

Abstract
The protein tyrosine phosphatase SHP-1 is a critical regulator of cytokine signaling and inflammation. Mice homozygous for a null allele at the SHP-1 locus have a phenotype of severe inflammation and are hyper-responsive to the TLR4 ligand LPS. TLR4 stimulation in the CNS has been linked to both neuropathic pain and sickness behaviors. To determine if reduction in SHP-1 expression affects LPS-induced behaviors, responses of heterozygous SHP-1-deficient (me/+) and wild-type (+/+) mice to LPS were measured. Chronic (4-week) treatment with LPS induced avoidant behaviors indicative of fear/anxiety in me/+, but not +/+, mice. These behaviors were correlated with a LPS-induced type 2 cytokine, cytokine receptor, and immune effector arginase profile in the brains of me/+ mice not found in +/+ mice. Me/+ mice also had a constitutively greater level of TLR4 in the CNS than +/+ mice. Additionally, me/+ mice displayed constitutively increased thermal sensitivity compared to +/+ mice, measured by the tail-flick test. Moreover, me/+ glial cultures were more responsive to LPS than +/+ glia. Therefore, the reduced expression of SHP-1 in me/+ imparts haploinsufficiency with respect to the control of CNS TLR4 and pain signaling. Furthermore, type 2 cytokines become prevalent during chronic TLR4 hyperstimulation in the CNS and are associated positively with behaviors that are usually linked to type 1 pro-inflammatory cytokines. These findings question the notion that type 2 immunity is solely anti-inflammatory in the CNS and indicate that type 2 immunity induces/potentiates CNS inflammatory processes.
Keywords: IL-4, IL-13, arginase, LPS
INTRODUCTION
The protein tyrosine phosphatase SHP-1 is a critical negative regulator of cytokine signaling and innate immunity. Research on SHP-1 has focused mainly on its inhibition of various Janus kinase (JAK)/signal-transducer and activator of transcription (STAT) pathways. SHP-1 limits the activation and activity of STATs, including STAT6 (Massa and Wu, 1996; Haque et al., 1998; Massa et al., 2000; Paling and Welham, 2002). Of particular interest is the regulation of STAT6-inducible arginase I expression by SHP-1 in CNS glia, and its effects on CNS innate immunity and inflammation (Bonaparte et al., 2006). However, it has also been demonstrated that SHP-1 regulates toll-like receptor (TLR) signaling. Lack of SHP-1 causes a greater, more prolonged activation of the transcription factor nuclear factor-κB (NF-κB) in both lymphocytes and glia in response to lipopolysaccharide (LPS) and other inducers of NF-κB (Khaled et al., 1998; Massa and Wu, 1998). This increased activation is caused by a more pronounced degradation of the NF-κB repressor IκB (Massa and Wu, 1998). Recently, it has been shown that in vitro, microglia from SHP-1-deficient (motheaten or me/me) mice have a greater type 1 pro-inflammatory response, as evidenced by increased production of tumor necrosis factor α (TNF-α), interleukin 1β (IL-1β) and inducible nitric oxide synthase (iNOS), after stimulation with the TLR4 agonist LPS (Zhao et al., 2006). In the CNS, multiple cell types express TLR4. TLR4 is present constitutively at relatively high levels in microglia. Although low in astrocytes in basal conditions, TLR4 concentration is upregulated by LPS stimulation (Bowman et al., 2003; Jack et al., 2005). Hence, the regulation of NF-κB and STAT6 signaling by SHP-1 is likely to be crucial in the control of glial innate immune responses in the CNS.
Motheaten mice suffer from severe inflammation/autoimmune disease and have a lifespan of <30 days. Therefore, they not amenable to studies of chronic inflammatory disease and associated behavioral abnormalities. By contrast, mice heterozygous at the SHP-1 locus (me/+) have normal lifespans and no overt phenotypic differences from wild-type animals. Me/+ mice, however, have 50% less expression of SHP-1 than wild types and are more susceptible to antigen-induced inflammatory/autoimmune diseases such as allergic airway inflammation, experimental autoimmune encephalomyelitis and experimental autoimmune myasthenia gravis (Deng et al., 2002; Kamata et al., 2003; Deng et al., 2003). Accordingly, me/+ leukocytes are hyper-responsive to various immune stimuli (Kamata et al., 2003). Thus, it appears that reduced expression of SHP-1 is responsible for a haploinsufficiency in the control of inflammatory responses that might involve both NF-κB and STAT6 signaling pathways.
Acute administration of LPS has long been known to induce a pattern of behavior called sickness behavior, which is characterized by fever, reduced food/water consumption, reduced activity and social avoidance in rodents (Dantzer et al., 1998). LPS also induces an increase in pain-related and hyperalgesic behaviors in rodents (Inceoglu et al., 2006). Recently, it has been demonstrated that nerve injury-induced neuropathic pain (measured by other avoidant behaviors) is also mediated through a TLR4-dependent mechanism (Tanga et al., 2005). Furthermore, this mechanism is dependent on TLR4 in CNS glia (Tanga et al., 2005). Hence, TLR4 stimulation in the CNS is associated with an increase of the avoidant behaviors representative of sickness behavior and pain. Like neuropathic pain, repeated administration of LPS induces a chronic state of TLR4-dependent CNS inflammation (Chakravarty and Herkenham, 2005). Because me/+ mice have reduced expression of SHP-1 and SHP-1 has been shown to negatively regulate TLR4 signaling, we reasoned that me/+ mice might display increased sensitivity to TLR4-induced behavioral abnormalities, including pain responses.
OBJECTIVE
To test if a reduction in SHP-1 increases sensitivity to TLR4 stimulation in the CNS and whether this might lead to behavioral and CNS inflammatory cytokine signaling abnormalities. Me/+ and +/+ mice will be treated with LPS chronically (twice a week for 4 weeks) before undergoing behavioral tests to measure behavioral alterations. Furthermore, the brains of LPS- and control-treated me/+ and +/+ mice will be analyzed for cytokines and related immune effectors to determine the type of LPS-induced immune response correlated with the behavioral changes. Untreated me/+ and +/+ mice will be tested for differences in pain sensation because pain and TLR4 have been positively associated. Glial cultures will also be treated with LPS to determine whether me/+ glia are intrinsically more responsive to LPS than +/+ glia. Hence, these experiments will determine whether me/+ mice have an increased CNS response to TLR4 stimulation and sensation of pain in vivo and increases in related behaviors.
METHODS
Mice
For in vivo LPS studies, 8−10-month old me/+ (C3FeLe.B6-a/a-Ptpn6 me/j, formerly known as C3FeLeB6-a/a Hcph me/j) and +/+ male and female mice were purchased from Jackson Laboratories (Bar Harbor). Animals were housed individually (to avoid fighting and injury) in 11″ × 7″ × 5′ cages at 22°C with 10% humidity and food and water ad libitum. Mice were maintained on a 12-hour light/dark cycle and allowed at least 1 week of acclimation in the animal room before any experimental manipulation. All animal procedures described in this study were approved by the Committee for the Humane Use of Animals (CHUA) at Upstate Medical University.
Adult glial culture
Adult (6−7 months) me/+ and +/+ female mice were euthanized by CO2 inhalation and spinal cords were harvested aseptically. Spinal cords were then homogenized and cell suspensions were dissociated from myelin debris with a 20% Percoll (Amersham) gradient. Cultures were grown in DMEM with 10% FBS and 50 μM 2-mercapto-ethanol for 2 weeks and were fed every 3 days. At day 14 the growth medium was removed and cultures treated with treatment media containing 0, 1, 4 and 20 μg ml−1 Salmonella minnesota re595 LPS (Sigma-Aldrich) in growth media. Media was harvested at 24 and 48 hours post-treatment. At 48 hours, cells were washed and lysed in RIPA buffer (Bonaparte et al., 2006) for protein concentration determination via the BCA Protein Assay Kit (Pierce).
Chronic LPS injection
To mimic a state of chronic inflammation, me/+ and +/+ mice were injected intraperitoneally twice a week for 4 weeks with either 0 or 50 μg of S. minnesota re595 LPS (Galanos et al., 1969) in 200 μl of sterile PBS. The LPS from S. minnesota re595 was chosen because it is an LPS from a rough strain and, therefore, contains no O-antigen. This LPS is mitogenic but not immunogenic, allowing for increased specificity of TLR4-mediated signaling in vivo. S. minnesota re595 LPS is less potent than LPS from wild-type strains (Werner-Felmayer et al., 1995) and so we used a higher dose than usual for wild-type LPS. This dose has been used previously for long-term in vivo LPS treatments in mice (Cavallo and Granholm, 1990; Cavallo and Granholm, 1991).
Three days after the last injection, mice (ten LPS-treated me/+, nine control me/+, six LPS-treated +/+ and control +/+) were subjected to a battery of behavioral tests (see below) over a 3-day period. All groups contained equal numbers of males and females except control me/+ (five females and four males). A subset of each group (four LPS-treated me/+ mice, four untreated control me/+ mice, three LPS-treated +/+ mice and three untreated control +/+ mice) were weighed to determine whether LPS treatment caused a loss in body weight relative to same-gender controls. After testing, mice were euthanized via CO2 asphyxiation, blood was drawn via cardiac puncture, mice were perfused with ice-cold PBS and brains were harvested. Serum was harvested from blood as described previously (Hudson et al., 2003).
Real-time RT PCR
Total RNA was isolated from brains and spinal cords using RNA STAT-60 reagent (Tel-Test, Inc.) per the manufacturer's protocol. RNA was quantified and assessed for purity spectrophotometrically. Total RNA (0.5 μg) was subjected to real-time RT-PCR using the Light Cycler-RNA Amplification Kit SYBR Green I (Roche Diagnostics) in a ABI Prism 7000 (Applied Biosystems) using the following primers: STAT6 forward, 5′-TGGTCCTGGTCCAATGAG-3′; STAT6 reverse, 5′-AGACGGCTTATCAGCGATG-3′: IL-10 receptor forward, 5′-CTGTTTCTAGCTGTGTGA-3′; IL-10 receptor reverse, 5′-CATTTGAGGCGTCTGTGC-3′; IL-13 receptor α1 forward, 5′-TCTTCTCCTCAAAAATGGTGCC-3′; IL-13 receptor α1 reverse, 5′-GGATTATGACTGCCACTGCGAC-3′. Serial dilutions of cDNA clones containing known copy numbers were used in each real-time RT PCR run to generate a standard curve relating copy number with threshold amplification cycle. Relative gene expression levels were calculated during the logarithmic amplification phase by determining the initial cDNA copy number using the standard curve. Amplification of each gene-specific fragment was confirmed by examination of melting peaks and by agarose gel electrophoresis. To control for loading, parallel examination of the housekeeping gene glyceraldehyde phosphate 3-dehydrogenase (GAPDH) was assessed in each sample using the following primers: forward primer, 5′-ACCACCATGGAGAAGGC-3′; reverse primer, 5′-GGCATGGACTGTGGTCATGA-3′.
Arginase activity
Brains were homogenized in a lysis buffer containing 50 mM Tris (pH 7.4), 1% NP-40, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1 mM Na3VO4 and 1 μg ml−1 aprotinin, leupeptin and pepstatin. Protein concentration of each lysate was then determined via the BCA Protein Assay Kit. To determine arginase activity, 25 μl of sample was added to an Eppendorf tube and mixed with 25 μl of 10 mM MnCl2 in 50 mM Tris-HCl (pH 7.5). Tubes were then incubated for 10 min at 55°C. Next, 50 μl of 0.5 M arginine was added to each tube and the tubes were incubated at 37°C for 75 min. After the incubation, 400 μl of stop solution containing 1 part H2SO4: 3 parts H3PO4: 7 parts H2O was added to each sample and the prepared standards. Standards were prepared by serially diluting a stock preparation of urea in 50 mM Tris-HCl (pH 7.5) to yield a standard range from 1500 to 23.4 μg ml21. To measure the amount of urea in each tube, 50 μl of 9% 1-phenyl-1,2-propanedione-2-oxime (Sigma-Aldrich) in 100% ethanol was added to each sample and standard and tubes were incubated at 100°C for 60 min. Tubes were then placed in the dark at 25°C for 30 min. Samples and standards (100 μl well−1) were then transferred in triplicate to a 96-well plate and optical density was read at 540 nm with a 690 nm correction. Sample concentrations were determined from the standard curve and normalized to Arginase Units via the following formula: urea produced (μg) / total protein (mg).
Cytokine ELISAs
Cytokines [IL-4, IL-10, IL-13, interferon γ (IFN-γ), TNF-α, IL-1β and IL-6] were measured using R&D Systems DuoSet ELISA kits (R&D Systems) following the manufacturer's protocol.
Behavioral tests
Light-dark box
Mice were placed in the dark side of a 20″ × 11″ glass box which was half-darkened and the time taken to enter the light side as well as the total time spent in the light side measured during a 10-min period (time-point measurements at 3, 5 and 10 min). This is a measurement of fear- and anxiety-like behavior, with the longer it takes to enter the light side and the less time spent in the light side indicates of fearful behavior (Rossi-George et al., 2004). Each mouse was tested once on day 2 of the testing schedule. The light-dark box was the first test performed on day 2.
Open-field chamber
Mice are placed in a circular chamber (28″ radius) divided into 5 cm × 5 cm squares and the total number of squares traveled in a 5-min period measured to determine exploratory behavior. More anxious/fearful mice explore less (Lalonde et al., 2003). Each mouse was tested once on day 3 of the testing schedule. The open-field chamber was the first test performed on day 3.
T-maze
Mice are placed in the ‘bottom’ of a 7-cm wide T-shaped maze with 17-cm arms in which one of the arms is blocked. The mouse is allowed to enter and remain in the unblocked arm for 1 min, after which it is moved back to the bottom of the T and both cross-arms now unblocked. The cross-arm entered along with the decision latency is recorded (Lalonde et al., 2003). This test measures spontaneous alternation, but the decision making time can also be used as a measure of anxiety and psychomotor slowing (Lalonde et al., 2003). Mice were tested once a day for 3 days. The T-maze was the last test performed on days 1−3.
Wire screen
Mice are placed on the bottom (with all four paws gripping) of a 25 cm × 25 cm wire-mesh screen that is suspended ∼1′ above a cushioned bench top. The time spent to reach the top of the screen and the ability to reach the top of the screen within 40 sec are recorded. This is a measure of motor skill, cerebellar function and fear/anxiety (Voikar et al., 2002). Mice were tested once a day for 3 days. The wire-screen test was the first test performed on day 1 and the second test performed on days 2 and 3.
Coat hanger
Mice are placed (gripping with their front paws) on a coat hanger that is suspended 2′ above a cushioned bench top. The ability to hang on the coat hanger for 60 sec, pull up onto the bar of the coat hanger, and traverse to the end of the bar are measured and scored. Each mouse is timed for the latency to fall off the bar and the ability to traverse to the end of the bar is scored as a latency of 2 min. Hence, a mouse that can traverse to the end of the bar would have an adjusted latency of 120 sec. The postural scoring used a modification of the method published by Voikar et al. (2002) as follows: 0, falls off bar in <10 sec; 1, hangs onto bar for 10 sec; 2, hangs onto bar for >10 sec with forepaws; 3, hangs onto bar for >10 sec with forepaws and at least one hind paw; 4, hangs onto bar for >10 sec with all 4 paws and wraps its tail around bar; 5, traverses to the end of the bar. This is test of motor skill and cerebellar function. Mice were tested once a day for 3 days. The coat hanger test was the second test performed on day 1 and the third test performed on days 2 and 3.
Tail flick-latency test
The tails of mice were placed in a 49°C water bath (∼1/3 of the tail submerged) and the latency to flick the tail is measured with a maximum time in the bath of 30 sec. Mice are also tested at 37°C for a maximum of 30 sec to see if any differences are caused by an inherent sensitivity to water at basal body temperature.
Serum corticosterone levels
To measure activation of the hypothalamic–pituitary–adrenal (HPA) axis, serum corticosterone (CORT) was measured with an EIA kit (Assay Designs, Inc.) following the manufacturer's protocol. The sensitivity of this assay is ∼27 pg ml−1 and the intra- and inter-assay variability ∼8 pg ml−1.
Statistical analysis
For glial culture cytokine analysis, three-way analysis of variance (ANOVA) with genotype, time and LPS dose as factors was performed followed by Student-Neuman-Keuls post hoc analyses. For glial culture and behavioral time point analyses, two-way ANOVA with genotype and LPS dose as factors was performed followed by Student-Neuman-Keuls post hoc analyses. To analyze the overall effects of LPS in the coat-hanger test within each genotype, two-way ANOVA with day and LPS dose as factors was utilized. For all other parametric comparisons, one-way ANOVA was performed followed by the Student-Neuman-Keuls method. For non-parametric comparisons, ANOVA on ranks followed by Dunn's post hoc analysis was performed. P<0.05 is considered statistically significant throughout.
RESULTS
Behaviors after chronic LPS administration
There were no differences in avoidant behaviors between untreated me/+ and +/+ mice. However, after 4 weeks administration of re595-LPS, LPS-treated me/+ mice showed profound signs of avoidant behavior that were not observed in +/+ mice. In the light-dark box, LPS-treated me/+ mice spent significantly less time in the light side of the box than untreated me/+ controls (at T = 3, 5 and 10 min; 10 min shown in Fig. 1A) and had significantly longer latencies to enter the light side (Fig. 1B) than untreated me/+ controls and +/+ animals. Similarly, whereas the T-maze results did not indicate differential effects of LPS on spontaneous alternation, there were significant differences in latency to enter a cross-arm, as LPS-treated me/+ mice had significantly longer latencies than same-genotype controls and LPS-treated +/+ animals on day 2 of testing (Fig. 1C) (day 1 are results similar but not significant and there were no differences on day 3, possibly because of habituation). Furthermore, LPS-treated me/+ mice had significantly less exploratory behavior than untreated me/+ controls in the open-field test, as measured by both horizontal exploration (squares crossed) at 3 min (Fig. 1D, 0−3 min) and vertical exploration (not shown). Interestingly, after the first 3 min of the open-field test, there were no differences amongst the groups in horizontal exploration (Fig. 1D, 3−5 min), suggesting that the differences found in the open field at the early time point were not caused by an overall reduction in locomotion in LPS-treated me/+ mice, but by increased fear and anxiety in the LPS-treated me/+ mice upon entering a novel environment.
Fig. 1. LPS-induced avoidant behaviors.
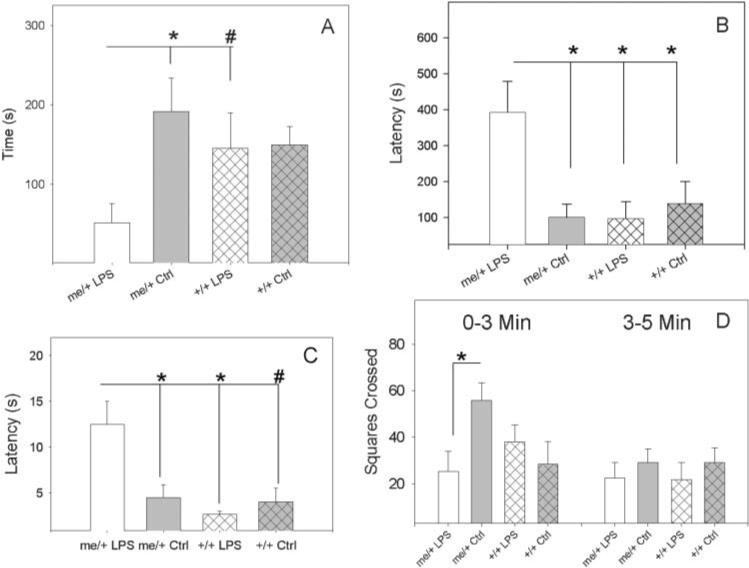
(A) Time spent in a light-dark box for LPS-treated and control (PBS) me/+ and +/+ mice after 4 weeks of biweekly injections of 50 μg of S. minnesota re595 LPS after 10 min. (B) Time taken to enter the light side of a light-dark box for LPS-treated and control (PBS) me/+ and +/+ mice. (C) Time taken to enter a cross-arm of a T-maze on day 2 of testing in LPS-treated and control (PBS) me/+ and +/+ mice. (D) Horizontal exploration (squares crossed) in an open-field chamber for LPS-treated and control (PBS) me/+ and +/+ mice. Statistical significance measured by two-way ANOVA followed by the Student-Neuman-Keuls method for time-point analyses and one-way ANOVA followed by the Student-Neuman-Keuls method for other measurements. *P<0.05, #P = 0.1.
To exclude the possibility that the increase in avoidant behaviors in the LPS-treated me/+ mice was caused by reduced motor ability, a test of motor strength/skill and coordination was conducted. LPS affected the performance of me/+ and +/+ mice similarly, as over 3 days of testing both genotypes displayed reduced ability to hang from a wire hanger, as measured by both latency to fall (Fig. 2A) and postural performance score (Fig. 2B), compared to PBS-treated controls. Although LPS affected both measures significantly over time and each individual testing day showed a similar trend, individual testing-day significance was only apparent on day 1 for postural scores and day 3 for adjusted latency. This indicates that the postural scores might be more sensitive to the LPS-induced motor impairments, and differences in adjusted latency are only apparent after the untreated controls become adapted to the test. The findings of the coat hanger are in contrast to the wire screen-climbing test, a test of both motor ability and fear/anxiety, in which over the 3-day testing period the LPS-treated me/+ mice performed significantly less well than same-genotype controls and LPS-treated +/+ animals (Fig. 2C).
Fig. 2. LPS-induced motor/coordination deficits.
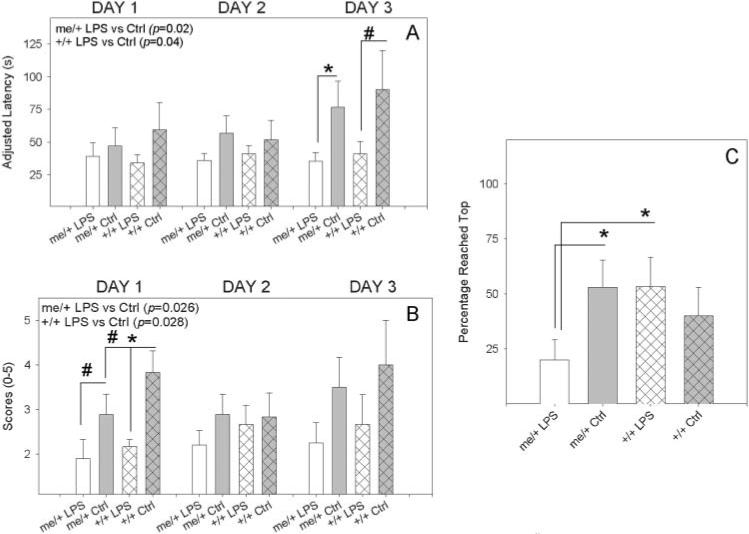
(A) Time taken to fall off a wire hanger for LPS-treated and control (PBS) me/+ and +/+ mice after 4 weeks of biweekly injections of 50 μg of S. minnesota re595 LPS, with a maximum hanging time of 60 sec. The ability to traverse to the end of a bar yielded an adjusted latency of 120 sec. (B) Postural scores of LPS-treated and control (PBS) me/+ and +/+ mice. (C) LPS-treated and control (PBS) me/+ and +/+ mice that reached the top of a wire screen in 40 sec. Statistical significance measured by two-way ANOVA followed by the Student-Neuman-Keuls method for multiple testing day analyses (shown in upper left corner of graph) and one-way ANOVA followed by the Student-Neuman-Keuls method for other measurements. For one-way ANOVA analyses, *P<0.05, #P = 0.1.
Although the average weight in LPS-treated mice was lower than that of untreated mice (me/+, 91.6 ± 0.07% of control; +/+, 97.3 ± 1.0% of control), LPS did not significantly reduce the body weight in either me/+ (P = 0.053) or +/+ (P = 0.7) mice. Nonetheless, the body weight results indicate that me/+ mice might be more responsive to LPS than wild types. In the subset of mice that were weighed, untreated me/+ mice weighed significantly more than untreated +/+ mice (me/+, 47.7 ± 1.3 g; +/+, 37.3 ± 6.4 g), but there were no significant differences between treated animals (not shown) and the increased body weight in me/+ mice did not negatively affect locomotion (Fig. 1).
Brain cytokine and immune effectors
Neither LPS-treated me/+ nor +/+ brains had significant increases relative to untreated mice in any of the three type 1 pro-inflammatory cytokines measured (TNF-α, IL-6 and IL-1β) (Fig. 3A–C). There was, however, a significant increase in serum CORT, which is induced by pro-inflammatory cytokine activity in the hypothalamus, in mice with an increase in avoidant behaviors (the LPS-treated me/+) compared to the other three groups (Fig. 3D). Furthermore, significant differences were found in the type 2 cytokines (Fig. 4A–C). LPS-treated me/+ mice had significantly higher brain levels of IL-13 and IL-10 than untreated me/+ mice and a substantial (insignificant) increase in IL-4. Furthermore, LPS-treated me/+ mice had significantly more IL-13, IL-4 and IL-10 than LPS-treated wild-type animals. LPS-treated me/+ brains also had significantly greater levels of the type 2 effector arginase activity than untreated me/+ control mice, whereas there were no significant increases in arginase in LPS-treated +/+ brains (Fig. 4D). Also, comparing untreated animals, untreated control me/+ mice had significantly more IL-4 than untreated +/+ mice (Fig. 4B). There were no significant differences between groups in the type 1 cytokine IFN-γ, although IFN-γ trended higher in LPS-treated me/+ brains (not shown).
Fig. 3. Brain pro-inflammatory cytokines and serum CORT.
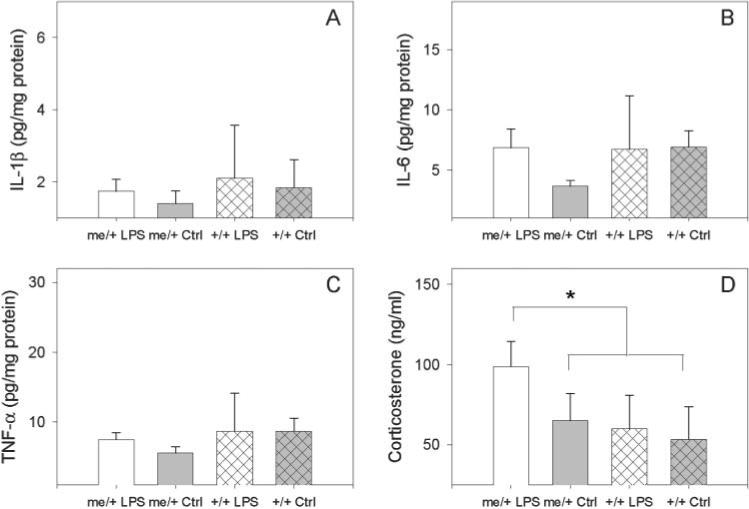
(A) IL-1β in the brains of LPS-treated and control (PBS) me/+ and +/+ mice after 4 weeks of biweekly injections of 50 μg of S. minnesota re595 LPS. (B) IL-6 levels in the brains of LPS-treated and control me/+ and +/+ mice. (C) TNF-α levels in the brains of LPS-treated and control me/+ and +/+ mice. (D) Serum corticosterone levels in behaviorally affected (LPS-treated me/+ mice) and unaffected (LPS-treated +/+ and control me/+ and +/+ mice). Cytokine levels are expressed as pg cytokine per mg total protein. Statistical significance was measured by one-way ANOVA followed by the Student-Neuman-Keuls method. *P<0.05.
Fig. 4. Brain type 2 cytokines and arginase activity.
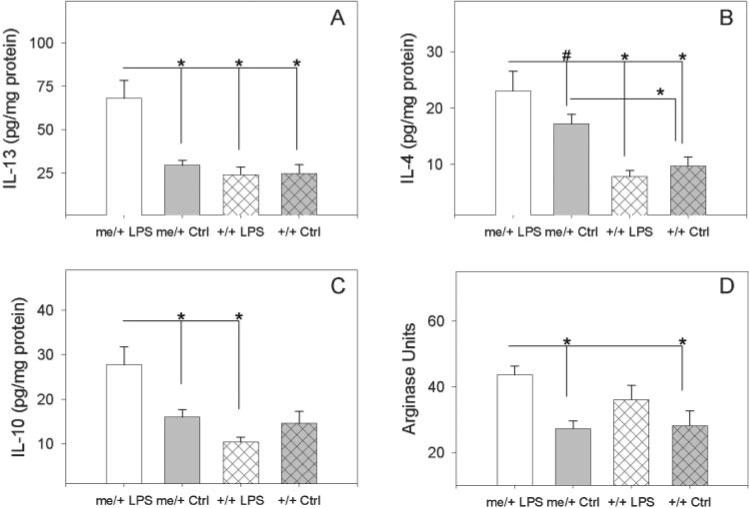
(A) IL-13 levels in the brains of LPS-treated me/+, untreated control (PBS) me/+, LPS-treated +/+ and untreated control +/+ mice after 4 weeks of biweekly injections of 50 μg of S. minnesota re595 LPS. (B) IL-4 levels in the brains of LPS-treated me/+, control me/+, LPS-treated +/+ and control +/+ mice. (C) IL-10 levels in the brains of LPS-treated me/+, control me/+, LPS-treated +/+ and control +/+ mice. (D) Arginase activity in the brains of LPS-treated me/+, control me/+, LPS-treated +/+ and control +/+ mice. Cytokine levels are expressed as pg cytokine per mg total protein and arginase activity is expressed as Arginase Units. 1 Arginase Unit = 1 μg urea produced per mg total protein. Statistical significance was measured via one-way ANOVA followed by the Student-Neuman-Keuls method. *P<0.05 and #P<0.1.
The increase in type 2 cytokines was mirrored by upregulation of type 2 cytokine receptor mRNA, as LPS-treated me/+ mice had significantly higher levels of IL-13Rα1 mRNA than control me/+ and +/+ mice and LPS-treated +/+ animals (Fig. 5A) and significantly greater IL-10R mRNA than control mice (Fig. 5B). mRNA encoding STAT6, the key signal transducer of both IL-4 and IL-13 signaling, was also upregulated in me/+ mice. STAT6 mRNA was ∼20-fold higher in untreated me/+ mice than in untreated +/+ animals (Fig. 5C), an increase that is similar to that in the bronchial epithelium of asthmatics (Mullings et al., 2001). Furthermore, the increase in STAT6 mRNA in me/+ brains was augmented significantly by chronic LPS treatment, but LPS did not significantly increase STAT6 mRNA in +/+ brains. There were no differences between groups in mRNA expression of the housekeeping gene GAPDH.
Fig. 5. Brain type 2 cytokine receptor and STAT6 mRNA expression.
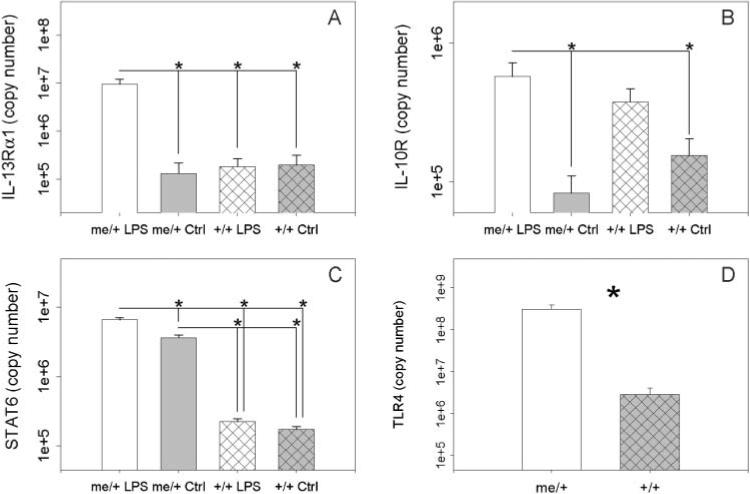
(A) Brain mRNA levels of IL-13Rα1 in of LPS-treated me/+, control (PBS) me/+,LPS-treated +/+ and control +/+ mice after 4 weeks of biweekly injections of 50 μg of S. minnesota re595 LPS. (B) Brain mRNA expression of IL-10R in LPS-treated me/+, control me/+, LPS-treated +/+ and control +/+ mice. (C) Brain mRNA expression of STAT6 in LPS-treated me/+, control me/+, LPS-treated +/+ and control +/+ mice. (D) Constitutive brain TLR4 mRNA expression in me/+ and +/+ mice. Statistical significance was measured via one-way ANOVA followed by the Student-Neuman-Keuls method. *P<0.05. There were no differences between groups in expression of the loading control GAPDH.
We also measured mRNA levels of TLR4 in the brain to determine if the basal expression of TLR4 differs in the CNS of me/+ mice and +/+ mice. The brains of untreated adult me/+ mice contained significantly more TLR4 mRNA than the brains of +/+ animals (Fig. 5D) indicating that me/+ mice might be constitutively more responsive to CNS TLR4 stimulation. Together, these data indicate both constitutive and LPS-induced differences in TLR4 and type 2 cytokine signaling pathways in me/+ and +/+ mice.
Adult glial culture
To determine if me/+ glia have the potential to contribute to the increased responsiveness seen in vivo, me/+ and +/+ glial cultures from adult CNS tissue were treated with LPS. As in vivo, there was significantly more arginase activity in LPS-treated me/+ glial cultures than +/+ cultures 48 hours after treatment (Fig. 6). To measure TLR4 signaling intensity, pro-inflammatory cytokine production was measured in response to LPS. LPS-treated me/+ glial cultures had significantly greater pro-inflammatory cytokine production than +/+ glial cultures (Fig. 7). Three-way ANOVA analysis indicated significantly greater secretion of IL-1β (P<0.001), IL-6 (P = 0.038) and TNF-α (P = 0.002). At individual time points, IL-1β was significantly greater in me/+ glial cultures at both 24 and 48 hours (Fig. 7A1,A2) whereas TNF-α was significantly higher in me/+ glia at 24 hours (Fig. 7C1). Also, both IL-1β and IL-6 significantly increased with time (both P<0.02) but TNF-α did not increase significantly after 24 hours. Nitric oxide production (measured at 1 and 20 μg ml−1) followed a similar trend, as measured by significantly greater nitrite produced by me/+ glial cultures than +/+ (P = 0.026) (not shown).
Fig. 6. Glial culture arginase activity.

The intracellular arginase activity of SHP-1 heterozygote (me/+) (open circles) and wild-type (+/+) (black squares) adult glial cultures 48 hours after treatment with 0, 1, 4 and 20 μg ml− LPS. Arginase activity is expressed as Arginase Units. 1 Arginase Unit = 1 μg urea produced per mg total protein. A pgenotype in the upper left corner indicates a significant difference between me/+ and +/+ via two-way ANOVA followed by the Student-Neuman-Keuls method. *P<0.05 and P<0.1 for individual doses.
Fig. 7. Glial culture pro-inflamamtory cytokine response to LPS.

(A) IL-1β levels in SHP-1 heterozygote (me/+) (open circles) and wild-type (+/+) (black squares) adult glial cultures treated with 0, 1, 4 and 20 μg ml LPS for 24 (A1) and 48 (A2) hours. (B) IL-6 levels in me/+ and +/+ glial culture supernatants at 24 (B1) and 48 (B2) hours. (C) TNF-α levels in me/+ and +/+ glial culture supernatants at 24 (C1) and 48 (C2) hours. All cytokine levels are shown as pg cytokine per mg of cell-lysate protein. A pgenotype in the upper left corner indicates a significant difference between me/+ and +/+ for that time point via two-way ANOVA followed by the Student-Neuman-Keuls method. *P<0.05 and P<0.1 for individual doses.
Tail-flick test
TLR4 and other components of innate immunity and neuronal development regulated by SHP-1 have been implicated in pain signaling via TRPV1 thermal receptors (Puntambekar et al., 2005; Marsh et al., 2003), so untreated me/+ and +/+ mice were tested by the tail-flick test in a 49°C water bath. Me/+ mice had a significantly reduced latency to tail-flick than +/+ animals on two consecutive days of testing, whereas there were no differences at 37°C (Fig. 8).
Fig. 8. Tail-flick test.
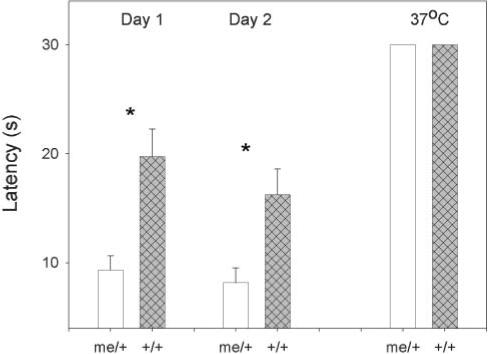
Time taken to tail-flick for untreated me/+ and +/+ mice. Mice were tested twice (24 hours apart) in a 49°C water bath and once in a 37°C water bath. Significance was measured via by Student's t-test.
CONCLUSIONS
• LPS-treated me/+ mice have avoidant-behavioral abnormalities not found in either untreated me/+ control or LPS-treated +/+ mice.
• These behavioral abnormalities are associated with increases in constitutive expression of TLR4 and both constitutive and LPS-induced type 2 cytokines and immune effectors.
• Me/+ glia are hyper-responsive to LPS as LPS induces significantly greater arginase activity and pro-inflammatory cytokine production in me/+ glia than in +/+ glia.
• Me/+ mice display thermal hyperalgesia in the tail-flick test compared to +/+ mice.
DISCUSSION
This report indicates that the reduced expression in SHP-1 in me/+ relates to a functional insufficiency in the control of TLR4 signaling and avoidant behaviors. The insufficiency in control of TLR4 signaling yields a hyper-responsivness to LPS that might relate to both an increase in TLR4 expression in the brains of me/+ mice and an increased receptor signal intensity (Massa and Wu, 1998). Interestingly, as the lack of SHP-1 has been linked to spontaneous inflammation and TLR4 stimulation increases TLR4 expression, the latter observation might reflect auto-regulation of TLR4 by endogenous ligands in me/+ mice (Bowman et al., 2003). Of potential interest, increased responsiveness to either an endogenous ligand of TLR4 or environmental, subclinical levels of endotoxin might be involved in the increased pain sensation in me/+ mice (see below). in vitro, hyperresponsiveness to LPS increases the pro-inflammatory response that is usually linked to LPS stimulation, as me/+ glia produce more IL-1β, IL-6 and TNF-α than +/+ glia after acute LPS treatment. However after prolonged LPS treatment in vivo, the brains of me/+ mice showed a distinctive skewing towards type 2 immunity that is not seen in LPS-treated +/+ mice. This skewing is evident with cytokines and immune effectors, cytokine receptors and signal transduction molecules. The increases in STAT6 expression and STAT6-inducible arginase activity in me/+ brains versus +/+ are intriguing because they mimic what occurs in the bronchial epithelium of asthmatics and in the airways in a murine model of asthma, respectively (Mullings et al., 2001; Yang et al., 2006). These increases indicate that me/+ mice might be predisposed to a type 2 inflammation in the CNS that is similar to the inflammation found in the lung of asthmatics.
The bias towards type 2 immunity in the CNS of LPS-treated me/+ mice is associated with a similar behavioral profile to that observed after acute LPS treatment (sickness behavior). This seems counter-intuitive because the behavioral effects of acute LPS treatment are mediated via type 1 pro-inflammatory cytokines (Bluthe et al., 2000a; Bluthe et al., 2000b; Connor et al., 2002). Many reports indicate that type 2 cytokines antagonize type 1 pro-inflammatory activity (Fenton et al., 1992; Miossec et al., 1992; Dokter et al., 1996; Nelson et al., 2003; Jenkins et al., 2004; Paintlia et al., 2006), therefore, it would seem a priori that, LPS-treated me/+ mice should have decreased avoidant behaviors. The latter notion is in accordance with reports indicating that administration of type 2 cytokines before LPS treatment reduces sickness behaviors (Bluthe et al., 2002). Alternatively, CNS administration of either IL-4 or IL-13 concurrent with systemic LPS treatment increases sickness behaviors, which indicates that, in the CNS, type 2 cytokines might either potentiate the effects of pro-inflammatory cytokines or act independently in a similar manner (Bluthe et al., 2001; Bluthe et al., 2002). These reports also indicate that CNS administration of either IL-4 or IL-13 alone is not sufficient for the induction of sickness behavior, indicating that a co-existing (or preexisting) pro-inflammatory response is necessary for ‘pro-sickness behavior’ effects of IL-4 and IL-13. The need for a co-existing pro-inflammatory response further indicates that the behavioral effects of IL-4 and IL-13 are mediated through potentiation of pro-inflammatory signals (Bluthe et al., 2001; Bluthe et al., 2002). Hence, although the levels of pro-inflammatory cytokines are not elevated in LPS-treated me/+ mice compared to +/+ and untreated me/+ mice, the presence of high IL-4 and IL-13 concentrations in LPS-treated me/+ mice might potentiate the effects of pro-inflammatory cytokines, resulting in a greater ‘pro-inflammatory signal’ and more avoidant-like behaviors. This notion is supported by the serum CORT levels. Activation of the HPA axis and the subsequent release of CORT are induced by pro-inflammatory cytokines acting in the hypothalamus (Blalock and Smith, 2006). The fact that there is increased CORT in the LPS-treated me/+ mice in the absence of increased pro-inflammatory cytokines in the brains of these mice indicates potentiation of the pro-inflammatory response in LPS-treated me/+ mice.
The type 2 immune effector arginase I is also reported to alter behavior (Liu et al., 2004). Both IL-4 and IL-13 induce the expression of arginase I through a STAT6-dependent mechanism in glia (Wei et al., 2000). One manner in which arginase might alter behavior is through the synthesis of the polyamines. Spermine, the end-product of the arginase-polyamine synthesis pathway, has many effects in the CNS. For example, spermine inhibits the N-methyl-D-aspartate (NMDA) receptor (Isa et al., 1996); NMDA-receptor stimulation decreases anxiety-like behaviors whereas inhibition increases such behavior (Kuzmin et al., 2005). Thus, the increased arginase activity in the CNS of LPS-treated me/+ mice might induce greater inhibition of NMDA receptors. Furthermore, metabotropic glutamate receptors, a set of receptors that is stimulated by spermine (Manev et al., 1993), are crucial in the mechanism of LPS-induced sickness behavior (Weiland et al., 2005). Therefore, increased arginase activity might potentiate the pro-inflammatory cytokine signaling involved in the manifestation of these avoidant behaviors.
As mentioned previously, it is possible that the increase in pain sensation in me/+ mice is related to the higher constitutive expression of TLR4 in these animals. For example, TLR4 is present on nociceptive neurons and it is believed that TLR4 might be involved in the sensation of pain during infections (Wadachi and Hargreaves, 2006). Furthermore, mast cells release the endogenous TLR4 ligand HSP70 (Mortaz et al., 2006) and mast cell degranulation, a process that is regulated by SHP-1, is implicated in thermal and inflammatory pain sensation (Zhang et al., 2003b; Drummond, 2004). Thus, greater expression of TLR4 in me/+ mice might lead to greater TLR4 triggering upon equivalent release of HSP70 by mast cells and, thus, increased pain sensation. It is also possible that the heightened expression of TLR4 is linked indirectly to increased pain sensation. Increased production of hydrogen peroxide by NADPH oxidase increases expression of the heat-sensing transient receptor potential (TRP) channel, TRPV1, and the nociceptive properities of nociceptive sensory neurons (Puntambekar et al., 2005). TLR4 stimulation induces the activation of subunits of NADPH oxidase, which enables production of more hydrogen peroxide (Lin et al., 2006). Hence, the higher constitutive levels of TLR4 in me/+ mice might allow greater hydrogen peroxide production, increased TRPV1 expression and activity, and sensitization to pain upon stimulation. In addition, TLR4 stimulation in me/+ mice also induces more arginase activity than in +/+ (Fig. 4D), which leads to increased polyamine synthesis, and polyamines, including spermine, also increase TRPV1 activity (Ahern et al., 2006).
The idea that SHP-1 controls TRPV1 expression goes beyond regulation of TLR4. It has been shown that nerve growth factor (NGF) induces the expression of TRPV1 in dorsal root ganglion neurons through the TrkA receptor (Puntambekar et al., 2005). SHP-1 is a TrkA phosphatase and a negative regulator of NGF signaling (Marsh et al., 2003). Thus, a reduction in SHP-1 might cause increased NGF signaling and increased TRPV1 expression in me/+ mice. Interestingly, the production of hydrogen peroxide is crucial for NGF-induced TRPV1 expression (Puntambekar et al., 2005). Thus, both TrkA and TLR4 might contribute to greater expression of TRPV1 via the same mechanism. It is also possible that TrkA controls the levels of TLR4 because the same NADPH oxidase / hydrogen peroxide mechanism that is involved in TRPV1 expression also controls TLR4 expression (Lin et al., 2006). NGF receptors have been found in several cell types in addition to neurons (including astrocytes, microglia and leukocytes) (Oderfeld-Nowak et al., 2003; Zhang et al., 2003a; Ahamed et al., 2004), so increased TrkA signaling might be responsible for the increase in both constitutive TLR4 expression that leads to greater LPS reactivity in the CNS and the increased pain sensation in me/+ mice.
Like the LPS model presented here, the pathogenesis of spinal nerve-injury induced neuropathic pain is known to involve chronic stimulation of TLR4 (although in neuropathic pain, the TLR4 ligand is endogenous). Thus, our results indicate that the immune mechanisms involved in neuropathic pain might be more complex than thought previously. It is well-accepted that the type 1 pro-inflammatory cytokines have crucial roles in the mechanism of neuropathic pain (Moalem and Tracey, 2006) and, as such, there have been attempts to curtail neuropathic pain via the forced induction and pharmacological delivery of type 2 mediators (Hao et al., 2006; Verri et al., 2006). However, our data indicates that this might be counter-productive in the CNS because during periods of chronic hyper-stimulation of TLR4, type 2 cytokines and immune effectors such as arginase I appear to be predominant and might contribute to the deleterious processes in the CNS. Furthermore, in the case of sickness behavior, only administration of IL-4 before LPS reduced the LPS-induced effects (Bluthe et al., 2002). Hence, the induction of a type 2 state, especially later in the pathogenesis (after the inflammatory state has been established) might be deleterious and exacerbate disease.
To date, most research has focused on how the type 1 pro-inflammatory cytokines are involved in the underlying mechanisms of fear/anxiety and pain. This is particularly so for TLR4-mediated processes such as LPS-induced behaviors and spinal nerve-injury induced neuropathic pain in which the acute response is type 1 pro-inflammatory in nature. However, the effects of chronic stimulation of TLR4 on these neurological processes are less well defined. We show here that in the setting of chronic TLR4 stimulation, increased responsiveness (me/+ mice) exacerbates the type 2 immune response in the CNS and that the predominant type 2 response is associated with similar behaviors to the ones associated with the type 1 pro-inflammatory response after acute stimulation of TLR4. This leads to the supposition that, although the initiation of chronic TLR4-stimulated neurological processes are caused by type 1 pro-inflammatory mediators, the long-term maintenance of these effects might be due to and/or enhanced by a later, TLR4-dependent type 2 response in the CNS that includes increased STAT6, arginase and polyamines. Thus, overlapping inflammatory activities of type 1 pro-inflammatory and type 2 cytokines in the CNS might be crucial in the pathogenesis of such diseases. Understanding this component of chronic, TLR4 stimulated, inflammatory processes in the CNS might be key to providing insight into mechanisms of excessive avoidant behavior and pain-related responses, and possible therapeutics.
REFERENCES
- Ahamed J, Venkatesha RT, Thangam EB, Ali H. C3a enhances nerve growth factor-induced NFAT activation and chemokine production in a human mast cell line, HMC-1. Journal of Immunology. 2004;172:6961–6968. doi: 10.4049/jimmunol.172.11.6961. [DOI] [PubMed] [Google Scholar]
- Ahern GP, Wang X, Miyares RL. Polyamines are potent ligands for the capsaicin receptor TRPV1. Journal of Biological Chemistry. 2006;281:8991–8995. doi: 10.1074/jbc.M513429200. [DOI] [PubMed] [Google Scholar]
- Blalock JE, Smith EM. Conceptual development of the immune system as a sixth sense. Brain Behavior and Immunity. 2007;21:23–33. doi: 10.1016/j.bbi.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Bluthe RM, Bristow A, Lestage J, Imbs C, Dantzer R. Central injection of interleukin-13 potentiates LPS-induced sickness behavior in rats. Neuroreport. 2001;12:3979–3983. doi: 10.1097/00001756-200112210-00025. [DOI] [PubMed] [Google Scholar]
- Bluthe RM, Laye S, Michaud B, Combe C, Dantzer R, Parnet P. Role of interleukin-1beta and tumour necrosis factor-alpha in lipopolysaccharide-induced sickness behaviour: a study with interleukin-1 type I receptor-deficient mice. European Journal of Neuroscience. 2000a;12:4447–4456. [PubMed] [Google Scholar]
- Bluthe RM, Lestage J, Rees G, Bristow A, Dantzer R. Dual effect of central injection of recombinant rat interleukin-4 on lipopolysaccharide-induced sickness behavior in rats. Neuropsychopharmacology. 2002;26:86–93. doi: 10.1016/S0893-133X(01)00305-0. [DOI] [PubMed] [Google Scholar]
- Bluthe RM, Michaud B, Poli V, Dantzer R. Role of IL-6 in cytokine-induced sickness behavior: a study with IL-6 deficient mice. Physiology and Behavior. 2000b;70:367–373. doi: 10.1016/s0031-9384(00)00269-9. [DOI] [PubMed] [Google Scholar]
- Bonaparte KL, Hudson CA, Wu C, Massa PT. Inverse regulation of inducible nitric oxide synthase (iNOS) and arginase I by the protein tyrosine phosphatase SHP-1 in CNS glia. Glia. 2006;53:827–835. doi: 10.1002/glia.20344. [DOI] [PubMed] [Google Scholar]
- Bowman CC, Rasley A, Tranguch SL, Marriott I. Cultured astrocytes express toll-like receptors for bacterial products. Glia. 2003;43:281–291. doi: 10.1002/glia.10256. [DOI] [PubMed] [Google Scholar]
- Cavallo T, Granholm NA. Bacterial lipopolysaccharide transforms mesangial into proliferative lupus nephritis without interfering with processing of pathogenic immune complexes in NZB/W mice. American Journal of Pathology. 1990;137:971–978. [PMC free article] [PubMed] [Google Scholar]
- Cavallo T, Granholm NA. Bacterial lipopolysaccharide induces long-lasting IgA deficiency concurrently with features of polyclonal B cell activation in normal and in lupus-prone mice. Clinical and Experimental Immunology. 1991;84:134–138. [PMC free article] [PubMed] [Google Scholar]
- Chakravarty S, Herkenham M. Toll-like receptor 4 on nonhematopoietic cells sustains CNS inflammation during endotoxemia, independent of systemic cytokines. Journal of Neuroscience. 2005;25:1788–1796. doi: 10.1523/JNEUROSCI.4268-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor TJ, O'Sullivan J, Nolan Y, Kelly JP. Inhibition of constitutive nitric oxide production increases the severity of lipopolysaccharide-induced sickness behaviour: a role for TNF-alpha. Neuroimmunomodulation. 2002;10:367–378. doi: 10.1159/000071478. [DOI] [PubMed] [Google Scholar]
- Dantzer R, Bluthe RM, Gheusi G, Cremona S, Laye S, Parnet P, et al. Molecular basis of sickness behavior. Annals of the New York Academy of Sciences. 1998;856:132–138. doi: 10.1111/j.1749-6632.1998.tb08321.x. [DOI] [PubMed] [Google Scholar]
- Deng C, Minguela A, Hussain RZ, Lovett-Racke AE, Radu C, Ward ES, et al. Expression of the tyrosine phosphatase SRC homology 2 domain-containing protein tyrosine phosphatase 1 determines T cell activation threshold and severity of experimental autoimmune encephalomyelitis. Journal of Immunology. 2002;168:4511–4518. doi: 10.4049/jimmunol.168.9.4511. [DOI] [PubMed] [Google Scholar]
- Deng C, Wu B, Yang H, Hussain RZ, Lovett-Racke AE, Christadoss P, et al. Decreased expression of Src homology 2 domain-containing protein tyrosine phosphatase 1 reduces T cell activation threshold but not the severity of experimental autoimmune myasthenia gravis. Journal of Neuroimmunology. 2003;138:76–82. doi: 10.1016/s0165-5728(03)00119-x. [DOI] [PubMed] [Google Scholar]
- Dokter WH, Koopmans SB, Vellenga E. Effects of IL-10 and IL-4 on LPS-induced transcription factors (AP-1, NF-IL6 and NF-kappa B) which are involved in IL-6 regulation. Leukemia. 1996;10:1308–1316. [PubMed] [Google Scholar]
- Drummond PD. The effect of cutaneous mast cell degranulation on sensitivity to heat. Inflammation Research. 2004;53:309–315. doi: 10.1007/s00011-004-1263-3. [DOI] [PubMed] [Google Scholar]
- Fenton MJ, Buras JA, Donnelly RP. IL-4 reciprocally regulates IL-1 and IL-1 receptor antagonist expression in human monocytes. Journal of Immunology. 1992;149:1283–1288. [PubMed] [Google Scholar]
- Galanos C, Luderitz O, Westphal O. A new method for the extraction of R lipopolysaccharides. European Journal of Biochemistry. 1969;9:245–249. doi: 10.1111/j.1432-1033.1969.tb00601.x. [DOI] [PubMed] [Google Scholar]
- Hao S, Mata M, Glorioso JC, Fink DJ. HSV-mediated expression of IL-4 in dorsal root ganglion neurons reduces neuropathic pain. Molecular Pain. 2006;2:6. doi: 10.1186/1744-8069-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque SJ, Harbor P, Tabrizi M, Yi T, Williams BR. Protein-tyrosine phosphatase Shp-1 is a negative regulator of IL-4 and IL-13-dependent signal transduction. Journal of Biological Chemistry. 1998;273:33893–33896. doi: 10.1074/jbc.273.51.33893. [DOI] [PubMed] [Google Scholar]
- Hudson CA, Cao L, Kasten-Jolly J, Kirkwood JN, Lawrence DA. Susceptibility of lupus-prone NZM mouse strains to lead exacerbation of systemic lupus erythematosus symptoms. Journal of Toxicology and Environmental Health A. 2003;66:895–918. doi: 10.1080/15287390306456. [DOI] [PubMed] [Google Scholar]
- Inceoglu B, Jinks SL, Schmelzer KR, Waite T, Kim IH, Hammock BD. Inhibition of soluble epoxide hydrolase reduces LPS-induced thermal hyperalgesia and mechanical allodynia in a rat model of inflammatory pain. Life Sciences. 2006;79:2311–2319. doi: 10.1016/j.lfs.2006.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isa T, Iino M, Ozawa S. Spermine blocks synaptic transmission mediated by Ca(2+)-permeable AMPA receptors. Neuroreport. 1996;7:689–692. doi: 10.1097/00001756-199602290-00002. [DOI] [PubMed] [Google Scholar]
- Jack CS, Arbour N, Manusow J, Montgrain V, Blain M, McCrea E, et al. TLR signaling tailors innate immune responses in human microglia and astrocytes. Journal of Immunology. 2005;175:4320–4330. doi: 10.4049/jimmunol.175.7.4320. [DOI] [PubMed] [Google Scholar]
- Jenkins K, Javadi M, Borghaei RC. Interleukin-4 suppresses IL-1-induced expression of matrix metalloproteinase-3 in human gingival fibroblasts. Journal of Periodontology. 2004;75:283–291. doi: 10.1902/jop.2004.75.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata T, Yamashita M, Kimura M, Murata K, Inami M, Shimizu C, et al. src homology 2 domain-containing tyrosine phosphatase SHP-1 controls the development of allergic airway inflammation. Journal of Clinical Investigation. 2003;111:109–119. doi: 10.1172/JCI15719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaled AR, Butfiloski EJ, Sobel ES, Schiffenbauer J. Functional consequences of the SHP-1 defect in motheaten viable mice: role of NF-kappa B. Cellular Immunology. 1998;185:49–58. doi: 10.1006/cimm.1998.1272. [DOI] [PubMed] [Google Scholar]
- Kuzmin A, Madjid N, Terenius L, Ogren SO, Bakalkin G. Big dynorphin, a prodynorphin-derived peptide produces NMDA receptor-mediated effects on memory, anxiolytic-like and locomotor behavior in mice. Neuropsychopharmacology. 2006;31:1928–1937. doi: 10.1038/sj.npp.1300959. [DOI] [PubMed] [Google Scholar]
- Lalonde R, Qian S, Strazielle C. Transgenic mice expressing the PS1-A246E mutation: effects on spatial learning, exploration, anxiety, and motor coordination. Behavioral Brain Research. 2003;138:71–79. doi: 10.1016/s0166-4328(02)00230-9. [DOI] [PubMed] [Google Scholar]
- Lin FY, Chen YH, Tasi JS, Chen JW, Yang TL, Wang HJ, et al. Endotoxin induces toll-like receptor 4 expression in vascular smooth muscle cells via NADPH oxidase activation and mitogen-activated protein kinase signaling pathways. Arteriosclerosis, Thrombosis and Vascular Biology. 2006;26:2630–2637. doi: 10.1161/01.ATV.0000247259.01257.b3. [DOI] [PubMed] [Google Scholar]
- Liu P, Smith PF, Appleton I, Darlington CL, Bilkey DK. Potential involvement of NOS and arginase in age-related behavioural impairments. Experimental Gerontology. 2004;39:1207–1222. doi: 10.1016/j.exger.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Manev RM, Gabellini N, Manev H. Polyamines modulate the function of transfected glutamate receptor mGluR1a. Neuroreport. 1993;4:830–832. doi: 10.1097/00001756-199306000-00059. [DOI] [PubMed] [Google Scholar]
- Marsh HN, Dubreuil CI, Quevedo C, Lee A, Majdan M, Walsh GS, et al. SHP-1 negatively regulates neuronal survival by functioning as a TrkA phosphatase. Journal of Cell Biology. 2003;163:999–1010. doi: 10.1083/jcb.200309036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massa PT, Saha S, Wu C, Jarosinski KW. Expression and function of the protein tyrosine phosphatase SHP-1 in oligodendrocytes. Glia. 2000;29:376–385. [PubMed] [Google Scholar]
- Massa PT, Wu C. The role of protein tyrosine phosphatase SHP-1 in the regulation of IFN-gamma signaling in neural cells. Journal of Immunology. 1996;157:5139–5144. [PubMed] [Google Scholar]
- Massa PT, Wu C. Increased inducible activation of NF-kappaB and responsive genes in astrocytes deficient in the protein tyrosine phosphatase SHP-1. Journal of Interferon and Cytokine Research. 1998;18:499–507. doi: 10.1089/jir.1998.18.499. [DOI] [PubMed] [Google Scholar]
- Miossec P, Briolay J, Dechanet J, Wijdenes J, Martinez-Valdez H, Banchereau J. Inhibition of the production of proinflammatory cytokines and immunoglobulins by interleukin-4 in an ex vivo model of rheumatoid synovitis. Arthritis Rheumatism. 1992;35:874–883. doi: 10.1002/art.1780350805. [DOI] [PubMed] [Google Scholar]
- Moalem G, Tracey DJ. Immune and inflammatory mechanisms in neuropathic pain. Brain Research. Brain Research Reviews. 2006;51:240–264. doi: 10.1016/j.brainresrev.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Mortaz E, Redegeld FA, Nijkamp FP, Wong HR, Engels F. Acetylsalicylic acid-induced release of HSP70 from mast cells results in cell activation through TLR pathway. Experimental Hematology. 2006;34:8–18. doi: 10.1016/j.exphem.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Mullings RE, Wilson SJ, Puddicombe SM, Lordan JL, Bucchieri F, Djukanovic R, et al. Signal transducer and activator of transcription 6 (STAT-6) expression and function in asthmatic bronchial epithelium. Journal of Allergy and Clinical Immunology. 2001;108:832–838. doi: 10.1067/mai.2001.119554. [DOI] [PubMed] [Google Scholar]
- Nelson G, Wilde GJ, Spiller DG, Kennedy SM, Ray DW, Sullivan E, et al. NF-kappaB signalling is inhibited by glucocorticoid receptor and STAT6 via distinct mechanisms. Journal of Cell Science. 2003;116:2495–2503. doi: 10.1242/jcs.00461. [DOI] [PubMed] [Google Scholar]
- Oderfeld-Nowak B, Zaremba M, Lipkowski AW, Kwiatkowska-Patzer B, Triaca V, Aloe L. High-affinity NGF receptor in the rat spinal cord during acute and chronic phases of experimental autoimmune encephalomyelitis: a possible functional significance. Archives of Italian Biology. 2003;141:103–116. [PubMed] [Google Scholar]
- Paintlia AS, Paintlia MK, Singh I, Singh AK. IL-4-induced peroxisome proliferator-activated receptor gamma activation inhibits NF-kappaB trans activation in central nervous system (CNS) glial cells and protects oligodendrocyte progenitors under neuroinflammatory disease conditions: implication for CNS-demyelinating diseases. Journal of Immunology. 2006;176:4385–4398. doi: 10.4049/jimmunol.176.7.4385. [DOI] [PubMed] [Google Scholar]
- Paling NR, Welham MJ. Role of the protein tyrosine phosphatase SHP-1 (Src homology phosphatase-1) in the regulation of interleukin-3-induced survival, proliferation and signalling. Biochemical Journal. 2002;368:885–894. doi: 10.1042/BJ20021054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puntambekar P, Mukherjea D, Jajoo S, Ramkumar V. Essential role of Rac1/NADPH oxidase in nerve growth factor induction of TRPV1 expression. Journal of Neurochemistry. 2005;95:1689–1703. doi: 10.1111/j.1471-4159.2005.03518.x. [DOI] [PubMed] [Google Scholar]
- Rossi-George A, LeBlanc F, Kaneta T, Urbach D, Kusnecov AW. Effects of bacterial superantigens on behavior of mice in the elevated plus maze and light-dark box. Brain Behavior and Immunity. 2004;18:46–54. doi: 10.1016/s0889-1591(03)00087-4. [DOI] [PubMed] [Google Scholar]
- Tanga FY, Nutile-McMenemy N, DeLeo JA. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proceedings of the National Academy of Sciences of the U.S.A. 2005;102:5856–5861. doi: 10.1073/pnas.0501634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verri WA, Jr, Cunha TM, Parada CA, Poole S, Cunha FQ, Ferreira SH. Hypernociceptive role of cytokines and chemokines: targets for analgesic drug development? Pharmacological Therapeutics. 2006;112:116–138. doi: 10.1016/j.pharmthera.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Voikar V, Rauvala H, Ikonen E. Cognitive deficit and development of motor impairment in a mouse model of Niemann-Pick type C disease. Behavioral Brain Research. 2002;132:1–10. doi: 10.1016/s0166-4328(01)00380-1. [DOI] [PubMed] [Google Scholar]
- Wadachi R, Hargreaves KM. Trigeminal nociceptors express TLR-4 and CD14: a mechanism for pain due to infection. Journal of Dentistry Research. 2006;85:49–53. doi: 10.1177/154405910608500108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei LH, Jacobs AT, Morris SM, Jr, Ignarro LJ. IL-4 and IL-13 upregulate arginase I expression by cAMP and JAK/STAT6 pathways in vascular smooth muscle cells. American Journal of Physiology. Cell Physiology. 2000;279:C248–C256. doi: 10.1152/ajpcell.2000.279.1.C248. [DOI] [PubMed] [Google Scholar]
- Weiland TJ, Anthony-Harvey-Beavis D, Voudouris NJ, Kent S. Metabotropic glutamate receptors mediate lipopolysaccharide-induced fever and sickness behavior. Brain Behavior and Immunity. 2006;20:233–245. doi: 10.1016/j.bbi.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Werner-Felmayer G, Baier-Bitterlich G, Fuchs D, Hausen A, Murr C, Reibnegger G, et al. Detection of bacterial pyrogens on the basis of their effects on gamma interferon-mediated formation of neopterin or nitrite in cultured monocyte cell lines. Clinical and Diagnostic Laboratory Immunology. 1995;2:307–313. doi: 10.1128/cdli.2.3.307-313.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Rangasamy D, Matthaei KI, Frew AJ, Zimmmermann N, Mahalingam S, et al. Inhibition of arginase I activity by RNA interference attenuates IL-13-induced airways hyperresponsiveness. Journal of Immunology. 2006;177:5595–5603. doi: 10.4049/jimmunol.177.8.5595. [DOI] [PubMed] [Google Scholar]
- Zhang J, Geula C, Lu C, Koziel H, Hatcher LM, Roisen FJ. Neurotrophins regulate proliferation and survival of two microglial cell lines in vitro. Experimental Neurology. 2003a;183:469–481. doi: 10.1016/s0014-4886(03)00222-x. [DOI] [PubMed] [Google Scholar]
- Zhang YH, Chen Y, Zhao ZQ. Resiniferatoxin reversibly blocks adjuvant-induced thermal hyperalgesia in the rat. European Journal of Pharmacology. 2003b;481:301–304. doi: 10.1016/j.ejphar.2003.09.053. [DOI] [PubMed] [Google Scholar]
- Zhao J, Brooks DM, Lurie DI. Lipopolysaccharide-activated SHP-1-deficient motheaten microglia release increased nitric oxide, TNF-alpha, and IL-1beta. Glia. 2006;53:304–312. doi: 10.1002/glia.20283. [DOI] [PubMed] [Google Scholar]