

Summary
Endocannabinoids (eCBs) mediate transient and long-lasting synaptic plasticity in several brain structures. In the dentate gyrus, activation of the type 1 cannabinoid receptor (CB1R) by exogenous ligands reportedly depresses excitatory synaptic transmission. However, direct evidence of eCB signaling at excitatory synapses in this region has been lacking. Here, we demonstrate that eCB release can be induced by a brief postsynaptic depolarization of dentate granule cells (DGCs), which potently and transiently suppresses glutamatergic inputs from mossy cell interneurons (MCs) but not from entorhinal cortex via the lateral and medial perforant paths. This input-specific depolarization-induced suppression of excitation (DSE) is calcium-dependent and can be modulated by agonists of cholinergic and group I metabotropic glutamate receptors. Inhibiting the synthesis of 2-arachidonoyl glycerol (2-AG), one of the most abundant eCBs in the brain, by diacyglycerol lipase (DGL) does not abolish DSE. Moreover, preventing the breakdown of anandamide, the other main eCB, does not potentiate DSE. Thus, eCB signaling underlying DSE in the dentate does not require DGL activity and is unlikely to be mediated by anandamide. Finally, we find that manipulations known to induce eCB-LTD at other central synapses do not trigger LTD at MCF-DGC synapses.
Keywords: endocannabinoids, CB1 receptors, DSE, mossy cells, excitatory synaptic transmission, glutamatergic inputs
Introduction
Endocannabinoids (eCB) have recently emerged as key mediators of short-term and long-term synaptic depression at both excitatory and inhibitory synapses in numerous brain structures (recently reviewed in Chevaleyre et al., 2006). Typically, the post-synaptic neuron releases eCBs in response either to depolarization or to bursts of presynaptic afferent activity that engage post-synaptic metabotropic glutamate receptors (Alger, 2002; Chevaleyre et al., 2006; Diana and Marty, 2004; Gerdeman and Lovinger, 2003; Kreitzer and Regehr, 2002). Once released, these messengers rapidly diffuse across the synaptic cleft and bind to type 1 cannabinoid receptors (CB1Rs) on nearby presynaptic terminals, resulting in a reduction of neurotransmitter release that can be transient (i.e. depolarization-induced suppression of inhibition or excitation, DSI/DSE) or long-lasting (i.e. eCB-mediated long-term depression or eCB-LTD). CB1Rs are widely distributed in the brain, and where the receptors are present, exogenous ligands reliably suppress synaptic transmission. In contrast, the magnitude and duration of eCB-mediated synaptic modulation varies widely at different CB1R-containing synapses.
The dentate gyrus has one of the highest levels of CB1R expression in the brain (Herkenham et al., 1990; Katona et al., 2006; Kawamura et al., 2006; Tsou et al., 1998). Exogenous cannabinoids have been shown to moderately reduce synaptic transmission at putative mossy cell fiber (MCF)-dentate granule cell (DGC) synapses (Monory et al., 2006), suggesting that mossy cell terminals may also be modulated by eCBs. However, very little is known about eCB signaling in this area and what potential role it may play. eCBs may have a significant functional impact on this system since, intriguingly, mossy cells excite, and are excited by, dentate granule cells (Buckmaster et al., 1992; Ratzliff et al., 2002; Scharfman, 1995). This positive feedback loop may predispose these cells to excitotoxic cell death. In fact, hilar mossy cells are one of the first cell types to be lost following experimentally-induced epileptiform activity (Buckmaster and Jongen-Relo, 1999; Cavazos et al., 1994), and their loss has been implicated in the development of epilepsy (Sloviter, 1994). In this context, the negative feedback provided by eCB signaling may be especially important. Indeed, recent work suggests that eCB signaling at excitatory synapses may play a protective role against epileptogenesis (Lutz, 2004; Monory et al., 2006; van der Stelt et al., 2002).
Here, we investigate whether excitatory synapses in the dentate gyrus can be modulated by eCBs. We make use of the finding that CB1Rs are highly expressed in the inner-most third of the molecular layer of the dentate gyrus (Katona et al., 2006; Kawamura et al., 2006; Monory et al., 2006; Tsou et al., 1998). This region, called the supragranular layer (SGL), receives excitatory associational/commissural axonal inputs primarily from hilar mossy cells (Buckmaster et al., 1992; Laurberg and Sorensen, 1981; Scharfman, 1995; Witter and Amaral, 2004). We recorded excitatory synaptic responses in DGCs while stimulating the mossy cell fibers (MCFs), the medial perforant path (MPP), or the lateral perforant path (LPP). We were able to induce eCB-mediated short-term synaptic suppression of the mossy cell synaptic input but not the perforant path inputs. Furthermore, we report that eCB-mediated short-term depression at MCF-DGC synapses can be enhanced by agonists of cholinergic and group I metabotropic glutamate receptors. eCB-LTD is not observed at this synapse, perhaps due to mechanistic differences with eCB-LTD-expressing synapses.
Materials and Methods
All animal experiments were carried in accordance with the National Institutes of Health guide for the care and use of laboratory animals. Acute hippocampal slices were prepared from postnatal day (P) 16-24 Wistar rats or from P28-30 CB1R-/- mice and CB1R+/+ C57BL/6J littermates. Mice were from our colony of Zimmer line CB1R-/- mice (Zimmer et al., 1999) and were maintained as heterozygotes and genotyped by PCR before use as described previously (Takahashi and Castillo, 2006). Animals were anesthetized with isofluorane, decapitated and their brains removed to a chilled cutting solution consisting of (in mM): 215 sucrose, 2.5 KCl, 26 NaHCO3, 1.6 NaH2PO4, 1 CaCl2, 4 MgCl2, 4 MgSO4 and 20 glucose. The brain was positioned in the cutting chamber with the anterior end facing downward and 400μm thick coronal slices were cut with a DTK-2000 vibrating microslicer (Dosaka EM Co., Ltd., Japan). Only caudal sections in which the dentate gyrus remained relatively intact along the longitudinal axis were used (Figure 1A). These longitudinal slices were stored at room temperature in holding solution containing (in mM) 62 NaCl, 2.5 KCl, 26 NaHCO3, 1.3 NaH2PO4, 1.75 CaCl2, 2 MgCl2, 2.15 MgSO4, 15 glucose and 107.5 sucrose. Thirty minutes post sectioning, slices were placed in extracellular recording solution containing: 124 NaCl, 2.5 KCl, 26 NaHCO3, 1 NaH2PO4, 2.5 CaCl2, 1.3 MgSO4 and 10 glucose. All solutions were saturated with 95% O2 and 5% CO2 (pH 7.4). Slices were incubated for at least 1 hr in the recording solution prior to experiments.
Fig. 1. Differential sensitivity of MCF and MPP inputs to group II mGluR agonist DGC-IV.

(A) Top, example of the typical longitudinal slices used in this study. Bottom, close-up of the region indicated by the white rectangular box in the top photograph showing the positioning of stimulating and recording pipettes. Schematic drawing of the organization of the different pathways in the molecular layer of the dentate gyrus are overlaid on the image. Stimulating electrodes were placed either proximal to the granule cell layer so as to activate fibers from mossy cells (MCF) or more distally (middle third of the molecular layer) to excite the medial perforant path (MPP). GC, granule cell; MC, mossy cell; LPP, lateral perforant path. (B) Time course of average excitatory responses demonstrating the inhibitory effect of DCG-IV on MPP (open squares) but not MCF (filled circles). Top, Representative EPSC traces from an individual experiment obtained at the time points in the time-course graph.
All experiments were conducted at 25 ± 1°C in a submersion-type recording chamber perfused at ~2 ml/min (Warner Instruments, LLC, Hamden, CT, USA). Whole-cell patch clamp experiments were performed in 100 μM picrotoxin (GABAA receptor antagonist) and 3 μM CGP 55845 (GABABR antagonist) to record isolated EPSCs. Field recordings were performed in 100 μM picrotoxin. To record AMPAR-mediated EPSCs, granule cells from the ventral blade of the dentate were voltage clamped at -70 mV using patch-type pipettes filled with intracellular solution containing (in mM): 131 Cs-Gluconate, 8 NaCl, 1 CaCl2, 10 EGTA, 10 glucose, 10 HEPES, 5 MgATP, 0.4 Na3GTP, pH = 7.2, 285 mmol/kg. In experiments with postsynaptic calcium chelators, 20 mM Cs-Gluconate and 10 mM EGTA were replaced with 20 mM BAPTA-K4 (osmolarity and pH were adjusted with CsOH). For experiments investigating spike-timing dependent plasticity, we used a K-based internal solution containing (in mM): 130 K-Gluconate, 10 NaCl, 0.2 EGTA, 10 HEPES, 5 MgATP, 0.4 Na3GTP, pH = 7.2, 285 mmol/kg. To record IPSCs from CA1 pyramidal cells, the postsynaptic cell was voltage clamped at 0 mV (using the same Cs-Gluconate intracellular solution as above) in the presence of 10 μM NBQX (AMPAR/KAR antagonist) and 25 μM D-APV (NMDAR antagonist). Series resistance (typically 15-30 MΩ) was monitored throughout the experiment with a -5 mV, 80 ms voltage step, and recordings with a greater than 10 % change in series resistance were excluded from analysis. Recording pipettes for field recordings were filled with 1 M NaCl.
Stimulating pipettes were filled with extracellular recording solution and placed in the SGL (<40 μm from the cell body layer) to stimulate the MCF and in the outer molecular layer (OML: <50 μm from the hippocampal fissure) to stimulate the LPP (Figure 1A). Given the possibility of crosstalk between the MCF and the more proximal MPP, we mainly focused on LPP transmission. Stimulation of specific inputs was confirmed using distinguishing electrophysiological and pharmacological properties of these synapses: The LPP input exhibits paired-pulse facilitation (PPF), while the MPP and MCF both exhibit paired-pulse depression (PPD). MPP, but not MCF, are sensitive to inhibition by group II metabotropic glutamate receptor (mGluR) agonists. Consistent with a previous report (Macek et al., 1996), we found that 1 μM DCG-IV strongly reduced MPP-EPSC by 74.7 ± 1.2 % at 10-15 minutes post drug application (n = 3, p<0.01; Figure 1B). In contrast, DCG-IV at this concentration had no effect on MCF-EPSC (102.9 ± 6.2 % of baseline, p>0.05). To reduce the likelihood of contamination when stimulating the LPP, we positioned the stimulating electrode as close to the hippocampal fissure as possible and stimulated with fine tipped (2.5-3.0 μm) patch-type electrodes at low stimulus intensity (21-25 V, 100 μs square-wave pulses).
Extracellular and whole-cell patch clamp recordings were performed using a Multiclamp 700B amplifier (Axon Instruments, Union City, CA, USA). Stimulation and acquisition were controlled by custom written software in Igor Pro 4.09A (Wavemetrics, Inc., Lake Oswego, OR, USA). In WIN 55,212-2 experiments, stimulation at each pathway was triggered every 20 s, and in DSE experiments, each pathway was stimulated every 6 s. Theta-burst stimulation consisted of a series of 10 bursts repeated 4 times. Each burst was comprised of five 200 Hz stimuli and the inter-burst interval was 200 ms. HFS consisted of 2 trains (20 s apart), each containing 100 pulses at 200 Hz. Spike-timing dependent plasticity was tested by pairing postsynaptic spikes with presynaptic stimulation (10 to 50 ms pre-post and post-pre pairing intervals). Twenty such pairings were typically given, the frequency of which ranged from 5 to 10 Hz. We also delivered bursts of coincident pre and postsynaptic activity (i.e., 5-10 pulses at 33.3-100 Hz), repeated four times at 100-500 ms inter-burst intervals. In DSE experiments, we initially tested depolarizing steps of different duration. We found that 1-sec depolarization from -70 mV to 0 mV was not sufficient to induce DSE, although a depolarization of 3-sec elicited strong DSE. Therefore, for the DSE experiments described in this paper, we used a 3-sec depolarizing voltage step.
All values are provided as the mean ± s.e.m. The paired-pulse ratio (PPR) is defined as the ratio of the amplitude of the second EPSC to the amplitude of the first EPSC. DSE magnitude is calculated as the percentage change between the mean of 10 consecutive EPSC traces preceding and the mean of 4 consecutive EPSC traces following depolarization. (EPSCs evoked at 0.17 Hz). The magnitude of drug effects is calculated as the percentage change between baseline (averaged excitatory responses for 10 to 15 minutes before drug application) and 10 to 15 minutes of stable responses at variable times (depending on the drug) post drug application. Statistical analysis was performed using both Student’s paired t-test and one-way ANOVA at the p<0.05 significance level in OriginPro 7.0 software (OriginLab Corporation, Northampton, MA); these tests yielded comparable p values.
Picrotoxin, CGP 55845, DCG-IV, WIN 55,212-2, Anandamide (AEA), DHPG, and physostigmine hemisulfate were purchased from Tocris-Cookson Inc. (Ellisville, MO). D-APV, NBQX and CNQX were obtained from Ascent Scientific (North Sumerset, UK). RHC-80267 was provided by Biomol International, LP (Plymouth Meeting, PA). URB597 was supplied by Cayman Chemical (Ann Arbor, MI), and carbachol and Orlistat (THL) were acquired from Sigma-Aldrich (St. Louis, MO). SR141716A was procured from the National Institute of Mental Health’s Chemical Synthesis and Drug Supply Program. WIN 55,212-2, SR141716A and URB597 were dissolved in DMSO and added to the bath. Total DMSO in the bath solution was maintained at 0.1% or below in all experiments.
Results
We first investigated whether exogenous activation of CB1Rs could suppress excitatory synaptic transmission onto DGCs. To this aim, we monitored AMPAR-EPSCs evoked by MCF or LPP stimulation before and after bath application of the CB1 receptor agonist WIN 55,212-2 (WIN) (5 μM) (Figure 2A). We found that 35-45 minutes following bath application of WIN the EPSC amplitude evoked by MCF stimulation decreased by 68 ± 7 % (n = 5, p<0.0001), while in the same cells EPSCs evoked by LPP stimulation were reduced by only 19 ± 9 % (n = 5, p=0.03). As previously described in other synapses, and consistent with a CB1R presynaptic action, the WIN-induced suppression of MCF inputs was associated with an increase in PPR (MCF: Control 0.77 ± 0.08, WIN 1.05 ± 0.09, p<0.05). In contrast, PPRs of LPP-EPSCs before and after WIN application were not significantly different (LPP: Control 1.23 ± 0.13, WIN 1.37 ± 0.11, p>0.1). Bath-applied WIN also reduced the slope of field EPSPs (fEPSPs) evoked by MCF stimulation by 76 ± 1 % (n = 3, p<0.0005) (Figure 2B). In contrast, fiber volley (FV) amplitude was unchanged (99 ± 3 % of baseline, p>0.5), suggesting that WIN does not affect MCF excitability. Finally, to confirm that WIN-depression is indeed mediated by CB1Rs, we performed extracellular field recordings in CB1+/+ and CB1-/- C57BL/6J mice (Figure 2C). We found that fEPSP slope in CB1+/+ animals decreased by ~56% when measured 35 to 50 min after WIN application (n = 2), whereas WIN had no effect on fEPSPs in CB1-/- animals (104 ± 3 % of baseline; n = 3, p>0.1). Together, these results show that functional CB1Rs are selectively expressed in mossy cell terminals, and that their activation leads to a decrease in glutamate release.
Fig. 2. Differential WIN 55,212-2 depression in the MCF and LPP inputs.
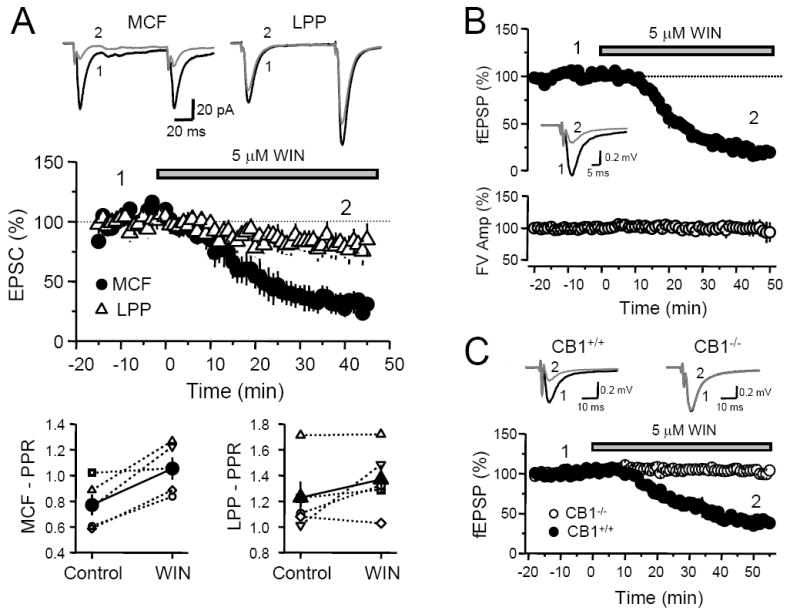
(A) Time course plot of average EPSC amplitudes upon application of WIN 55,212-2 in rats. MCF-EPSC (filled circles) depressed more significantly than LPP-EPSC (open triangles) following WIN. Top, Representative EPSC traces from an individual experiment obtained at the time points indicated. Each trace was obtained by averaging 30 individual traces that comprise a 10-minute period of recording. Bottom, Plot of the average PPR before and after WIN application. PPR of MCF-EPSCs significantly increased while PPR of LPP-EPSCs did not. Open symbols represent the PPR of individual experiments and filled symbols indicate the mean PPR averaged from all experiments. (B) Time course plot of average MCF fEPSP slope (top) and FV amplitude (bottom) upon application of WIN in rats. As in whole-cell recordings, WIN potently depressed MCF fEPSP slope, but had no effect on the FV. Inset, Representative fEPSP traces from an individual experiment before and after WIN application. Each trace was obtained by averaging 30 individual traces that comprise 10 minutes of recording. (C) Time course of average fEPSP slope upon application of WIN 55,212-2 in CB1+/+ and CB1-/- mice. WIN depressed MCF-fEPSP in wild type mice to a similar extent as in rats. The inhibitory effect of WIN was abolished in mice lacking CB1Rs. Top, Representative fEPSP traces (averaged from 30 individual traces) from single experiments obtained at the time points indicated.
Having established that MCFs express CB1 receptors, we next sought to determine whether DGCs are capable of producing enough eCBs to functionally engage them. A previous study looking at inhibitory synapses reported that DGCs can release eCBs (Isokawa and Alger, 2005), while another similar work obtained the opposite result (Chen et al., 2003). We re-examined this issue at excitatory synapses and found that a 3-second postsynaptic depolarization of DGCs from −70 to 0 mV triggered a transient (<120 s), 40 ± 3% depression of MCF-EPSC (n = 13; Figure 3A). In the same cells, postsynaptic depolarization only resulted in a weak 6 ± 1% depression of LPP-EPSCs. Consistent with a presynaptic effect, DSE of the MCF input was accompanied by an increase in PPR from 0.77 ± 0.05 before to 0.88 ± 0.05 immediately after postsynaptic depolarization (n = 7, p<0.05; Figure 3B). In contrast, PPR of the LPP synaptic responses was not significantly different before and after depolarization (1.09 ± 0.03 and 1.12 ± 0.03, respectively, p>0.05). Because LPP-DGC synapses are located farther from the cell body than MCF-DGC synapses, a potential confound for the lack of DSE at this synapse may be an inability to sufficiently depolarize distal dendrites. Therefore, we also examined DSE in the MPP-DGC synapse, which are located closer to the soma. In two cells, while the MCF-DGC synapse showed strong DSE (~49 % depression), MPP-EPSCs were unchanged following depolarization (~97 % of baseline, data not shown).
Fig. 3. DSE at MCF-GC but not LPP-GC synapse.
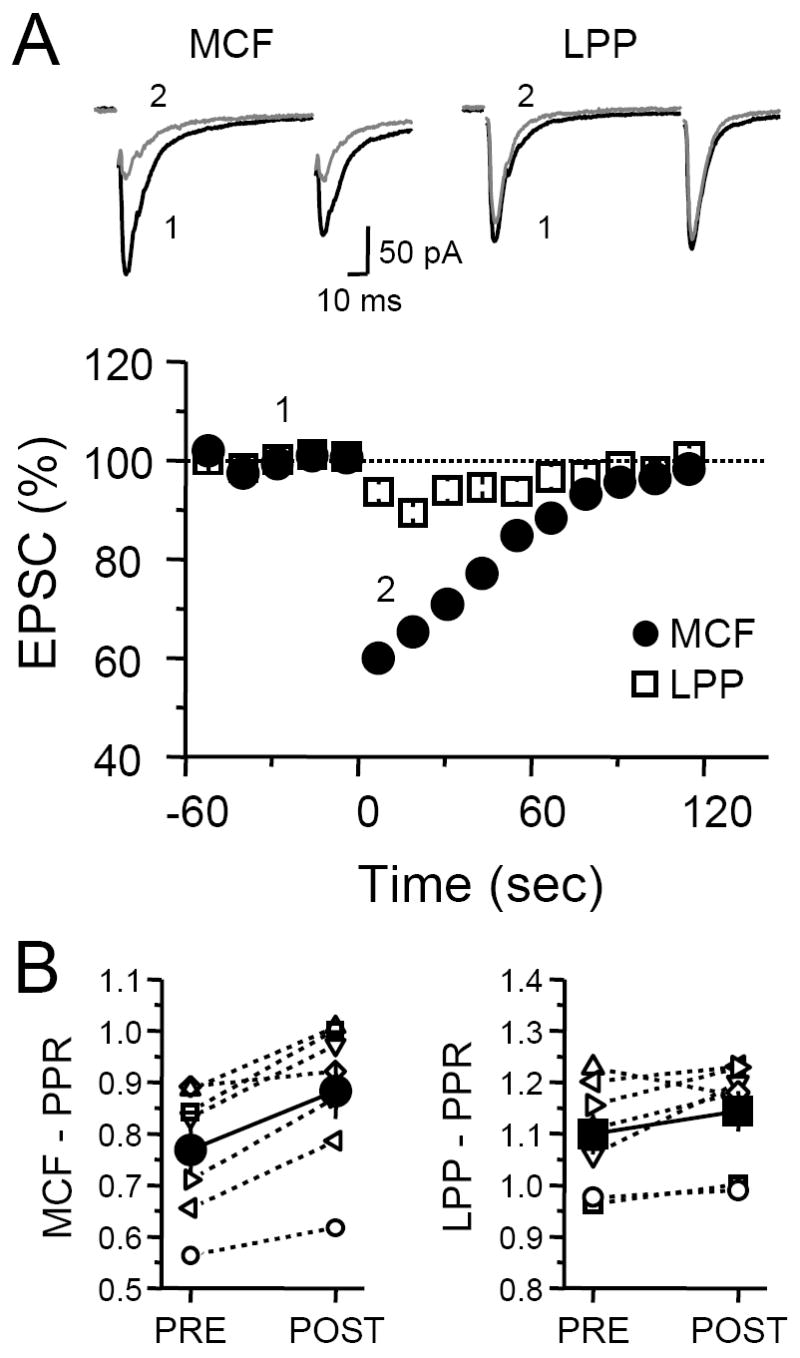
(A) Time course of average EPSC amplitudes before and after 3-sec postsynaptic depolarization of the granule cell. Depolarization transiently depressed MCF responses while having negligible effect on LPP responses. Top, Representative EPSC traces from a single experiment. EPSC traces designated as Pre were generated by averaging the 10 individual traces just prior to depolarization. Post EPSC traces were generated by averaging the first 4 individual traces post depolarization. (B) PPR plot before and after postsynaptic depolarization. On average, PPR of MCF-EPSCs (filled circles) significantly increased but PPR of LPP-EPSCs (filled squares) did not.
To test whether DSE in the MCF was indeed mediated by eCBs, we first obtained baseline DSE and then bath applied the CB1R antagonist SR141716A (SR). Wash-in of 4 μM SR clearly reduced the magnitude of DSE (Control 46 ± 4 %, SR 10 ± 5 % depression, n = 4, p<0.01; Figure 4A). However, SR application did not abolish the small depression (5 ± 3 %) observed following depolarization in the LPP (data not shown), suggesting that this effect was not mediated by eCBs. Consistent with the requirement of postsynaptic calcium rise for DSI/DSE in other synapses (Diana and Marty, 2004; Kreitzer and Regehr, 2002), inclusion of the fast calcium chelator BAPTA (20 mM) into the recording pipette completely blocked MCF DSE (Figure 4B). Thus, our results clearly show that DGCs are capable of releasing sufficient eCBs to induce DSE at least at MCF-DCG synapses.
Fig. 4. DSE at the MCF-GC synapse is eCB-mediated and calcium-dependent.
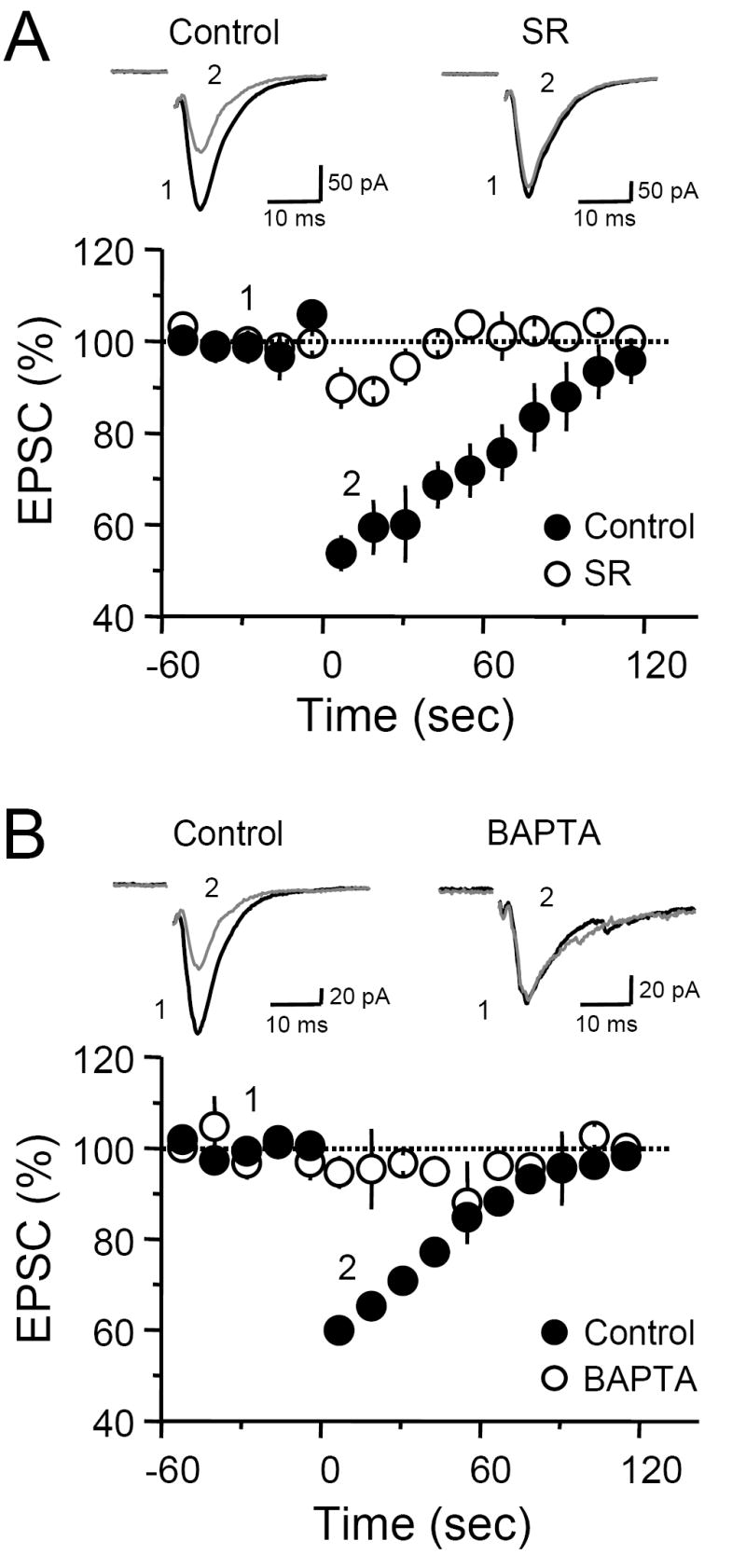
(A) Time course plot of DSE before and after SR wash-in. Blocking CB1R activation almost completely abolished DSE. Top, Representative EPSC traces, constructed by averaging 10 (Pre) or 4 (Post) individual traces, from a single experiment. (B) Time course plot of average MCF-EPSC amplitudes in control (filled circles) and in cells loaded with 20 mM BAPTA (open circles). Inclusion of this calcium chelator completely abolished DSE. Top, Representative EPSC traces from a single experiment that were obtained as in A.
Cholinergic projections from the medial septal nucleus directly terminate on interneurons in the hilus as well as in the SGL (Frotscher and Leranth, 1985; Mrzljak and Goldman-Rakic, 1993). Moreover, specific muscarinic acetylcholine receptor (mAChR) subtypes have been found to be differentially and highly expressed in the SGL (Rouse and Levey, 1997). The presence of the cholinergic inputs and mAChRs raises the possibility that DSE at MCF-DGC synapses may be subject to cholinergic modulation. Activation of mAChRs has been shown to enhance eCB release in the CA1 region (Kim et al., 2002; Martin and Alger, 1999) and hippocampal cell cultures (Ohno-Shosaku et al., 2003). We found that bath application of the non-selective cholinergic agonist carbachol (CCh) at a very low concentration of 500 nM potentiated DSE (Control 45 ± 6 %, CCh 57 ± 3 %, n = 5, p<0.05; Figure 5A) while slightly depressing basal transmission (84 ± 7 % of baseline, n = 6, p <0.05, not shown). To examine whether endogenous ACh could potentiate DSE, we bath-applied the inhibitor of acetylcholine esterase (AChE) physostigmine, a manipulation expected to increase cholinergic tone by blocking the breakdown of endogenous ACh. Wash-in of 2 μM physostigmine did not affect the magnitude of DSE (Control 43 ± 6 %, physostigmine 44.8 ± 5 %, n = 7, p>0.5; Figure 5B). However, the amount of time required for full recovery following DSE increased from about 2 min to >4min. At 120 s post depolarization, EPSC amplitudes in control and in physostigmine were 96 ± 2 % and 80 ± 3 % of baseline, respectively (n = 5, p<0.001). Thus, ACh, either constitutively present or released upon stimulation in the SGL, may facilitate DSE expression.
Fig. 5. Activation of either mAChRs or mGluRs facilitates DSE at the MCF-GC synapse.
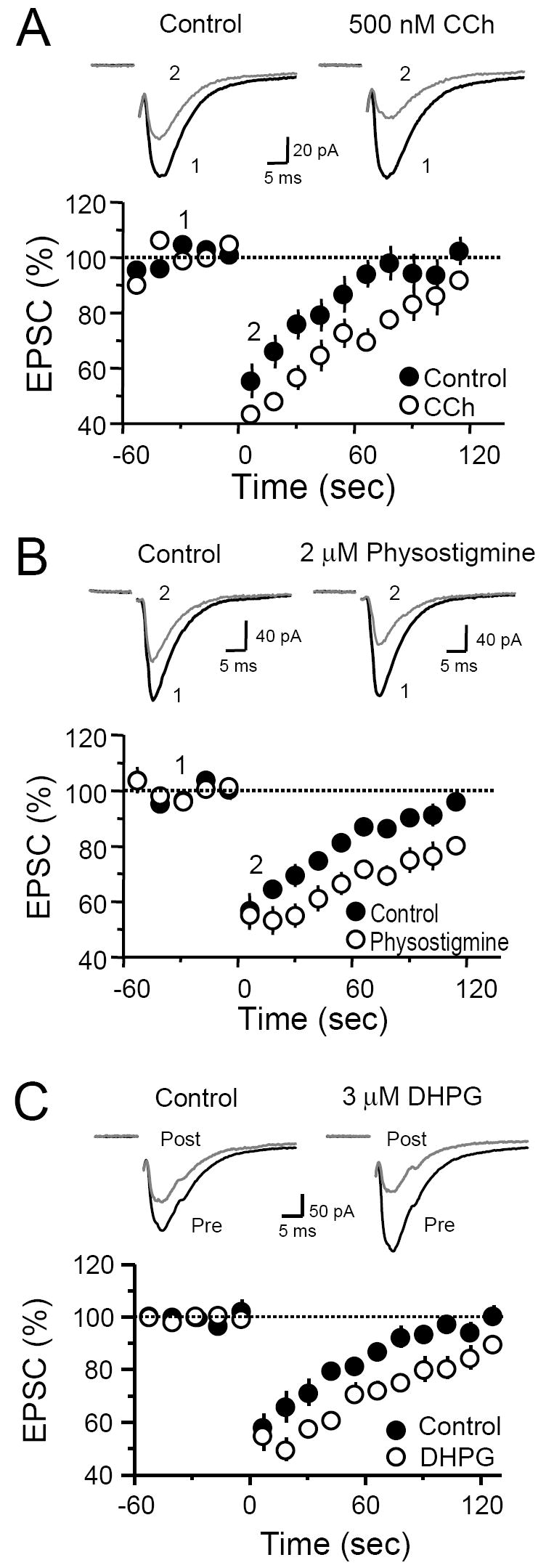
(A-C) Time course plots of DSE before and after bath application of 500 nM CCh (A), 2 μM physostigmine (B) and 3 μM DHPG (C). Top of each panel (A-C), representative EPSC traces from single experiments.
Activation of metabotropic glutamate receptors (mGluRs) has previously been shown to enhance hippocampal DSI (Ohno-Shosaku et al., 2002; Varma et al., 2001). Given the high expression of mGluR1 and mGluR5 in the SGL (Ferraguti et al., 1998; Shigemoto et al., 1997), we next examined whether activation of these receptors with the group I mGluR agonist DHPG could also enhance MCF DSE. Bath application of 3 μM DHPG did not affect MCF basal transmission (98 ± 9 % of baseline, n = 3, p>0.5) and did not augment the magnitude of DSE (Control 42 ± 5 %; DHPG 45 ± 6 %, n = 3, p>0.1). However, this low concentration of DHPG significantly prolonged DSE. In the presence of DHPG, MCF-EPSCs recovered to baseline >3 min following postsynaptic depolarization, compared to <2 min prior to DHPG application. At 120 s post depolarization, EPSC amplitudes in control and in DHPG were 102 ± 3 % and 90 ± 2 % of baseline, respectively (n = 3, p<0.05). These results show that MCF DSE can also be modulated by group I mGluR activation.
Among the several eCBs that have been identified in the brain, 2-arachidonoyl glycerol (2-AG), is one of the most abundant (Freund et al., 2003; Kogan and Mechoulam, 2006; Piomelli, 2003), and reportedly mediates DSI in cerebellum, striatum (Szabo et al., 2006) and presumably in hippocampus as well (Hashimotodani et al., 2007; Kim and Alger, 2004; Makara et al., 2005). The main enzyme responsible for 2-AG production, diacylglycerol lipase (DGL), is highly expressed in the SGL (Katona et al., 2006), suggesting that 2-AG may also be involved in DSE at the MCF-DGC synapse. To block DGL activity, we loaded the recording pipette with 50 μM RHC80267. However, DSE remained normal with this manipulation (44.7 ± 4 % depression, n = 4, p>0.05; Figure 6A). We also tested another chemically unrelated DGL inhibitor, Orlistat (Bisogno et al., 2003; Szabo et al., 2006). Loading 4 μM Orlistat in the postsynaptic cell also failed to block DSE (46 ± 6 % depression, n = 4, p>0.05; Figure 6B), strongly suggesting that DSE at the MCF-DGC synapse is independent of DGL activity. To confirm that Orlistat is effective in blocking DGL activity, we made use of the observation that eCB-LTD at inhibitory synapses in the hippocampal CA1 region (I-LTD) requires DGL activity (Chevaleyre and Castillo, 2003). As expected, loading CA1 pyramidal cells with 4 μM Orlistat blocked I-LTD induced by theta-burst stimulation (TBS) in stratum radiatum. IPSC amplitude 20-30 minutes following TBS in control (n = 3) and Orlistat-loaded (n = 4) pyramidal cells were 75 ± 1 % and 98 ± 6 % baseline (data not shown). Thus, if 2-AG mediates DSE at MFC-DGC synapses, this eCB is likely to be produced or released in a DGL-independent manner. This observation is consistent with previous reports in hippocampus (Chevaleyre and Castillo, 2003) and cerebellum (Safo and Regehr, 2005; but see Szabo et al., 2006). Finally, we investigated whether anandamide (AEA), the other main eCB found in the brain (Freund et al., 2003; Kogan and Mechoulam, 2006; Piomelli, 2003), may mediate DSE at the MCF-DGC synapse. If AEA mediates DSE, blocking AEA hydrolysis with the fatty acid amide hydrolase (FAAH) inhibitor URB597 (Kathuria et al., 2003) should facilitate DSE. We first tested whether URB597 was sufficient to block FAAH activity in brain slices as has been reported previously (Kim and Alger, 2004; Szabo et al., 2006). In our hands, 1 μM URB597 bath application for 30 minutes modestly but significantly enhances AEA-induced depression of inhibitory transmission in the CA1 region (Control 22 ± 4 % depression, n = 4; URB597 39 ± 5 % depression, n = 3, p<0.05). Next, we examined the effect of inhibiting FAAH on DSE. Bath application of 1 μM URB597 for 30 min did not affect DSE (Control 40 ± 4 %, URB597 39 ± 8 %, n = 4, p>0.05) (Figure 6C). Thus, it is unlikely that anandamide mediates DSE at MCF-DGC synapses.
Fig. 6. Inhibiting DGL or FAAH activity does not block DSE at the MCF-GC synapse.
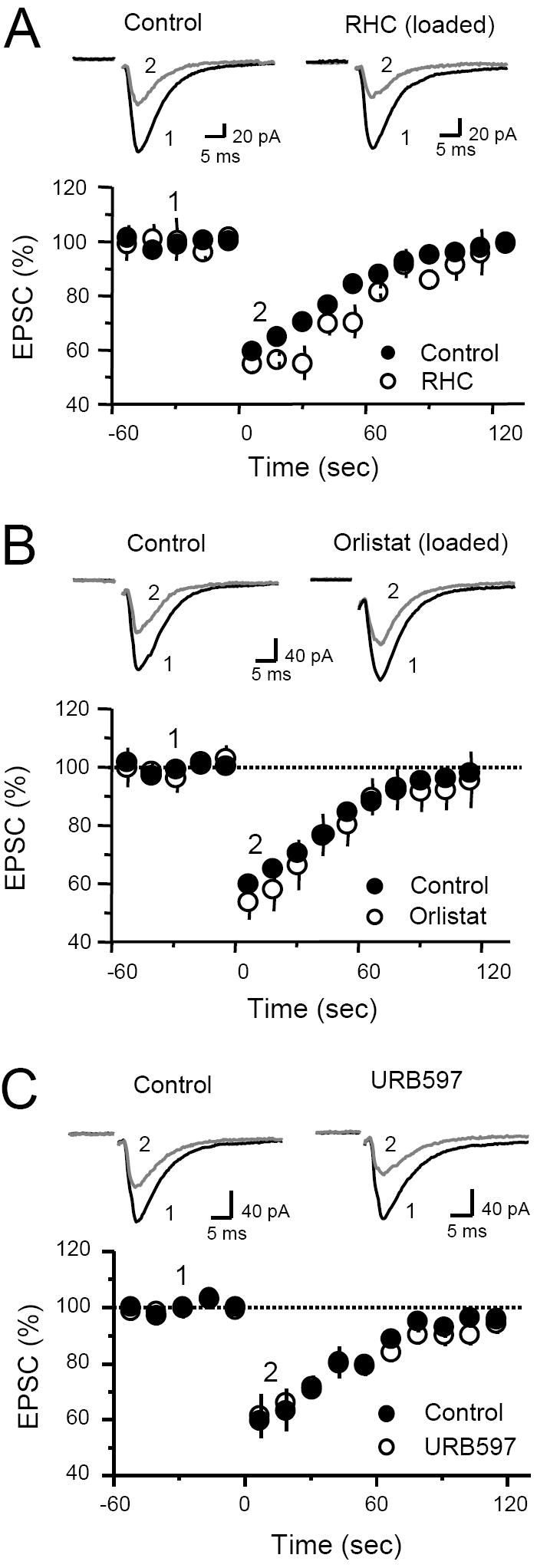
(A) Time course of average MCF-EPSC amplitudes in control (filled circle) and in cells loaded with 50 μM RHC80267. Inclusion of RHC80267 does not block DSE. Right, Representative EPSC traces from a single experiment before (average of 10 individual traces) and after (average of 4 individual traces) depolarization. (B) Time course of average MCF-EPSC amplitudes in control (filled circles) and in cells loaded with 4 μM Orlistat (open circles). Inclusion of Orlistat does not abolish DSE. Right, Representative EPSC traces from a single experiment before and after depolarization. (C) Time course of DSE before (filled circles) and after (open circles) bath application of 1 μM URB597. Blocking FAAH activity does not affect DSE. Top, Representative EPSC traces from a single experiment before and after depolarization.
eCBs are known to mediate both short and long-term synaptic plasticity (Chevaleyre et al., 2006). We next tested whether bursts of presynaptic activity (i.e. TBS) known to induce eCB-LTD at other synapses can also trigger LTD at MCF-DGC synapses. To avoid the potential induction of “classical” NMDAR-dependent forms of plasticity (i.e. LTP/LTD) (Malenka and Bear, 2004), these experiments were performed in the presence of 25 μM d-APV, a manipulation that does not affect the induction eCB-LTD at most synapses (Chevaleyre et al., 2006). We found that TBS did not induce LTD of MCF transmission (n = 4; Figure 7A). Several other stimulation protocols were also employed with similar results (see methods), such as high frequency stimulation (n = 6) and pairing presynaptic activity and postsynaptic spiking (n = 6). Because eCB-LTD may require NMDAR activity at some synapses (Bender et al., 2006; Sjostrom et al., 2003), TBS and spike-timing experiments were also repeated without d-APV (n = 4 and 6, respectively), but eCB-LTD was still not observed (data not shown). Many variables can influence whether plasticity can be triggered by synaptic stimulation. For example, glutamate release during presynaptic repetitive stimulation may have been insufficient to engage eCB signaling. Given previous findings that group I mGluR agonists can trigger eCB release and induce LTD in several brain structures (Azad et al., 2004; Chevaleyre and Castillo, 2003; Edwards et al., 2006; Robbe et al., 2002), we sought to bypass glutamate release and induce eCB-LTD pharmacologically. The group I mGluR agonist DHPG (50 μM) induced a transient depression of MCF transmission (Fig. 7B). However, this depression was not significantly affected by the CB1R antagonist (DHPG 22 ± 8 % depression, n = 9, DHPG+SR 13 ± 5 % depression, n = 4, p>0.05), and did not survive DHPG washout (94 ± 5 % of baseline 20-30 min after washout; n = 9; p>0.05; Figure 7B).
Fig. 7. MCF inputs do not exhibit eCB-mediated LTD.
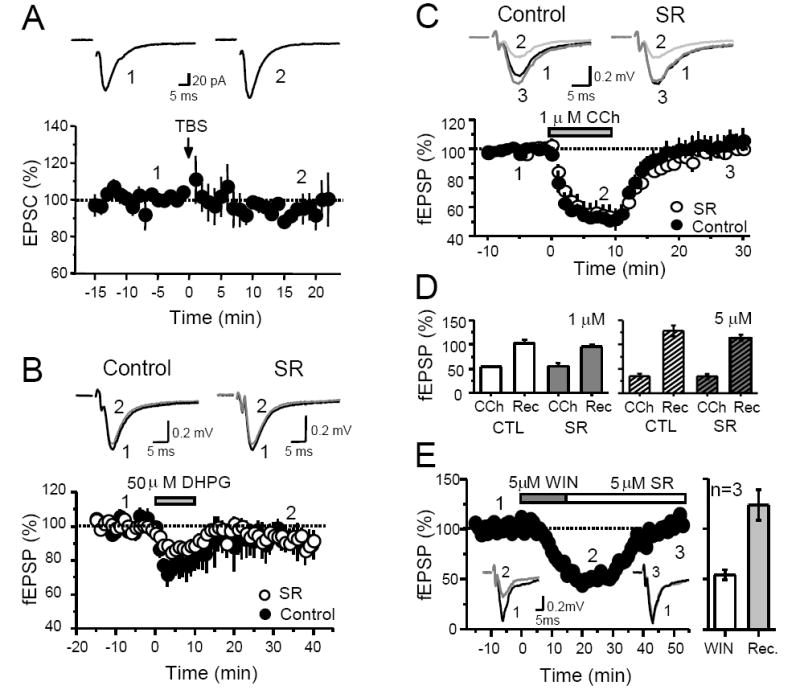
(A) Time course plot of average EPSC amplitudes before and after theta-burst stimulation (TBS) at the MCF-GC synapse. Top, Representative EPSC traces from a single experiment obtained at the time points indicated. Each trace was obtained by averaging 20 individual traces. (B) Time course plot of MCF fEPSP slope before and after DHPG application in control (filled circles) and in slices pre-incubated with SR (open circles). Top, Representative EPSC traces from single experiments obtained at the time points indicated. Each trace was obtained by averaging 30 individual traces. (C) Time course plot of the effect of CCh on MCF fEPSP slope in control slices (filled circles) and in slices pre-incubated with SR (open circles). Top, Representative EPSC traces from single experiments obtained at the time points indicated. Each trace was obtained by averaging 15 individual traces. (D) Summary plot of the effect of CCh on MCF-fEPSP slope at two different concentrations. 5 μM CCh depresses fEPSP to a greater extent than 1 μM CCh. However, this effect is CB1R-independent, and synaptic responses completely recover upon washout of CCh. (E) Time course plot of the effect of transient WIN application on fEPSP slope from one single experiment. WIN was bath applied for 15 minutes and washed out in the presence of SR. Activation of CB1Rs is not sufficient to trigger LTD of MCF inputs. Inset, fEPSP traces averaged from 30 individual traces at the time points indicated by numbers. Right, summary plot of WIN depression (WIN) and recovery of fEPSPs (Rec) from 3 experiments.
Because of the cholinergic influence on MCF DSE (Figure 5A,B) as well as the finding that mAChR activation can induce eCB release in CA1 (Edwards et al., 2006), we wondered whether a transient stimulation of mAChRs could trigger LTD of MCF transmission. Application of 1 μM CCh for 10 min depressed MCF-fEPSP to a slightly higher extent than 50 μM DHPG (Figure 7C, 7D). However, MCF-fEPSPs fully recovered upon CCh washout. Surprisingly, SR did not affect the magnitude of CCh-mediated suppression (CCh 46 ± 1 % depression; CCh+SR 45 ± 7 % depression, n =3, p>0.05), suggesting that, unlike the CA1 area (Edwards et al., 2006), this suppression is CB1R-independent. We also tried a higher dose of CCh (5 μM) and obtained similar results (Figure 7D). In conclusion, our results suggest that activation of AChRs or group I mGluRs alone does not promote significant eCB signaling at MFC-DGC synapses.
Activation of CB1Rs has been reported to be sufficient for the induction of eCB-LTD in dorsal striatum (Kreitzer and Malenka, 2005; Singla et al., 2007). To test this possibility at MCF-DCG synapses we monitored MCF-fEPSPs while stimulating MCF inputs at 0.05 Hz and bath applied 5 μM WIN for 15 min. We then washed out WIN in the presence of 5 μM SR (Figure 7E). Although MCF-fEPSPs were transiently suppressed in the presence of WIN, these responses completely recovered 35 to 60 minutes after terminating CB1R activation with SR. Thus, activation of CB1Rs is not sufficient to induce LTD in the MCF-DGC synapses.
Discussion
In this study we investigated the role of eCBs in regulating excitatory synaptic transmission in the dentate gyrus. We report that inputs from hilar mossy cells (e.g. MCF), but not from entorhinal cortex (i.e. MPP and LPP), can be regulated by eCBs. This finding is entirely consistent with the anatomical localization and functional expression of CB1Rs in the dentate gyrus. Previous studies have shown a stronger CB1R immunolabeling in SGL (the MCF synaptic field) than in the dentate molecular layer (the MPP and LPP synaptic field) (Katona et al., 2006; Kawamura et al., 2006; Monory et al., 2006). In addition, here we show that exogenous activation of CB1Rs produces a much stronger suppression of MCF than LPP synaptic transmission (Fig. 2A). Together, these observations support the notion that in the dentate gyrus, eCBs modulate excitatory transmission in a synapse type-specific manner. Given that we performed our experiments in young rats and young mice at ~25 °C, it is formally possible that eCBs may mediate a stronger effect on LPP/MPP inputs in older animals or at more physiological temperatures.
Another important finding of our study is that eCBs may mediate short-term (e.g. DSE) but not long-term plasticity (e.g. LTD) at MCF-DGC synapses. In other brain structures, previous work has extensively shown that synapses can undergo either short-term, long-term, or both forms of eCB-mediated plasticity (Alger, 2002; Chevaleyre et al., 2006; Diana and Marty, 2004; Gerdeman and Lovinger, 2003; Kreitzer and Regehr, 2002). The reason for the diverse duration of eCB actions is not entirely understood but it may be related to variable eCB production and CB1R efficiency across different synapse types (Chevaleyre et al., 2006). Previous studies have also demonstrated that transient application of a group I mGluR agonist can induce a CB1R-dependent, long-lasting depression of synaptic transmission in hippocampus (Chevaleyre and Castillo, 2003; Edwards et al., 2006), nucleus accumbens (Robbe et al., 2002) and amygdala (Azad et al., 2004). However, the same pharmacological manipulation does not trigger eCB-LTD at MCF-DGC synapses (Fig. 7B). Presumably, activation of mGluRs at these synapses might not be efficiently coupled to the synthesis and release of eCBs as it is in other structures. The ineffectiveness of mGluR activation to trigger eCB release may explain why DHPG application was unable to trigger eCB-LTD at MCF-DGC synapses.
It is worth noting that the WIN-mediated suppression of MCF transmission is fully abolished in CB1R-/- mice, further supporting the hypothesis that the CB1R is the major cannabinoid receptor mediating cannabinoid-dependent suppression of glutamatergic synapses in the CNS (Domenici et al., 2006; Katona et al., 2006; Kawamura et al., 2006; Takahashi and Castillo, 2006). In addition, the WIN-suppression of MCF transmission that we observe here is significantly larger than has been reported by others. In a previous paper (Monory et al., 2006), the same concentration of WIN only produced a 20-25% suppression of synaptic transmission in mice, whereas we observed >55% depression in both rats and mice. A potential source for this discrepancy may arise from some contamination of nearby MPP inputs. Although we did not directly assess the effects of WIN on MPP synaptic transmission, based on the uniform labeling in the outer two-thirds of the molecular layer (Katona et al., 1999), and lack of DSE (this study), the degree of depression following WIN in the MPP may be rather small, similar to that found in LPP inputs. Another variable which may contribute to the difference in the WIN depression may be the age of animals used. Whereas we used mice at about 1 month of age, Monory et al., 2006 used older mice (2-5 months old).
In contrast to the robust suppression of WIN at MCF synapses, WIN produced a barely significant effect at LPP-DGC synapses (Fig. 2A). Consistent with this observation, we were unable to induce DSE at perforant-path inputs (both LPP and MPP) following 3-sec depolarization of DGCs. An obvious explanation for the difference in DSE expression between the MCF and perforant-path synapses may be the disproportionate distribution of CB1Rs that are present at their presynaptic terminals. Alternatively, there may be comparable numbers of CB1Rs at all theses synapses but the coupling between CB1R and downstream effectors such as voltage-gated calcium channels may be less efficient in the perforant-path inputs. Another possibility is that release of eCBs could differ in the distal dendrites due to differences in the localization of the eCB synthetic machinery or in the ability to engage that machinery by depolarization, either because the magnitude of depolarization is smaller because of space clamp issues as discussed earlier, or because the calcium transient elicited by depolarization is smaller.
Cholinergic neurons from the medial septal nucleus send profuse projections to the SGL of the dentate gyrus (Frotscher and Leranth, 1985; Mrzljak and Goldman-Rakic, 1993). Such localization so close in proximity to the MCF-DGC synapse puts the cholinergic inputs in an ideal position to modulate synaptic transmission at this synapse. Consistent with previous reports showing that mAChR activation can increase eCB release at inhibitory synapses in acute slices of the CA1 region of hippocampus (Kim et al., 2002), and in cultured hippocampal neurons (Ohno-Shosaku et al., 2003), we report that exogenous activation of mAChRs facilitates DSE in dentate gyrus. Furthermore, our physostigmine experiments demonstrate that endogenous ACh can increase cholinergic tone and enhance DSE at the MCF-DGC synapse. Given that CCh application by itself does not lead to direct production of eCBs (Figure 7C), mAChR activation most likely modulates DSE by either enhancing calcium increase following postsynaptic depolarization or facilitating the efficacy with which calcium induces eCB release. Because eCBs may also modulate inhibitory inputs to DGCs (Isokawa and Alger, 2005), it will be important to see whether cholinergic inputs to the dentate can differentially regulate excitatory and inhibitory synaptic transmission via eCB signaling. At any rate, the modulation of eCB signaling by cholinergic inputs to the dentate raises the intriguing possibility that the two systems may affect learning and memory processes by interacting synergistically to regulate synaptic transmission in the dentate gyrus and thus the entire hippocampal formation.
The axonal projections from mossy cells travel long distances and innervate many granule cells (Buckmaster et al., 1996; Scharfman et al., 2003). Consequently, hilar mossy cells have been thought to mediate the coordination and integration of neuronal activity from different populations of DGCs along the septotemporal axis (Amaral and Witter, 1989). In addition to sending excitatory inputs to these cells, hilar mossy cells also receive reciprocal excitatory connections from them (Ratzliff et al., 2002). This positive feedback circuit makes mossy cells particularly vulnerable to reverberating neuronal activity and/or hyperactivity, as has been shown in experimentally induced epileptic seizures (Buckmaster and Jongen-Relo, 1999; Cavazos et al., 1994). Perhaps, DSE at MCF-DGC synapses helps to keep this reinforcing circuit in check and maintain excitatory transmission within safe operating levels.
Acknowledgments
We thank Kanji Takahashi, Vivien Chevaleyre and Boris Heifets for critical reading of the manuscript. CB1+/- mice of the Zimmer line are a gift of Andreas Zimmer (University of Bonn) and were obtained from George Kunos and Sandor Batkai (NIH/NIAAA). We thank Kanji Takahashi for his help in maintaining the Zimmer line and genotyping. This study was supported by NIH/NIDA, the Pew Biomedical Program and Institutional Postdoctoral Training Grant T32 NS 07439.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alger BE. Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol. 2002;68:247–286. doi: 10.1016/s0301-0082(02)00080-1. [DOI] [PubMed] [Google Scholar]
- Amaral DG, Witter MP. The three-dimensional organization of the hippocampal formation: a review of anatomical data. Neuroscience. 1989;31:571–591. doi: 10.1016/0306-4522(89)90424-7. [DOI] [PubMed] [Google Scholar]
- Azad SC, Monory K, Marsicano G, Cravatt BF, Lutz B, Zieglgansberger W, Rammes G. Circuitry for associative plasticity in the amygdala involves endocannabinoid signaling. J Neurosci. 2004;24:9953–9961. doi: 10.1523/JNEUROSCI.2134-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender VA, Bender KJ, Brasier DJ, Feldman DE. Two coincidence detectors for spike timing-dependent plasticity in somatosensory cortex. J Neurosci. 2006;26:4166–4177. doi: 10.1523/JNEUROSCI.0176-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno T, Howell F, Williams G, Minassi A, Cascio MG, Ligresti A, Matias I, Schiano-Moriello A, Paul P, Williams EJ, Gangadharan U, Hobbs C, Di Marzo V, Doherty P. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol. 2003;163:463–468. doi: 10.1083/jcb.200305129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckmaster PS, Jongen-Relo AL. Highly specific neuron loss preserves lateral inhibitory circuits in the dentate gyrus of kainate-induced epileptic rats. J Neurosci. 1999;19:9519–9529. doi: 10.1523/JNEUROSCI.19-21-09519.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckmaster PS, Strowbridge BW, Kunkel DD, Schmiege DL, Schwartzkroin PA. Mossy cell axonal projections to the dentate gyrus molecular layer in the rat hippocampal slice. Hippocampus. 1992;2:349–362. doi: 10.1002/hipo.450020403. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Wenzel HJ, Kunkel DD, Schwartzkroin PA. Axon arbors and synaptic connections of hippocampal mossy cells in the rat in vivo. J Comp Neurol. 1996;366:271–292. doi: 10.1002/(sici)1096-9861(19960304)366:2<270::aid-cne7>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Cavazos JE, Das I, Sutula TP. Neuronal loss induced in limbic pathways by kindling: evidence for induction of hippocampal sclerosis by repeated brief seizures. J Neurosci. 1994;14:3106–3121. doi: 10.1523/JNEUROSCI.14-05-03106.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Ratzliff A, Hilgenberg L, Gulyas A, Freund TF, Smith M, Dinh TP, Piomelli D, Mackie K, Soltesz I. Long-term plasticity of endocannabinoid signaling induced by developmental febrile seizures. Neuron. 2003;39:599–611. doi: 10.1016/s0896-6273(03)00499-9. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron. 2003;38:461–472. doi: 10.1016/s0896-6273(03)00235-6. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci. 2006;29:37–76. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- Diana MA, Marty A. Endocannabinoid-mediated short-term synaptic plasticity: depolarization-induced suppression of inhibition (DSI) and depolarization-induced suppression of excitation (DSE) Br J Pharmacol. 2004;142:9–19. doi: 10.1038/sj.bjp.0705726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domenici MR, Azad SC, Marsicano G, Schierloh A, Wotjak CT, Dodt HU, Zieglgansberger W, Lutz B, Rammes G. Cannabinoid receptor type 1 located on presynaptic terminals of principal neurons in the forebrain controls glutamatergic synaptic transmission. J Neurosci. 2006;26:5794–5799. doi: 10.1523/JNEUROSCI.0372-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DA, Kim J, Alger BE. Multiple mechanisms of endocannabinoid response initiation in hippocampus. J Neurophysiol. 2006;95:67–75. doi: 10.1152/jn.00813.2005. [DOI] [PubMed] [Google Scholar]
- Ferraguti F, Conquet F, Corti C, Grandes P, Kuhn R, Knopfel T. Immunohistochemical localization of the mGluR1beta metabotropic glutamate receptor in the adult rodent forebrain: evidence for a differential distribution of mGluR1 splice variants. J Comp Neurol. 1998;400:391–407. [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83:1017–1066. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- Frotscher M, Leranth C. Cholinergic innervation of the rat hippocampus as revealed by choline acetyltransferase immunocytochemistry: a combined light and electron microscopic study. J Comp Neurol. 1985;239:237–246. doi: 10.1002/cne.902390210. [DOI] [PubMed] [Google Scholar]
- Galante M, Diana MA. Group I metabotropic glutamate receptors inhibit GABA release at interneuron-Purkinje cell synapses through endocannabinoid production. J Neurosci. 2004;24:4865–4874. doi: 10.1523/JNEUROSCI.0403-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdeman GL, Lovinger DM. Emerging roles for endocannabinoids in long-term synaptic plasticity. Br J Pharmacol. 2003;140:781–789. doi: 10.1038/sj.bjp.0705466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Kano M. Presynaptic monoacylglycerol lipase activity determines basal endocannabinoid tone and terminates retrograde endocannabinoid signaling in the hippocampus. J Neurosci. 2007;27:1211–1219. doi: 10.1523/JNEUROSCI.4159-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, de Costa BR, Rice KC. Cannabinoid receptor localization in brain. Proc Natl Acad Sci U S A. 1990;87:1932–1936. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isokawa M, Alger BE. Retrograde endocannabinoid regulation of GABAergic inhibition in the rat dentate gyrus granule cell. J Physiol. 2005;567:1001–1010. doi: 10.1113/jphysiol.2005.094219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- Katona I, Sperlagh B, Sik A, Kafalvi A, Vizi ES, Mackie K, Freund TF. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci. 1999;19:4544–4558. doi: 10.1523/JNEUROSCI.19-11-04544.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Urban GM, Wallace M, Ledent C, Jung KM, Piomelli D, Mackie K, Freund TF. Molecular composition of the endocannabinoid system at glutamatergic synapses. J Neurosci. 2006;26:5628–5637. doi: 10.1523/JNEUROSCI.0309-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura Y, Fukaya M, Maejima T, Yoshida T, Miura E, Watanabe M, Ohno-Shosaku T, Kano M. The CB1 cannabinoid receptor is the major cannabinoid receptor at excitatory presynaptic sites in the hippocampus and cerebellum. J Neurosci. 2006;26:2991–3001. doi: 10.1523/JNEUROSCI.4872-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Alger BE. Inhibition of cyclooxygenase-2 potentiates retrograde endocannabinoid effects in hippocampus. Nat Neurosci. 2004;7:697–698. doi: 10.1038/nn1262. [DOI] [PubMed] [Google Scholar]
- Kim J, Isokawa M, Ledent C, Alger BE. Activation of muscarinic acetylcholine receptors enhances the release of endogenous cannabinoids in the hippocampus. J Neurosci. 2002;22:10182–10191. doi: 10.1523/JNEUROSCI.22-23-10182.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogan NM, Mechoulam R. The chemistry of endocannabinoids. J Endocrinol Invest. 2006;29:3–14. [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci. 2005;25:10537–10545. doi: 10.1523/JNEUROSCI.2959-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Retrograde signaling by endocannabinoids. Curr Opin Neurobiol. 2002;12:324–330. doi: 10.1016/s0959-4388(02)00328-8. [DOI] [PubMed] [Google Scholar]
- Laurberg S, Sorensen KE. Associational and commissural collaterals of neurons in the hippocampal formation (hilus fasciae dentatae and subfield CA3) Brain Res. 1981;212:287–300. doi: 10.1016/0006-8993(81)90463-7. [DOI] [PubMed] [Google Scholar]
- Lutz B. On-demand activation of the endocannabinoid system in the control of neuronal excitability and epileptiform seizures. Biochem Pharmacol. 2004;68:1691–1698. doi: 10.1016/j.bcp.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Macek TA, Winder DG, Gereau RWt, Ladd CO, Conn PJ. Differential involvement of group II and group III mGluRs as autoreceptors at lateral and medial perforant path synapses. J Neurophysiol. 1996;76:3798–3806. doi: 10.1152/jn.1996.76.6.3798. [DOI] [PubMed] [Google Scholar]
- Makara JK, Mor M, Fegley D, Szabo SI, Kathuria S, Astarita G, Duranti A, Tontini A, Tarzia G, Rivara S, Freund TF, Piomelli D. Selective inhibition of 2-AG hydrolysis enhances endocannabinoid signaling in hippocampus. Nat Neurosci. 2005;8:1139–1141. doi: 10.1038/nn1521. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Martin LA, Alger BE. Muscarinic facilitation of the occurrence of depolarization-induced suppression of inhibition in rat hippocampus. Neuroscience. 1999;92:61–71. doi: 10.1016/s0306-4522(98)00745-3. [DOI] [PubMed] [Google Scholar]
- Monory K, Massa F, Egertova M, Eder M, Blaudzun H, Westenbroek R, Kelsch W, Jacob W, Marsch R, Ekker M, Long J, Rubenstein JL, Goebbels S, Nave KA, During M, Klugmann M, Wolfel B, Dodt HU, Zieglgansberger W, Wotjak CT, Mackie K, Elphick MR, Marsicano G, Lutz B. The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron. 2006;51:455–466. doi: 10.1016/j.neuron.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrzljak L, Goldman-Rakic PS. Low-affinity nerve growth factor receptor (p75NGFR)- and choline acetyltransferase (ChAT)-immunoreactive axons in the cerebral cortex and hippocampus of adult macaque monkeys and humans. Cereb Cortex. 1993;3:133–147. doi: 10.1093/cercor/3.2.133. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Matsui M, Fukudome Y, Shosaku J, Tsubokawa H, Taketo MM, Manabe T, Kano M. Postsynaptic M1 and M3 receptors are responsible for the muscarinic enhancement of retrograde endocannabinoid signalling in the hippocampus. Eur J Neurosci. 2003;18:109–116. doi: 10.1046/j.1460-9568.2003.02732.x. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Shosaku J, Tsubokawa H, Kano M. Cooperative endocannabinoid production by neuronal depolarization and group I metabotropic glutamate receptor activation. Eur J Neurosci. 2002;15:953–961. doi: 10.1046/j.1460-9568.2002.01929.x. [DOI] [PubMed] [Google Scholar]
- Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- Ratzliff AH, Santhakumar V, Howard A, Soltesz I. Mossy cells in epilepsy: rigor mortis or vigor mortis? Trends Neurosci. 2002;25:140–144. doi: 10.1016/s0166-2236(00)02122-6. [DOI] [PubMed] [Google Scholar]
- Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ. Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. PNAS. 2002;99:8384–8388. doi: 10.1073/pnas.122149199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouse ST, Levey AI. Muscarinic acetylcholine receptor immunoreactivity after hippocampal commissural/associational pathway lesions: evidence for multiple presynaptic receptor subtypes. J Comp Neurol. 1997;380:382–394. [PubMed] [Google Scholar]
- Safo PK, Regehr WG. Endocannabinoids control the induction of cerebellar LTD. Neuron. 2005;48:647–659. doi: 10.1016/j.neuron.2005.09.020. [DOI] [PubMed] [Google Scholar]
- Scharfman HE. Electrophysiological evidence that dentate hilar mossy cells are excitatory and innervate both granule cells and interneurons. J Neurophysiol. 1995;74:179–194. doi: 10.1152/jn.1995.74.1.179. [DOI] [PubMed] [Google Scholar]
- Scharfman HE, Sollas AL, Berger RE, Goodman JH. Electrophysiological evidence of monosynaptic excitatory transmission between granule cells after seizure-induced mossy fiber sprouting. J Neurophysiol. 2003;90:2536–2547. doi: 10.1152/jn.00251.2003. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Kinoshita A, Wada E, Nomura S, Ohishi H, Takada M, Flor PJ, Neki A, Abe T, Nakanishi S, Mizuno N. Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J Neurosci. 1997;17:7503–7522. doi: 10.1523/JNEUROSCI.17-19-07503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singla S, Kreitzer AC, Malenka RC. Mechanisms for synapse specificity during striatal long-term depression. J Neurosci. 2007;27:5260–5264. doi: 10.1523/JNEUROSCI.0018-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron. 2003;39:641–654. doi: 10.1016/s0896-6273(03)00476-8. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. The functional organization of the hippocampal dentate gyrus and its relevance to the pathogenesis of temporal lobe epilepsy. Ann Neurol. 1994;35:640–654. doi: 10.1002/ana.410350604. [DOI] [PubMed] [Google Scholar]
- Szabo B, Urbanski MJ, Bisogno T, Di Marzo V, Mendiguren A, Baer WU, Freiman I. Depolarization-induced retrograde synaptic inhibition in the mouse cerebellar cortex is mediated by 2-arachidonoylglycerol. J Physiol. 2006;577:263–280. doi: 10.1113/jphysiol.2006.119362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi KA, Castillo PE. The CB1 cannabinoid receptor mediates glutamatergic synaptic suppression in the hippocampus. Neuroscience. 2006;139:795–802. doi: 10.1016/j.neuroscience.2006.01.024. [DOI] [PubMed] [Google Scholar]
- Tsou K, Brown S, Sanudo-Pena MC, Mackie K, Walker JM. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience. 1998;83:393–411. doi: 10.1016/s0306-4522(97)00436-3. [DOI] [PubMed] [Google Scholar]
- van der Stelt M, Veldhuis WB, Maccarrone M, Bar PR, Nicolay K, Veldink GA, Di Marzo V, Vliegenthart JF. Acute neuronal injury, excitotoxicity, and the endocannabinoid system. Mol Neurobiol. 2002;26:317–346. doi: 10.1385/MN:26:2-3:317. [DOI] [PubMed] [Google Scholar]
- Varma N, Carlson GC, Ledent C, Alger BE. Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus. J Neurosci. 2001;21:RC188. doi: 10.1523/JNEUROSCI.21-24-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witter MP, Amaral DG. Hippocampal formation. In: Paxinos G, editor. The rat nervous system. Elsevier Academic Press; San Diego: 2004. pp. 635–704. [Google Scholar]
- Zimmer A, Zimmer AM, Hohmann AG, Herkenham M, Bonner TI. Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc Natl Acad Sci U S A. 1999;96:5780–5785. doi: 10.1073/pnas.96.10.5780. [DOI] [PMC free article] [PubMed] [Google Scholar]