mM were considered diabetic. The experimental groups comprised control and diabetic rats treated with or without fidarestat (16 mg kg−1 day−1, in the diet). The treatments were started immediately after induction of diabetes. The duration of treatment was 6 weeks.
Anesthesia, euthanasia, and tissue sampling
The animals were sedated by CO2 and immediately killed by cervical dislocation. Both kidneys were rapidly isolated, blotted with fine filter paper to remove any accompanying blood, and weighed. The left kidney was frozen in liquid nitrogen for subsequent measurements of glucose, sorbitol pathway intermediates, and nitrosylated and poly(ADP-ribosyl)ated protein abundance. The right kidney was fixed in formalin and later used for assessment of nitrotyrosine and poly(ADP-ribose) by immunohistochemistry.
Human mesangial cell culture
Human mesangial cells were cultured in the commercial mesangial cell medium containing 5.55 mM glucose, according to manufacturer's instructions. Passages 4 and 5 were used for all experiments.
Specific methods
Metabolic studies
Glucose, sorbitol, and fructose concentrations in renal cortex were assessed spectrofluorometrically, by enzymatic procedures with hexokinase/glucose 6-phosphate dehydrogenase, sorbitol dehydrogenase, and fructose dehydrogenase as described [25,41,43].
Immunohistochemical studies
All immunohistochemical samples were coded and examined by a single investigator in a blinded fashion. Microphotographs of stained kidneys were taken with a Zeiss Axiolab microscope equipped with a Fuji HC-300C digital camera.
NT immunoreactivity
Kidneys were fixed in 4% paraformaldehyde in PBS and 5 μm sections were prepared from paraffin embedded tissues. Endogenous peroxidase was quenched with 0.3% H2O2 in 60% methanol for 15 min. The sections were incubated overnight with 1:1000–1:2000 dilution of primary anti-NT antibody. In control measurements, tissues were incubated with the primary antibody in the presence of 10 mM NT. Specific labeling was detected with a biotin-conjugated goat anti-rabbit IgG and avidin-biotin peroxidase complex both supplied in the Vector Elite kit (Vector Laboratories, Burlingame, CA). Color was developed using Ni-diaminobenzidine substrate kit (Vector Laboratories). The sections were counterstained with hematoxylin-eosin, dehydrated, and mounted in Permount. The photomicrographs shown are representative sections (n = 4–12) for each experimental group. The intensity of staining was graded from 1 to 4 (1, no staining; 2, faint; 3, moderate; 4, intense). Average immunohistochemistry scores were calculated for each group.
Poly(ADP-ribose) immunoreactivity
Paraffin sections (5 μm) were loaded onto polylysine-coated slides (Fisher, Atlanta, GA), deparaffinized, and rehydrated. Optimal staining was achieved with an antigen retrieval method which was performed in 10 mmol/L citric acid for 15 min. Endogenous peroxidase was quenched with 0.3% H2O2 in 60% methanol for 15 min. Sections were blocked with 2% normal goat serum at room temperature for 1–2 h, and were incubated overnight with 1:250–1:500 dilution of primary anti-poly(ADP)-ribose antibody (generous gift from Tulip Biolabs Inc.). Specific labeling was detected with a biotin-conjugated goat anti-chicken IgG and avidin–biotin peroxidase complex (Vector Laboratories, Inc.). The enzymatic reaction product was enhanced with nickel cobalt to give a black precipitate, and the sections were counterstained with hematoxylin-eosin, dehydrated, and mounted in Permount. Positive controls included formalin-fixed, paraffin-embedded tissues from LPS-treated rats. Negative controls included elimination of the primary antibody. The photomicrographs shown are representative sections (n = 4–10) for each experimental group. The intensity of staining was graded from 1 to 4 (1, no staining; 2, faint; 3, moderate; 4, intense). Average immunohistochemistry scores were calculated for each group.
Western blot analysis
Western blot analyses of nitrosylated and poly(ADP-ribosyl) ated proteins in the renal cortex
To assess nitrosylated and poly(ADP-ribosyl)ated proteins by Western blot analysis, 50 mg of renal cortex samples was transferred to an extraction buffer (1:10 wt/vol) containing 50 mM Tris-HCl, pH 7.2, 150 mM NaCl, 0.1% sodium dodecyl sulfate, 1% NP-40, 5 mM EDTA, 1 mM EGTA, 1% sodium deoxycholate, and the protease/ phosphatase inhibitors leupeptin (10 μg/ml), aprotinin (20 μg/ml), benzamidine (10 mM), phenylmethylsulfonyl fluoride (1 mM), and sodium orthovanadate (1 mM), and homogenized on ice. The homogenate was sonicated (3 × 5 s) and centrifuged at 14,000g for 20 min. All the afore-noted steps were performed at 4°C. The lysates (20 μg protein) were mixed with equal volume of 2× sample-loading buffer containing 62.5 mM Tris-HCl, pH 6.8, 2% sodium dodecyl sulfate, 5% β-mercaptoethanol, 10% glycerol, and 0.025% bromophenol blue and fractionated in 5–17% SDS-PAGE in an electrophoresis cell (Mini-Protean III; Bio-Rad Laboratories, Richmond, CA.). Electrophoresis was conducted at 15 mA constant current for stacking, and at 25 mA for protein separation. Gel contents were electrotransferred (250 mA, 2 h) to nitrocellulose membranes using Mini Trans-Blot cell (Bio-Rad Laboratories) and Western transfer buffer (25 mM Tris-HCl, pH 8.3, 192 mM glycine, and 20% (v/v) methanol) [48]. Free binding sites were blocked in 2% (w/v) BSA in 20 mM Tris-HCl buffer, pH 7.5, containing 150 mM NaCl and 0.05% Tween 20, for 1 h, after which nitrotyrosine or poly(ADP-ribose) antibodies were applied for 2 h, for detection of nitrosylated and poly(ADP-ribosyl)ated proteins, respectively. The horseradish peroxidase-conjugated secondary antibody was then applied for 1 h. After extensive washing, protein bands detected by the antibodies were visualized with the BM Chemiluminescence Blotting Substrate (POD) (Roche, Indianapolis, IN). The total content of all nitrosylated and poly(ADP-ribosyl)ated proteins was quantified by densitometry (Quantity One 4.5.0 software, Bio-Rad Laboratories). Membranes were then stripped in the 62.5 mM Tris-HCl, pH 6.7, buffer containing 2% SDS and 100 mM β-mercaptoethanol, and reprobed with β-actin antibody to confirm equal protein loading.
Western blot analyses of AR, PARP-1, and nitrosylated and poly(ADP-ribosyl)ated proteins in human mesangial cells
To assess AR and PARP-1 proteins by Western blot analysis, human mesangial cells were cultured for 24 h in commercial media containing (1) 5.55 mM D-glucose; (2) 30 mM D-glucose; (3) 30 mM D-glucose plus 10 μM fidarestat. Cells were lysed in the 2× sample buffer containing 62.5 mM Tris-HCl, pH 6.8, 2% sodium dodecyl sulfate, 5% β-mercaptoethanol, 10% glycerol, and 0.025 % bromophenol blue. Western blot analyses of cell lysates (100 μg protein) were performed as described above. AR expression was verified with four different antibodies and two AR proteins (see Materials and methods).
To assess nitrosylated and poly(ADP-ribosyl)ated proteins, human mesangial cells were cultured for 24 h under the conditions described above. An additional group of cells cultured in commercial media containing 30 mM L-glucose has been added. Cells lysates containing 100 μg protein were used for Western blot analyses.
Statistical analysis
The results are expressed as means ± SE. Data were subjected to equality of variance F test, and then to log transformation, if necessary, before one-way analysis of variance. Where overall significance (p < 0.05) was attained, individual between-group comparisons were made using the Student-Newman-Keuls multiple range test. Significance was defined at p < 0.05. When between-group variance differences could not be normalized by log transformation (datasets for body weights and plasma glucose), the data were analyzed by the nonparametric Kruskal-Wallis one-way analysis of variance, followed by the Bonferroni/Dunn test for multiple comparisons.
Results
The final body weights were comparably lower in untreated and fidarestat-treated diabetic rats than in the control group (Table 1). The final blood glucose concentrations were similarly elevated in untreated and fidarestat-treated diabetic rats compared with the control rats.
Table 1.
Body weights and blood glucose concentrations in control and diabetic rats with and without fidarestat treatment
| Body weight (g)
|
Blood glucose
(mmol/L) |
||
|---|---|---|---|
| Initial a | Final | ||
| Control (n = 10) | 285 ± 7 | 483 ± 10 | 5.08 ± 0.21 |
| Diabetic (n = 15) | 288 ± 7 | 350 ± 7 ** | 18.8 ± 0.56 ** |
| Diabetic + Fidarestat (n = 10) | 293 ± 6 | 342 ± 12 ** | 19.4 ± 0.62 ** |
Data are means ± SE.
Before induction of STZ diabetes.
Significantly different from controls (p < 0.01).
Kidney weights were increased in diabetic rats compared with controls (2.41 ± 0.059 vs 1.79 ± 0.061 g, p < 0.01), and this increase was slightly, but significantly, reduced by fidarestat (2.2 ± 0.088 g, p < 0.01 vs controls, and p < 0.05 vs untreated diabetic group).
Renal cortex glucose, sorbitol, and fructose concentrations were increased in diabetic rats compared with controls (Table 2). Fidarestat treatment did not affect glucose concentrations, but essentially normalized sorbitol and fructose concentrations in diabetic rats.
Table 2.
Sorbitol pathway intermediate concentrations a in renal cortex of control rats and diabetic rats with and without fidarestat treatment
| Glucose | Sorbitol | Fructose | |
|---|---|---|---|
| C (n = 8) | 5.12 ± 0.41 | 0.24 ± 0.03 | 0.41 ± 0.06 |
| D (n = 10) | 32.0 ± 3.2 ** | 0.77 ± 0.03 ** | 1.21 ± 0.1 ** |
| D + F (n = 10) | 34.5 ± 4.2 ** | 0.32 ± 0.03 ## | 0.56 ± 0.1 ## |
Mean ± SE. C, control; D, diabetic; F, fidarestat.
Expressed in μmol/g wet weight.
Significantly different from controls (p < 0.01).
Significantly different from untreated diabetic rats (p < 0.01).
NT immunoreactivities were increased in glomeruli and tubuli of the renal cortex of diabetic rats compared with controls, and this increase in both compartments was markedly reduced by fidarestat treatment (Fig. 1, Table 3). In a similar fashion, diabetes-associated increase in poly(ADP-ribose) immunoreactivity in glomeruli and tubuli of the renal cortex was less manifest in the diabetic rats treated with fidarestat compared with untreated diabetic rats (Fig. 2, Table 3).
Fig. 1.
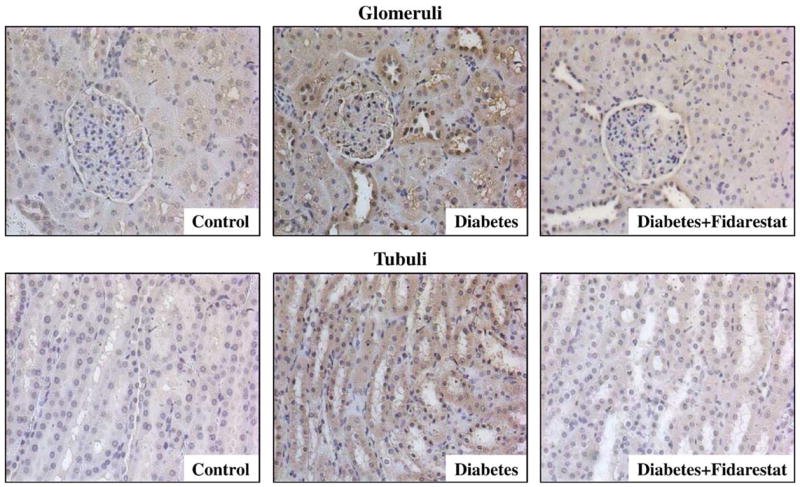
Representative microphotographs of immunohistochemical staining of nitrotyrosine in glomerular and tubular compartments of the renal cortex in control rats, diabetic rats, and diabetic rats treated with fidarestat (n = 4–12 per group). Magnification ×400.
Table 3.
Immunohistochemistry scores for nitrotyrosine and poly(ADP-ribose) immunoreactivities in renal cortex of control rats and diabetic rats with and without fidarestat treatment
| Nitrotyrosine | Poly(ADP-ribose) | |
|---|---|---|
| Glomeruli | ||
| C | 1.0 ± 0.0 (n = 6) | 2.25 ± 0.63 (n = 4) |
| D | 3.3 ± 0.17 (n = 9) | 3.75 ± 0.22 (n = 5) |
| D + F | 1.5 ± 0.19 (n = 12) | 2.25 ± 0.25 (n = 4) |
| Tubuli | ||
| C | 1.8 ± 0.3 (n = 4) | 2.0 ± 0.41 (n = 4) |
| D | 3.3 ± 0.22 (n = 9) | 2.8 ± 0.58 (n = 5) |
| D + F | 2.4 ± 0.16 (n = 10) | 1.8 ± 0.20 (n = 5) |
Mean ± SE. C, control; D, diabetic; F, fidarestat. The number of stained slides per group is indicated in parentheses.
Fig. 2.
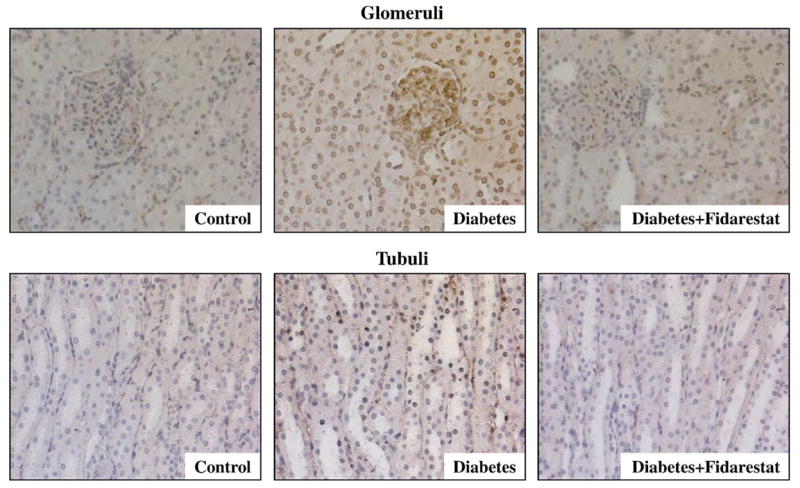
Representative microphotographs of immunohistochemical staining of poly(ADP-ribose) in glomerular and tubular compartments of the renal cortex in control rats, diabetic rats, and diabetic rats treated with fidarestat (n = 4–5 per group). Magnification ×400.
Renal cortex nitrosylated protein abundance assessed by Western blot analysis was increased ~2-fold in the untreated diabetic group compared with control group (Figs. 3A and B), and this increase was reduced by fidarestat to the levels that were not significantly different from those in either control (p = 0.16) or diabetic rats (p = 0.11). In a similar fashion, renal cortex poly(ADP-ribosyl)ated protein abundance was increased 1.8-fold in the untreated diabetic group compared with control group (p < 0.01, Figs. 4A and B). This increase was essentially prevented in diabetic rats treated with fidarestat (p < 0.05 vs untreated diabetic group).
Fig. 3.
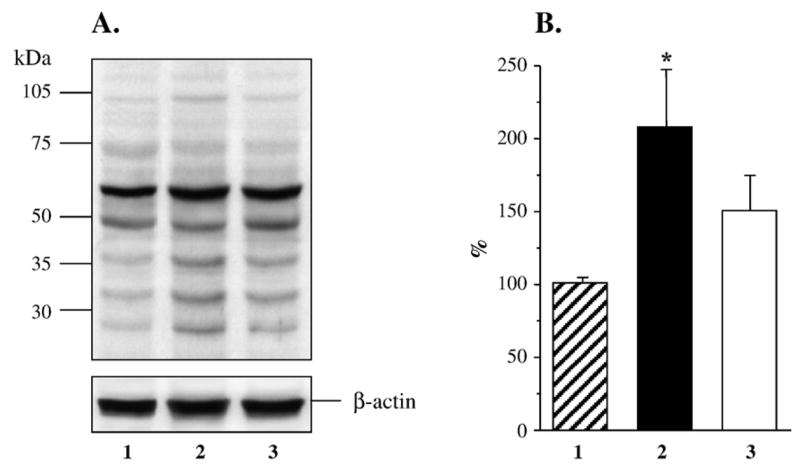
(A) Representative Western blot analysis of renal cortex nitrosylated proteins. Equal protein loading was confirmed with β-actin antibody. Lane 1, control group; lane 2, diabetic group; lane 3, diabetic group treated with fidarestat. (B) Total nitrosylated protein content in renal cortex of control rats (1, n = 6), diabetic rats (2, n = 6), and diabetic rats treated with fidarestat (3, n = 4). The data are expressed as means ± SE. Total nitrosylated protein content in control rats is taken as 100%. *p < 0.05 vs control group.
Fig. 4.
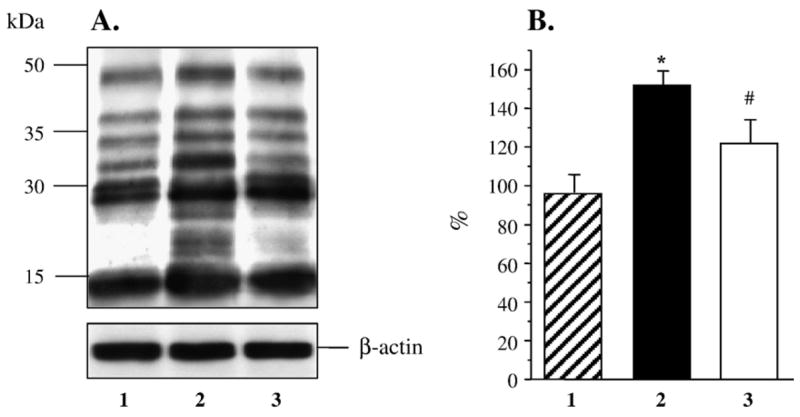
(A) Representative Western blot analysis of renal cortex poly(ADP-ribosyl)ated proteins. Equal protein loading was confirmed with β-actin antibody. Lane 1, control group; lane 2, diabetic group, lane 3, diabetic group treated with fidarestat. (B) Total poly(ADP-ribosyl)ated protein content in renal cortex of control rats (1, n = 5), diabetic rats (2, n = 5), and diabetic rats treated with fidarestat (3, n = 5). The data are expressed as means ± SE. Total nitrosylated protein content in control rats is taken as 100%. ** p < 0.01 vs control group; #p < 0.05 vs diabetic group.
Both AR and PARP-1 were abundantly expressed in human mesangial cells, and none of these variables were affected by either hyperglycemia or fidarestat treatment (Fig. 5). Nitrosylated protein abundance was increased in human mesangial cells cultured in 30 mM D-glucose compared with those cultured in 5.55 mM glucose, but not in cells cultured in 30 mM L-glucose, or 30 mM D-glucose plus 10 μM fidarestat (Figs. 6A and B). Poly(ADP-ribosyl)ated protein content was increased in human mesangial cells cultured in 30 mM glucose compared with those cultured in 5.55 mM glucose (Figs. 7A and B). No accumulation of poly(ADP-ribosyl)ated proteins was found in human mesangial cells cultured in 30 mM L-glucose, or 30 mM D-glucose plus 10 μM fidarestat.
Fig. 5.
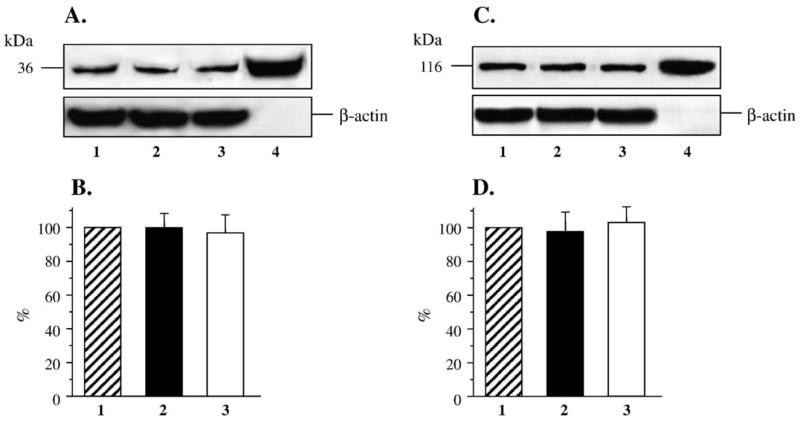
(A) Representative Western blot analyses of human mesangial cell AR protein expression. Equal protein loading was confirmed with β-actin antibody. Lane 1, 5.55 mM glucose; lane 2, 30 mM glucose; lane 3, 30 mM glucose + 10 μM fidarestat, lane 4, human AR. (B) AR protein content in human mesangial cells cultured in 5.55 mM glucose, 30 mM glucose, and 30 mM glucose + 10 μM fidarestat (mean ± SE, n = 5 per condition). AR protein content in cells cultured in 5.55 mM glucose is taken as 100%. (C) Representative Western blot analysis of human mesangial cell PARP-1 protein expression. Equal protein loading was confirmed with β-actin antibody. Lane 1, 5.55 mM glucose; lane 2, 30 mM glucose; lane 3, 30 mM glucose +10 μM fidarestat; lane 4, PARP-1 positive control. (D) Total PARP-1 protein content in human mesangial cells cultured in 5.55 mM glucose (1), 30 mM glucose (2), and 30 mM glucose + 10 μM fidarestat (3, mean ± SE, n = 5 per condition). Total poly(ADP-ribosyl)ated protein content in cells cultured in 5.55 mM glucose is taken as 100%.
Fig. 6.
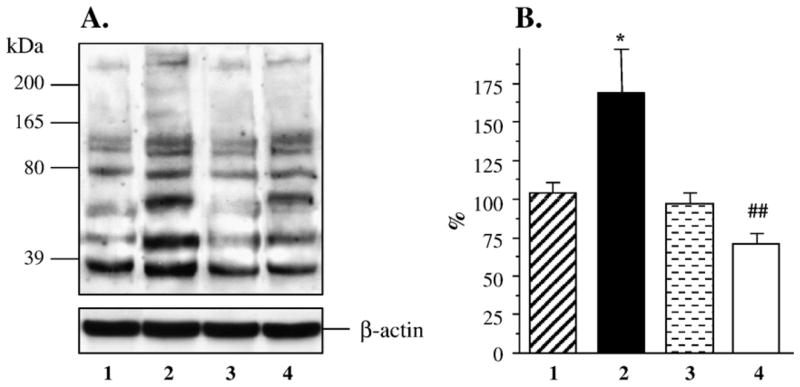
(A) Representative Western blot analysis of human mesangial cell nitrosylated proteins. Equal protein loading was confirmed with β-actin antibody. Lane 1, 5.55 mM glucose; lane 2, 30 mM D-glucose; lane 3, 30 mM L-glucose; lane 4, 30 mM D-glucose + 10 μM fidarestat. (B) Total nitrosylated protein content in human mesangial cells cultured in 5.55 mM glucose (1), 30 mM D-glucose (2), 30 mM L-glucose (3), and 30 mM glucose + 10 μM fidarestat (4, mean ± SE, n = 4 per condition). Total nitrosylated protein content in cells cultured in 5.55 mM glucose is taken as 100%. *p < 0.05 vs cells cultured in 5.55 mM glucose; #p < 0.05 vs cells cultured in 30 mM D-glucose.
Fig. 7.
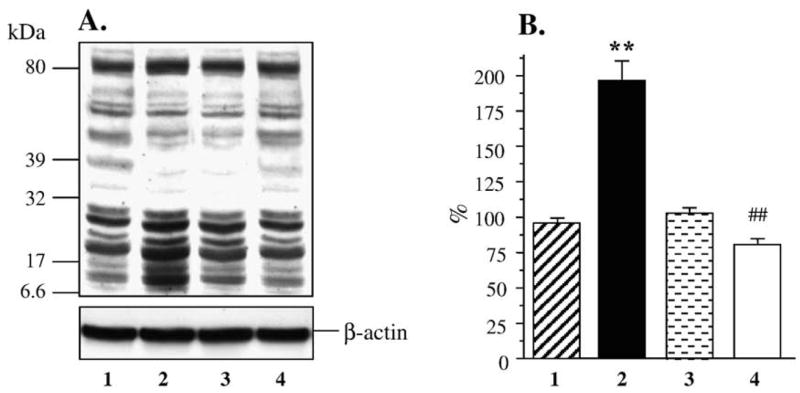
(A) Representative Western blot analysis of human mesangial cell poly(ADP-ribosyl)ated proteins. Equal protein loading was confirmed with β-actin antibody. Lane 1, 5.55 mM glucose; lane 2, 30 mM glucose; lane 3, 30 mM L-glucose; and lane 4, 30 mM glucose + 10 μM fidarestat. (B) Total poly(ADP-ribosyl)ated protein content in human mesangial cells cultured in 5.55 mM glucose (1), 30 mM glucose (2), 30 mM L-glucose (3), and 30 mM glucose + 10 μM fidarestat (4, mean ± SE, n = 4 per condition). Total poly(ADP-ribosyl)ated protein content in cells cultured in 5.55 mM glucose is taken as 100%. **p < 0.01 vs cells cultured in 5.55 mM glucose; ##p < 0.01 vs cells cultured in 30 mM glucose.
Discussion
Our results provide evidence of clearly manifest nitrosative stress in early experimental DN. Increased immunoreactivity of NT (a marker of peroxynitrite-induced injury) in both glomerular and tubular compartments of the renal cortex of STZ-diabetic rats in the present study is consistent with elevated renal nitrosylated protein content (assessed by Western blot analysis) in another report [49]. Furthermore, increased NT immunoreactivity has also been documented in vasculature of diabetic rats [50] as well as kidneys, myocardium, and vasculature of human subjects with diabetes mellitus [51–53]. Studies of the contribution of two components needed for peroxynitrite formation, i.e., superoxide anion radicals and nitric oxide, to diabetes-associated nitrosative stress generated contradictory data [49,54]. One group found that the early (2-week) stage of STZ-induced diabetes provokes accelerated rat renal cortical superoxide anion production in a setting of normal nitric oxide synthase isoform protein level and distribution [49]. Others reported increased immunoreactivities of the enzymes generating both superoxide (p47phox component of NAD(P)H oxidase) and nitric oxide (endothelial, but not neuronal nitric oxide synthase) in the renal cortex of rats with similar duration of STZ-diabetes [54].
The present study also revealed clearly manifest poly(ADP-ribose) accumulation in both glomerular and tubular compartments of the renal cortex of STZ-diabetic rats with a 6-week duration of diabetes. Furthermore, PARP appeared abundantly expressed in human mesangial cells that displayed increased levels of poly(ADP-ribosyl)ated proteins at the early (24 h) stage of exposure to high glucose. These findings suggest an important role of PARP activation in diabetic renal disease, e.g., in human subjects.
Oxidative-nitrosative stress and PARP activation in tissue sites for diabetic complications are caused primarily by hyper-glycemia [36,55], although other factors, e.g., elevated fatty acid [56] and angiotensin II [18,20] concentrations may also contribute to free radical and peroxynitrite generation and downregulation of antioxidative defense. Oxidative-nitrosative stress and resultant PARP activation are interrelated with several other hyperglycemia-initiated mechanisms. In particular, advanced glycation end-products (AGE) generate free radicals during interaction with their receptors [57]. PKC activation promotes phosphorylation/activation of NAD(P)H oxidase [58], a superoxide-generating enzyme, which is particularly important in the vasculature, thus leading to oxidative-nitrosative stress and consequent PARP activation [59]. In turn, oxidative-nitrosative stress and PARP activation divert the glycolytic flux toward the formation of diacylglycerol, an activator of PKC, and methylglyoxal, an AGE precursor [36]. The afore-noted interactions among glycation, oxidative-nitrosative stress, and protein kinase C activation are supported by several lines of evidence, i.e., (1) induction of free radical generation by AGE [57]; (2) inhibition of systemic oxidative stress by the PKC β isoform inhibitor LY333531 [59]; and (3) inhibition of endothelial methylglyoxal formation and PKC activation by inhibitors of mitochondrial superoxide formation as well as the PARP inhibitor PJ34 [36].
The relation between oxidative-nitrosative stress/PARP activation and the most important “upstream” mechanism in the pathogenesis of diabetic complications, i.e., increased AR activity, is less clear. On the one hand, neither antioxidants including the most potent superoxide scavenger dihydrolipoic acid (formed intracellularly from DL-α-lipoic acid) nor PARP inhibitors decrease sorbitol pathway intermediate accumulation in the diabetic retina [32,43] and peripheral nerve [41,60]. On the other hand, structurally diverse ARIs counteract oxidative-nitrosative stress in diabetic lens [61,62], nerve [55,63], retina [45], aorta [55,64], and vasa nervorum [55] as well as high-glucose-exposed endothelial cells [55,65]. Furthermore, our group has recently demonstrated that AR inhibition counteracts PARP activation in the diabetic nerve and retina as well as high-glucose-exposed Schwann cells [55]. All these findings suggest that sorbitol pathway hyperactivity precedes and provides a major contribution to oxidative-nitrosative stress and resultant PARP activation in, at least, several tissue sites for diabetic complications.
The only study addressing a relation between diabetes-associated renal sorbitol pathway hyperactivity and oxidative injury suggests that AR plays a protective role in neutralization of the lipid peroxidation products 4-hydroxyalkenals, and, therefore, AR inhibition in the diabetic kidney exacerbates, rather than counteracts, oxidative injury [66]. However, this contradicts numerous reports of (1) alleviation, rather than aggravation, of oxidative-nitrosative stress by AR inhibitors in other tissues of diabetic animals as discussed above, and (2) beneficial effects of both AR inhibitors and antioxidants on experimental DN [4,5,14,15]. Furthermore, several groups demonstrated that prevalence of DN depends on (1) erythrocyte AR content, and/or (2) frequency of the z-2 allele of the AR gene, in human subjects with diabetes mellitus [4,67,68]. Homozygosity for the z-2 allele in Type 1 (insulin-dependent) diabetes is associated with an increased expression of the AR gene and DN [67]. In Type 2 (non-insulin-dependent diabetes), the z-2 allele is also associated with an increased AR activity and nephro-retinopathy [68]. Furthermore, homozygosity for the z-2 allele was recently reported to be associated with classic diabetic glomerulopathy manifested by overexpression of TGF-β1, mesangial cell transdifferentiation by expression of α-smooth muscle actin, and aberrant deposition of collagen type IY, fibronectin, and laminin [69]. These findings agree with (1) clearly manifest AR protein expression in human mesangial cells in the present study, and (2) reduction of high-glucose-induced TGF-β overexpression by two structurally diverse ARIs, epalrestat and sorbinil, in cultured human mesangial and methothelial cells in two other reports [70,71]. Note that AR has also been localized in several other cell types playing an important role in DN, i.e., glomerular podocytes and endothelial cells and tubular cells [72].
Our findings provide the first evidence of the key role of increased AR activity in diabetes-associated nitrosative stress and PARP activation in both glomerular and tubular compartments of the renal cortex. Furthermore, AR is involved in high-glucose-induced accumulation of poly(ADP-ribosyl)ated proteins in human mesangial cells. The latter occurs due to increased PARP catalytic activity, but not protein expression which remained unchanged by either hyperglycemia or fidarestat treatment. The latter is consistent with the current knowledge on PARP as an abundant constitutively expressed nuclear enzyme with very minor, if any, transcriptional regulation [37].
Increased AR activity can lead to diabetes-associated oxidative-nitrosative stress via several mechanisms (Fig. 8). Studies in other tissues indicate that increased AR activity and resulting osmotic stress and/or NADPH deficiency are responsible for downregulation of antioxidative defense provided by both nonenzymatic antioxidants, i.e., GSH, ascorbate [55,61–63], and taurine [73], and antioxidative defense enzymes (superoxide dismutase, catalase, glutathione peroxidase, glutathione reductase, glutathione transferase) [45]. In addition, increased AR activity contributes to oxidative-nitrosative stress indirectly, via other pathways, such as nonenzymatic glycation and activation of PKC. In particular, AR is involved in generation of fructose, a 10 times more potent glycation agent than glucose, as well as other AGE precursors, i.e., methylglyoxal and 3-deoxyglucosone [74,75]. ARI treatment suppresses formation of the AGE pentosidine and carboxymethyllysine [76,77] known to generate oxidative stress via interaction with their receptors [57]. Taking into consideration that diabetic kidney accumulates AGE [13], it is quite plausible that increased AR activity contributes to renal oxidative stress by promoting nonenzymatic glycation. Several groups have reported that increased AR activity leads to PKC activation in glomerular mesangial cells [78] and vasculature [79,80]. The latter probably occurs due to AR-dependent decrease in free cytosolic NAD+/NADH ratio and associated diversion of the glycolytic flux toward the increased formation of α-glycerophosphate and diacylglycerol, a PKC activator. As discussed above, PKC is required for phosphorylation (activation) of NAD(P)H oxidase, the superoxide generating enzyme [58]. Therefore, increased AR activity can also contribute to superoxide formation via activation of PKC. Furthermore, as we have recently demonstrated, AR-mediated PARP activation exacerbates oxidative-nitrosative stress by contributing to superoxide production, and lipid peroxidation, probably via NF- κB-mediated upregulation of inducible nitric oxide synthase, endothelin-1, and inflammatory cytokines [59]. However, the mechanisms linking increased AR and oxidative-nitrosative stress in the kidney require detailed specific studies.
Fig. 8.
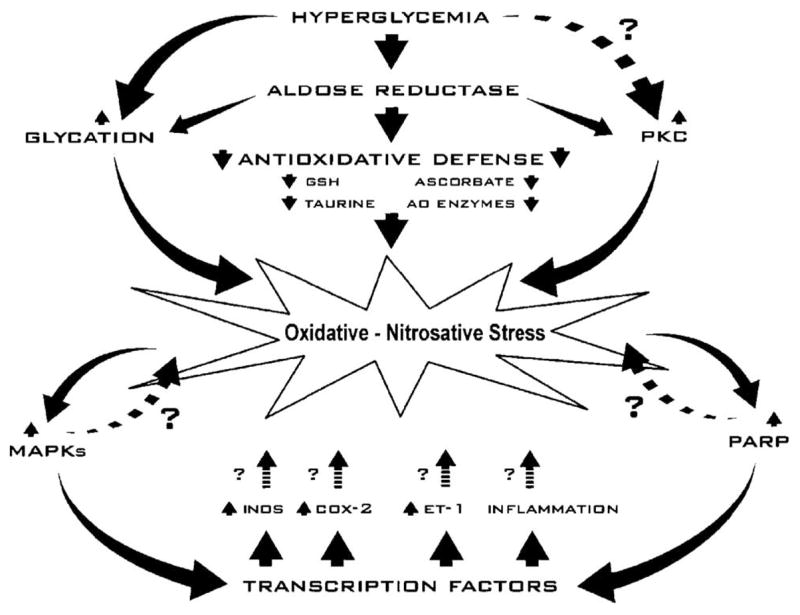
Links among increased AR activity, oxidative-nitrosative stress, poly(ADP-ribose) polymerase activation, and its downstream consequences in the pathogenesis of diabetic nephropathy.
In conclusion, our experiments generated evidence of the important role of increased AR activity in diabetes-associated oxidative-nitrosative stress and PARP activation in both glomerular and tubular compartments of the renal cortex of rats with early STZ-diabetes. We also revealed the presence of both AR and PARP in human mesangial cells, a key cell target in human diabetic glomerulopathy and DN in general. Furthermore, early accumulation of poly(ADP-ribosyl)ated proteins in response to high glucose in these cells was blunted by an ARI treatment, thus suggesting that increased AR activity can be implicated in the pathogenesis of human DN via PARP activation. The results reveal new beneficial properties of fidarestat, thus justifying a detailed study of this specific, potent, and low-toxic ARI in experimental, and, potentially, human DN.
Acknowledgments
The study was supported by the National Institutes of Health Grant 1R21DK070720-01, grants from American Diabetes Association and Juvenile Diabetes Research Foundation (all to Irina G. Obrosova), and the Juvenile Diabetes Research Foundation Center for the Study of Complications of Diabetes Grant 4-200-421 (to I. G.Obrosova and M.J. Stevens). The authors are grateful to Drs. D. Carper, R.L. Sorenson, and J.M. Petrash for their gifts of AR antibodies, and human AR enzyme.
Abbreviations
- DN
diabetic nephropathy
- AR
aldose reductase
- PKC
protein kinase C
- AA
ascorbate
- TGF-β
transforming growth factor-β
- PARP
poly(ADP-ribose) polymerase
- ARI
AR inhibitor
- STZ
streptozotocin
- NT
nitrotyrosine
- AGE
advance glycation end-products
- SDS-PAGE
sodium dodecyl sulfate—polyacrylamide gel electrophoresis
References
- 1.Nelson RG, Knowler WC, Pettitt DJ, Bennett PH. Diabetes in America. 2. Bethesda, Maryland: National Institutes of Diabetes and Digestive and Kidney Diseases, Chapter 16; 1995. Kidney diseases in diabetes; pp. 349–385. NIH Publication No.95-1468. [Google Scholar]
- 2.The Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy. N Engl J Med. 2000;342:381–389. doi: 10.1056/NEJM200002103420603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33) Lancet. 1998;352:837–853. [PubMed] [Google Scholar]
- 4.Oates PJ, Mylari BL. Aldose reductase inhibitors: therapeutic implications for diabetic complications. Expert Opin Invest Drugs. 1999;8:2095–2119. doi: 10.1517/13543784.8.12.2095. [DOI] [PubMed] [Google Scholar]
- 5.Schrijvers BF, De Vriese AS, Flyvbjerg A. From hyperglycemia to diabetic kidney disease: the role of metabolic, hemodynamic, intracellular factors and growth factors/cytokines. Endocr Rev. 2004;25:971–1010. doi: 10.1210/er.2003-0018. [DOI] [PubMed] [Google Scholar]
- 6.Forbes JM, Soulis T, Thallas V, Panagiotopoulos S, Long DM, Vasan S, Wagle D, Jerums G, Cooper ME. Renoprotective effects of a novel inhibitor of advanced glycation. Diabetologia. 2001;44:108–114. doi: 10.1007/s001250051587. [DOI] [PubMed] [Google Scholar]
- 7.Berlanga J, Cibrian D, Guillen I, Freyre F, Alba JS, Lopez-Saura P, Merino N, Aldama A, Quintela AM, Triana ME, Montequin JF, Ajamieh H, Urquiza D, Ahmed N, Thornalley PJ. Methylglyoxal administration induces diabetes-like microvascular changes and perturbs the healing process of cutaneous wounds. Clin Sci (Lond) 2005;109:83–95. doi: 10.1042/CS20050026. [DOI] [PubMed] [Google Scholar]
- 8.Koya D, Jirousek MR, Lin YW, Ishii H, Kuboki K, King GL. Characterization of protein kinase C beta isoform activation on the gene expression of transforming growth factor-beta, extracellular matrix components, and prostanoids in the glomeruli of diabetic rats. J Clin Invest. 1997;100:115–126. doi: 10.1172/JCI119503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kikkawa R, Koya D, Haneda M. Progression of diabetic nephropathy. Am J Kidney Dis. 2003;41(3 Suppl 1):S19–S21. doi: 10.1053/ajkd.2003.50077. [DOI] [PubMed] [Google Scholar]
- 10.Schleicher ED, Weigert C. Role of the hexosamine biosynthetic pathway in diabetic nephropathy. Kidney Int Suppl. 2000;77:S13–S18. doi: 10.1046/j.1523-1755.2000.07703.x. [DOI] [PubMed] [Google Scholar]
- 11.Kim YS, Reddy MA, Lanting L, Adler SG, Natarajan R. Differential behavior of mesangial cells derived from 12/15-lipoxygenase knockout mice relative to control mice. Kidney Int. 2003;64:1702–1714. doi: 10.1046/j.1523-1755.2003.00286.x. [DOI] [PubMed] [Google Scholar]
- 12.Kim YS, Xu ZG, Reddy MA, Li SL, Lanting L, Sharma K, Adler SG, Natarajan R. Novel interactions between TGF-{beta}1 actions and the 12/15-lipoxygenase pathway in mesangial cells. J Am Soc Nephrol. 2005;16:352–362. doi: 10.1681/ASN.2004070568. [DOI] [PubMed] [Google Scholar]
- 13.Babaei-Jadidi R, Karachalias N, Ahmed N, Battah S, Thornalley PJ. Prevention of incipient diabetic nephropathy by high-dose thiamine and benfotiamine. Diabetes. 2003;52:2110–2120. doi: 10.2337/diabetes.52.8.2110. [DOI] [PubMed] [Google Scholar]
- 14.Melhem MF, Craven PA, DeRubertis FR. Effects of dietary supplementation of alpha-lipoic acid on early glomerular injury in diabetes mellitus. J Am Soc Nephrol. 2001;12:124–133. doi: 10.1681/ASN.V121124. [DOI] [PubMed] [Google Scholar]
- 15.Melhem MF, Craven PA, Liachenko J, DeRubertis FR. Alpha-lipoic acid attenuates hyperglycemia and prevents glomerular mesangial matrix expansion in diabetes. J Am Soc Nephrol. 2002;13:108–116. doi: 10.1681/ASN.V131108. [DOI] [PubMed] [Google Scholar]
- 16.Craven PA, Melhem MF, Phillips SL, DeRubertis FR. Overexpression of Cu2+/Zn2+ superoxide dismutase protects against early diabetic glomerular injury in transgenic mice. Diabetes. 2001;50:2114–2125. doi: 10.2337/diabetes.50.9.2114. [DOI] [PubMed] [Google Scholar]
- 17.DeRubertis FR, Craven PA, Melhem MF, Salah EM. Attenuation of renal injury in db/db mice overexpressing superoxide dismutase: evidence for reduced superoxide-nitric oxide interactions. Diabetes. 2004;53:762–768. doi: 10.2337/diabetes.53.3.762. [DOI] [PubMed] [Google Scholar]
- 18.Gorin Y, Kim NH, Feliers D, Bhandari B, Choudhury GG, Abboud HE. Angiotensin II activates Akt/protein kinase B by an arachidonic acid/redox-dependent pathway and independent of phosphoinositide 3-kinase. FASEB J. 2001;15:1909–1920. doi: 10.1096/fj..01-0165com. [DOI] [PubMed] [Google Scholar]
- 19.Lee HB, Yu MR, Yang Y, Jiang Z, Ha H. Reactive oxygen species-regulated signaling pathways in diabetic nephropathy. J Am Soc Nephrol. 2003;14:S241–S245. doi: 10.1097/01.asn.0000077410.66390.0f. [DOI] [PubMed] [Google Scholar]
- 20.Gorin Y, Ricono JM, Wagner B, Kim NH, Bhandari B, Choudhury GG, Abboud HE. Angiotensin II-induced ERK1/ERK2 activation and protein synthesis are redox-dependent in glomerular mesangial cells. Biochem J. 2004;381(Pt 1):231–239. doi: 10.1042/BJ20031614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horie K, Miyata T, Maeda K, Miyata S, Sugiyama S, Sakai H, van Ypersole de Strihou C, Monnier VM, Witztum JL, Kurokawa K. Immunohistochemical colocalization of glycoxidation products and lipid peroxidation products in diabetic renal glomerular lesions. Implication for glycoxidative stress in the pathogenesis of diabetic nephropathy. J Clin Invest. 1997;100:2995–3004. doi: 10.1172/JCI119853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dobashi K, Asayama K, Hayashibe H, Uchida N, Kobayashi M, Kawaoi A, Kato K. Effect of diabetes mellitus induced by streptozotocin on renal superoxide dismutases in the rat. A radioimmunoassay and immunohistochemical study. Virchows Arch B Cell Pathol Incl Mol Pathol. 1991;60:67–72. doi: 10.1007/BF02899529. [DOI] [PubMed] [Google Scholar]
- 23.Schnackenberg CG, Wilcox CS. The SOD mimetic tempol restores vasodilation in afferent arterioles of experimental diabetes. Kidney Int. 2001;59:1859–1864. doi: 10.1046/j.1523-1755.2001.0590051859.x. [DOI] [PubMed] [Google Scholar]
- 24.Onozato ML, Tojo A, Goto A, Fujita T, Wilcox CS. Oxidative stress and nitric oxide synthase in rat diabetic nephropathy: effects of ACEI and ARB. Kidney Int. 2002;61:186–194. doi: 10.1046/j.1523-1755.2002.00123.x. [DOI] [PubMed] [Google Scholar]
- 25.Obrosova IG, Fathallah L, Liu E, Nourooz-Zadeh J. Early oxidative stress in the diabetic kidney: effect of DL-alpha-lipoic acid. Free Radic Biol Med. 2003;34:186–195. doi: 10.1016/s0891-5849(02)01195-4. [DOI] [PubMed] [Google Scholar]
- 26.Thuraisingham RC, Nott CA, Dodd SM, Yaqoob MM. Increased nitrotyrosine staining in kidneys from patients with diabetic nephropathy. Kidney Int. 2000;57:1968–1972. doi: 10.1046/j.1523-1755.2000.00046.x. [DOI] [PubMed] [Google Scholar]
- 27.Haneda M, Koya D, Kikkawa R. Cellular mechanisms in the development and progression of diabetic nephropathy: activation of the DAG-PKC-ERK pathway. Am J Kidney Dis. 2001;38(4 Suppl 1):S178–S181. doi: 10.1053/ajkd.2001.27438. [DOI] [PubMed] [Google Scholar]
- 28.Lee FT, Cao Z, Long DM, Panagiotopoulos S, Jerums G, Cooper ME, Forbes JM. Interactions between angiotensin II and NF-kappaB-dependent pathways in modulating macrophage infiltration in experimental diabetic nephropathy. J Am Soc Nephrol. 2004;15:2139–2151. doi: 10.1097/01.ASN.0000135055.61833.A8. [DOI] [PubMed] [Google Scholar]
- 29.Mezzano S, Aros C, Droguett A, Burgos ME, Ardiles L, Flores C, Schneider H, Ruiz-Ortega M, Egido J. NF-kappaB activation and overexpression of regulated genes in human diabetic nephropathy. Nephrol Dial Transplant. 2004;19:2505–2512. doi: 10.1093/ndt/gfh207. [DOI] [PubMed] [Google Scholar]
- 30.Ha H, Lee HB. Oxidative stress in diabetic nephropathy: basic and clinical information. Curr Diab Rep. 2001;1:282–287. doi: 10.1007/s11892-001-0047-1. [DOI] [PubMed] [Google Scholar]
- 31.Wolf G. New insights into the pathophysiology of diabetic nephropathy: from haemodynamics to molecular pathology. Eur J Clin Invest. 2004;34:785–796. doi: 10.1111/j.1365-2362.2004.01429.x. [DOI] [PubMed] [Google Scholar]
- 32.Obrosova IG, Minchenko AG, Marinescu V, Fathallah L, Kennedy A, Stockert CM, Frank RN, Stevens MJ. Antioxidants attenuate early up regulation of retinal vascular endothelial growth factor in streptozotocin-diabetic rats. Diabetologia. 2001;44:1102–1110. doi: 10.1007/s001250100631. [DOI] [PubMed] [Google Scholar]
- 33.Flyvbjerg A, Dagnaes-Hansen F, De Vriese AS, Schrijvers BF, Tilton RG, Rasch R. Amelioration of long-term renal changes in obese type 2 diabetic mice by a neutralizing vascular endothelial growth factor antibody. Diabetes. 2002;51:3090–3094. doi: 10.2337/diabetes.51.10.3090. [DOI] [PubMed] [Google Scholar]
- 34.Senthil D, Choudhury GG, McLaurin C, Kasinath BS. Vascular endothelial growth factor induces protein synthesis in renal epithelial cells: a potential role in diabetic nephropathy. Kidney Int. 2003;64:468–479. doi: 10.1046/j.1523-1755.2003.00135.x. [DOI] [PubMed] [Google Scholar]
- 35.Ha HC, Hester LD, Snyder SH. Poly(ADP-ribose) polymerase-1 dependence of stress-induced transcription factors and associated gene expression in glia. Proc Natl Acad Sci USA. 2002;99:3270–3275. doi: 10.1073/pnas.052712399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Du X, Matsumura T, Edelstein D, Rossetti L, Zsengeller Z, Szabo C, Brownlee M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest. 2003;112:1049–1057. doi: 10.1172/JCI18127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jagtap P, Szabo C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov. 2005;4:421–440. doi: 10.1038/nrd1718. [DOI] [PubMed] [Google Scholar]
- 38.Garcia Soriano F, Virag L, Jagtap P, Szabo E, Mabley JG, Liaudet L, Marton A, Hoyt DG, Murthy KG, Salzman AL, Southan GJ, Szabo C. Diabetic endothelial dysfunction: the role of poly(ADP-ribose) polymerase activation. Nat Med. 2001;7:108–113. doi: 10.1038/83241. [DOI] [PubMed] [Google Scholar]
- 39.Pacher P, Liaudet L, Soriano FG, Mabley JG, Szabo E, Szabo C. The role of poly(ADP-ribose) polymerase activation in the development of myocardial and endothelial dysfunction in diabetes. Diabetes. 2002;51:514–521. doi: 10.2337/diabetes.51.2.514. [DOI] [PubMed] [Google Scholar]
- 40.Obrosova IG, Li F, Abatan OI, Forsell MA, Komjati K, Pacher P, Szabo C, Stevens MJ. Role of poly(ADP-ribose) polymerase activation in diabetic neuropathy. Diabetes. 2004;53:711–720. doi: 10.2337/diabetes.53.3.711. [DOI] [PubMed] [Google Scholar]
- 41.Li F, Szabo C, Pacher P, Southan GJ, Abatan OI, Charniauskaya T, Stevens MJ, Obrosova IG. Evaluation of orally active poly(ADP-ribose) polymerase inhibitor in streptozotocin-diabetic rat model of early peripheral neuropathy. Diabetologia. 2004;47:710–717. doi: 10.1007/s00125-004-1356-0. [DOI] [PubMed] [Google Scholar]
- 42.Zheng L, Szabo C, Kern TS. Poly(ADP-ribose) polymerase is involved in the development of diabetic retinopathy via regulation of nuclear factor-kappaB. Diabetes. 2004;53:2960–2967. doi: 10.2337/diabetes.53.11.2960. [DOI] [PubMed] [Google Scholar]
- 43.Obrosova IG, Minchenko AG, Frank RN, Seigel GM, Zsengeller Z, Pacher P, Stevens MJ, Szabo C. Poly(ADP-ribose) polymerase inhibitors counteract diabetes- and hypoxia-induced retinal vascular endothelial growth factor overexpression. Int J Mol Med. 2004;14:55–64. [PubMed] [Google Scholar]
- 44.Minchenko AG, Stevens MJ, White L, Abatan OI, Komjati K, Pacher P, Szabo C, Obrosova IG. Diabetes-induced overexpression of endothelin-1 and endothelin receptors in the rat renal cortex is mediated via poly(ADP-ribose) polymerase activation. FASEB J. 2003;17:1514–1516. doi: 10.1096/fj.03-0013fje. [DOI] [PubMed] [Google Scholar]
- 45.Obrosova IG, Minchenko AG, Vasupuram R, White L, Abatan OI, Kumagai AK, Frank RN, Stevens MJ. Aldose reductase inhibitor fidarestat prevents retinal oxidative stress and vascular endothelial growth factor overexpression in streptozotocin-diabetic rats. Diabetes. 2003;52:864–871. doi: 10.2337/diabetes.52.3.864. [DOI] [PubMed] [Google Scholar]
- 46.Kato N, Yashima S, Suzuki T, Nakayama Y, Jomori T. Long-term treatment with fidarestat suppresses the development of diabetic retinopathy in STZ-induced diabetic rats. J Diab Complications. 2003;17:374–379. doi: 10.1016/s1056-8727(02)00193-9. [DOI] [PubMed] [Google Scholar]
- 47.Price SA, Agthong S, Middlemas AB, Tomlinson DR. Mitogen-activated protein kinase p38 mediates reduced nerve conduction velocity in experimental diabetic neuropathy: interactions with aldose reductase. Diabetes. 2004;53:1851–1856. doi: 10.2337/diabetes.53.7.1851. [DOI] [PubMed] [Google Scholar]
- 48.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure, some applications. 1979. Biotechnology. 1992;24:145–149. [PubMed] [Google Scholar]
- 49.Ishii N, Patel KP, Lane PH, Taylor T, Bian K, Murad F, Pollock JS, Carmines PK. Nitric oxide synthesis and oxidative stress in the renal cortex of rats with diabetes mellitus. J Am Soc Nephrol. 2001;12:1630–1639. doi: 10.1681/ASN.V1281630. [DOI] [PubMed] [Google Scholar]
- 50.Stadler K, Jenei Vvon; Bolcshazy G, Somogyi A, Jakus J. Increased nitric oxide levels as an early sign of premature aging in diabetes. Free Radic Biol Med. 2003;35:1240–1251. doi: 10.1016/s0891-5849(03)00499-4. [DOI] [PubMed] [Google Scholar]
- 51.Thuraisingham RC, Nott CA, Dodd SM, Yaqoob MM. Increased nitrotyrosine staining in kidneys from patients with diabetic nephropathy. Kidney Int. 2000;57:1968–1972. doi: 10.1046/j.1523-1755.2000.00046.x. [DOI] [PubMed] [Google Scholar]
- 52.Frustaci A, Kajstura J, Chimenti C, Jakoniuk I, Leri A, Maseri A, Nadal-Ginard B, Anversa P. Myocardial cell death in human diabetes. Circ Res. 2000;87:1123–1132. doi: 10.1161/01.res.87.12.1123. [DOI] [PubMed] [Google Scholar]
- 53.Szabo C, Zanchi A, Komjati K, Pacher P, Krolewski AS, Quist WC, LoGerfo FW, Horton ES, Veves A. Poly(ADP-Ribose) polymerase is activated in subjects at risk of developing type 2 diabetes and is associated with impaired vascular reactivity. Circulation. 2002;106:2680–2686. doi: 10.1161/01.cir.0000038365.78031.9c. [DOI] [PubMed] [Google Scholar]
- 54.Onozato ML, Tojo A, Goto A, Fujita T, Wilcox CS. Oxidative stress and nitric oxide synthase in rat diabetic nephropathy: effects of ACEI and ARB. Kidney Int. 2002;61:186–194. doi: 10.1046/j.1523-1755.2002.00123.x. [DOI] [PubMed] [Google Scholar]
- 55.Obrosova IG, Pacher P, Szabo C, Zsengeller Z, Hirooka H, Stevens MJ, Yorek MA. Aldose reductase inhibition counteracts oxidative-nitrosative stress and poly(ADP-ribose) polymerase activation in tissue sites for diabetes complications. Diabetes. 2005;54:234–242. doi: 10.2337/diabetes.54.1.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cacicedo JM, Benjachareowong S, Chou E, Ruderman NB, Ido Y. Palmitate-induced apoptosis in cultured bovine retinal pericytes: roles of NAD(P)H oxidase, oxidant stress, and ceramide. Diabetes. 2005;54:1838–4185. doi: 10.2337/diabetes.54.6.1838. [DOI] [PubMed] [Google Scholar]
- 57.Thornalley PJ. Glycation in diabetic neuropathy: characteristics, consequences, causes, and therapeutic options. Int Rev Neurobiol. 2002;50:37–57. doi: 10.1016/s0074-7742(02)50072-6. [DOI] [PubMed] [Google Scholar]
- 58.Inoguchi T, Sonta T, Tsubouchi H, Etoh T, Kakimoto M, Sonoda N, Sato N, Sekiguchi N, Kobayashi K, Sumimoto H, Utsumi H, Nawata H. Protein kinase C-dependent increase in reactive oxygen species (ROS) production in vascular tissues of diabetes: role of vascular NAD(P) H oxidase. J Am Soc Nephrol. 2003;14(Suppl 3):S227–S232. doi: 10.1097/01.asn.0000077407.90309.65. [DOI] [PubMed] [Google Scholar]
- 59.Abiko T, Abiko A, Clermont AC, Shoelson B, Horio N, Takahashi J, Adamis AP, King GL, Bursell SE. Characterization of retinal leukostasis and hemodynamics in insulin resistance and diabetes: role of oxidants and protein kinase-C activation. Diabetes. 2003;52:829–837. doi: 10.2337/diabetes.52.3.829. [DOI] [PubMed] [Google Scholar]
- 60.Stevens MJ, Obrosova I, Cao X, Van Huysen C, Greene DA. Effects of DL-alpha-lipoic acid on peripheral nerve conduction, blood flow, energy metabolism, and oxidative stress in experimental diabetic neuropathy. Diabetes. 2000;49:1006–1015. doi: 10.2337/diabetes.49.6.1006. [DOI] [PubMed] [Google Scholar]
- 61.Obrosova IG, Fathallah L. Evaluation of an aldose reductase inhibitor on lens metabolism, ATPases and antioxidative defense in streptozotocin-diabetic rats: an intervention study. Diabetologia. 2000;43:1048–1055. doi: 10.1007/s001250051488. [DOI] [PubMed] [Google Scholar]
- 62.Yorek MA, Coppey LJ, Gellett JS, Davidson EP, Lund DD. Effect of fidarestat and alpha-lipoic acid on diabetes-induced epineurial arteriole vascular dysfunction. Exp Diab Res. 2004;5:123–135. doi: 10.1080/15438600490277824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Obrosova IG, Van Huysen C, Fathallah L, Cao XC, Greene DA, Stevens MJ. An aldose reductase inhibitor reverses early diabetes-induced changes in peripheral nerve function, metabolism, and antioxidative defense. FASEB J. 2002;16:123–125. doi: 10.1096/fj.01-0603fje. [DOI] [PubMed] [Google Scholar]
- 64.Gupta S, Chough E, Daley J, Oates P, Tornheim K, Ruderman NB, Keaney JF., Jr Hyperglycemia increases endothelial superoxide that impairs smooth muscle cell Na+-K+-ATPase activity. Am J Physiol Cell Physiol. 2002;282:C560–C566. doi: 10.1152/ajpcell.00343.2001. [DOI] [PubMed] [Google Scholar]
- 65.El-Remessy AB, Abou-Mohamed G, Caldwell RW, Caldwell RB. High glucose-induced tyrosine nitration in endothelial cells: role of eNOS uncoupling and aldose reductase activation. Invest Ophthalmol Vis Sci. 2003;44:3135–3143. doi: 10.1167/iovs.02-1022. [DOI] [PubMed] [Google Scholar]
- 66.Rittner HL, Hafner V, Klimiuk PA, Szweda LI, Goronzy JJ, Weyand CM. Aldose reductase functions as a detoxification system for lipid peroxidation products in vasculitis. J Clin Invest. 1999;103:1007–1013. doi: 10.1172/JCI4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Heesom AE, Hibberd ML, Millward A, Demaine AG. Polymorphism in the 5′-end of the aldose reductase gene is strongly associated with the development of diabetic nephropathy in type I diabetes. Diabetes. 1997;46:287–291. doi: 10.2337/diab.46.2.287. [DOI] [PubMed] [Google Scholar]
- 68.Wang Y, Ng MC, Lee SC, So WY, Tong PC, Cockram CS, Critchley JA, Chan JC. Phenotypic heterogeneity and associations of two aldose reductase gene polymorphisms with nephropathy and retinopathy in type 2 diabetes. Diabetes Care. 2003;26:2410–2415. doi: 10.2337/diacare.26.8.2410. [DOI] [PubMed] [Google Scholar]
- 69.Zhao HL, Tong PC, Lai FM, Tomlinson B, Chan JC. Association of glomerulopathy with the 5′-end polymorphism of the aldose reductase gene and renal insufficiency in type 2 diabetic patients. Diabetes. 2004;53:2984–2991. doi: 10.2337/diabetes.53.11.2984. [DOI] [PubMed] [Google Scholar]
- 70.Ishii H, Tada H, Isogai S. An aldose reductase inhibitor prevents glucose-induced increase in transforming growth factor-beta and protein kinase C activity in cultured mesangial cells. Diabetologia. 1998;41:362–364. doi: 10.1007/s001250050916. [DOI] [PubMed] [Google Scholar]
- 71.Wong TY, Phillips AO, Witowski J, Topley N. Glucose-mediated induction of TGF-beta 1 and MCP-1 in mesothelial cells in vitro is osmolality and polyol pathway dependent. Kidney Int. 2003;63:1404–1416. doi: 10.1046/j.1523-1755.2003.00883.x. [DOI] [PubMed] [Google Scholar]
- 72.Ludvigson MA. Sorenson RLImmunohistochemical localization of aldose reductase: II Rat eye and kidney. Diabetes. 1980;29:450–459. doi: 10.2337/diab.29.6.450. [DOI] [PubMed] [Google Scholar]
- 73.Stevens MJ, Lattimer SA, Kamijo M, Van Huysen C, Sima AA, Greene DA. Osmotically-induced nerve taurine depletion and the compatible osmolyte hypothesis in experimental diabetic neuropathy in the rat. Diabetologia. 1993;36:608–614. doi: 10.1007/BF00404069. [DOI] [PubMed] [Google Scholar]
- 74.Phillips SA, Mirrlees D, Thornalley PJ. Modification of the glyoxalase system in streptozotocin-induced diabetic rats. Effect of the aldose reductase inhibitor Statil. Biochem Pharmacol. 1993;46:805–811. doi: 10.1016/0006-2952(93)90488-i. [DOI] [PubMed] [Google Scholar]
- 75.Beisswenger PJ, Howell SK, Nelson RG, Mauer M, Szwergold BS. Alpha-oxoaldehyde metabolism and diabetic complications. Biochem Soc Trans. 2003;31:1358–1363. doi: 10.1042/bst0311358. [DOI] [PubMed] [Google Scholar]
- 76.Nagaraj RH, Prabhakaram M, Ortwerth BJ, Monnier VM. Suppression of pentosidine formation in galactosemic rat lens by an inhibitor of aldose reductase. Diabetes. 1994;43:580–586. doi: 10.2337/diab.43.4.580. [DOI] [PubMed] [Google Scholar]
- 77.Hamada Y, Nakamura J, Naruse K, Komori T, Kato K, Kasuya Y, Nagai R, Horiuchi S, Hotta N. Epalrestat, an aldose reductase ihibitor, reduces the levels of Nepsilon-(carboxymethyl)lysine protein adducts and their precursors in erythrocytes from diabetic patients. Diab Care. 2000;23:1539–1544. doi: 10.2337/diacare.23.10.1539. [DOI] [PubMed] [Google Scholar]
- 78.Keogh RJ, Dunlop ME, Larkins RG. Effect of inhibition of aldose reductase on glucose flux, diacylglycerol formation, protein kinase C, and phospholipase A2 activation. Metabolism. 1997;46:41–47. doi: 10.1016/s0026-0495(97)90165-7. [DOI] [PubMed] [Google Scholar]
- 79.Yamagishi S, Uehara K, Otsuki S, Yagihashi S. Differential influence of increased polyol pathway on protein kinase C expressions between endoneurial and epineurial tissues in diabetic mice. J Neurochem. 2003;87:497–507. doi: 10.1046/j.1471-4159.2003.02011.x. [DOI] [PubMed] [Google Scholar]
- 80.Nakamura J, Kasuya Y, Hamada Y, Nakashima E, Naruse K, Yasuda Y, Kato K, Hotta N. Glucose-induced hyperproliferation of cultured rat aortic smooth muscle cells through polyol pathway hyperactivity. Diabetologia. 2001;44:480–487. doi: 10.1007/s001250051646. [DOI] [PubMed] [Google Scholar]