

Abstract
Heart failure is the major cause of hospitalization, morbidity and mortality worldwide. Previous experimental and clinical studies have suggested that there is an increased production of reactive oxygen species (ROS: superoxide, hydrogen peroxide, hydroxyl radical) both in animals and in patients with acute and chronic heart failure. The possible source of increased ROS in the failing myocardium include xanthine and NAD(P)H oxidoreductases, cyclooxygenase, the mitochondrial electron transport chain and activated neutrophils among many others. The excessively produced nitric oxide (NO) derived from NO synthases (NOS) has also been implicated in the pathogenesis of chronic heart failure (CHF). The combination of NO and superoxide yields peroxynitrite, a reactive oxidant, which has been shown to impair cardiac function via multiple mechanisms. Increased oxidative and nitrosative stress also activates the nuclear enzyme poly(ADP-ribose) polymerase (PARP), which importantly contributes to the pathogenesis of cardiac and endothelial dysfunction associated with myocardial infarction, chronic heart failure, diabetes, atherosclerosis, hypertension, aging and various forms of shock. Recent studies have demonstrated that pharmacological inhibition of xanthine oxidase derived superoxide formation, neutralization of peroxynitrite or inhibition of PARP provide significant benefit in various forms of cardiovascular injury. This review discusses the role of oxidative/nitrosative stress and downstream pathways in various forms of cardiomyopathy and heart failure.
INTRODUCTION
Various pathophysiological conditions including acute and chronic ischemic heart disease resulting from altered coronary artery circulation or infarction, cardiomyopathies, myocarditis, pressure overload and defects in genes encoding contractile apparatus, intercellular matrix, cytoskeleton or mitochondrial proteins eventually lead to impaired myocardial function [reviewed in: 1–8]. The deleterious way of the progression of cardiovascular dysfunction to heart failure is complex and involves activation of numerous secondary pathways (neurohormones, neuropeptides, cytokines, inducible nitric oxide synthase (iNOS), oxidative/nitrosative stress, matrix metalloproteinases (MMPs), and the nuclear enzyme poly(ADP-ribose) polymerase (PARP)), leading to abnormalities in various signaling processes and cardiac receptors, calcium homeostasis, contractile protein desensitization, besides structural alterations such as cardiac and vascular remodeling with hypertrophy, fibrosis, cardiac dilation and necrosis. There is ample evidence that development of heart failure is also associated with generalized vascular endothelial dysfunction [9–14]. The adverse cardiac remodeling and increased peripheral resistance further aggravate heart failure [1–14].
There is accumulating evidence for an important role of xanthine oxidase and/or NAD(P)H oxidase derived super-oxide generation, peroxynitrite formation and PARP activation in the development and progression of endothelial and cardiac dysfunction associated with various forms of myocardial injury and heart failure [reviewed in 7,8]. This review focuses on the role of oxidative-nitrosative stress-PARP pathway in various forms of cardiomyopathies and heart failure, and on novel promising therapeutic strategies offered by inhibition of superoxide generation and PARP or neutralization of peroxynitrite.
INCREASED REACTIVE OXYGEN SPECIES (ROS) FORMATION IN HEART FAILURE
Experimental and clinical studies have suggested an increased production of reactive oxygen species (ROS: super-oxide, hydrogen peroxide, hydroxyl radical) both in animals and patients with acute and chronic heart failure [1–15]. Plasma lipid peroxidation is increased in patients with dilated cardiomyopathy, and it positively correlates with the severity of symptoms [16–18]. Also, there is an inverse correlation between lipid peroxidation parameters and cardiac performance (ejection fraction, exercise capacity). Pericardial levels of 8-iso-PGF2_ (an isoprostane, which is a bio-marker of increased ROS production in vivo) also closely correlate with end-systolic and end-diastolic diameters of the left ventricle and with the functional severity of heart failure in patients [17]. Furthermore, there is a significant positive correlation between myocardial ROS levels and left ventricular contractile dysfunction in various experimental models [1–8].
Myocardial ROS generation has been shown to be triggered by repetitive episodes of ischemia/reperfusion, by an increased level of inflammatory cytokines (e.g. TNF-α, IL-6), by catecholamine auto-oxidation and/or during prostaglandin biosynthesis. Impaired antioxidant defense mechanisms can also increase ROS production within the myocardium [reviewed in 7]. The possible sources of increased production of superoxide, hydrogen peroxide and hydroxyl radical in failing myocardium are multiple, including xanthine oxidase (XO), NAD(P)H oxidase, cyclooxygenases, the mitochondrial electron transport chain activity, activated neutrophils, nitric oxide synthases (NOS), and auto-oxidation of certain tissue metabolites [3,7].
Inspite of the experimental data supporting the role of ROS in development of the structural and functional changes in the myocardium during the progression of the heart failure, the results of relatively few controlled clinical trials with antioxidants such as vitamin C, α-tocopherol (vitamin E) and co-enzyme Q10 are controversial. In some trials, antioxidants reduced plasma MDA concentration, in patients an indicator of oxidative stress, and showed improvement in symptoms and in myocardial function, but in others, they exerted only mild or no effects on symptoms, ejection fraction and exercise capacity [1, 7].
Importantly, some drugs that have currently been effectively used in the treatment of heart failure, including angiotensin-converting-enzyme inhibitors, and statins, the β-adrenergic antagonist carvedilol, may have important anti-oxidant properties. There is also evidence that the β-adrenergic antagonist metoprolol, which is not an antioxidant, can also decrease oxidative stress in patients with heart failure, raising the possibility that the effective treatment by decreasing cardiac overload could indirectly reduce oxidative stress [1].
INCREASED XANTHINE OXIDASE AND NAD(P)H OXIDASE DERIVED SUPEROXIDE GENERATION IN HEART FAILURE
Since the introduction of the concept of ischemia-reperfusion injury in the early 1980’s [19,20], several lines of evidence support the pathophysiological role of XO-derived ROS generation, and show beneficial effects of XO inhibitors against myocardial ischemia-reperfusion.
Numerous studies during the past two decades reported that the administration of allopurinol or oxypurinol has beneficial effects on the ischemic and reperfused hearts by the reduction of the reperfusion-induced arrhythmias and infarct size in diverse rat, rabbit, canine and pig models [8]. At the same time, other groups could not confirm the protective effect of allopurinol or oxypurinol in various models of myocardial ischemia and reperfusion, especially in response to more severe periods of ischemia [8]. Additional studies have also reported direct free radical scavenging effects of allopurinol or oxypurinol in isolated hearts unrelated to the inhibition of XO.
Nevertheless, it is now generally accepted that XO is functional in the mammalian heart including the human heart [8,21]. Because substrate accumulation does occur during ischemia, it likely results in the production of free radicals during the reperfusion stage [22]. Although the mechanism of action remained unclear, follow-up work confirmed the cardioprotective effects of allopurinol, and extended it to hypoxic storage and cardioplegia conditions [23,24], cardiopulmonary surgery [25,26] and acute cardiac allograft rejection [27].
Recent work has implicated a role for XO-related oxidant species in the pathogenesis of chronic heart failure [CHF; overviewed in 8]. Studies in isolated hearts have demonstrated that the progressive development of contractile failure is associated with increased myocardial XO levels, which contributes to an enhancement of oxidative stress in the heart [28,29]. Similarly, in heart failure models induced by pacing in the dog or by coronary ligation in rats, increased myocardial XO activity or levels were found, with subsequent increases in oxidative stress in the heart [30–32]. In canine pacing-induced CHF, allopurinol decreased the myocardial oxygen consumption and improved cardiac contractility and mechanical efficiency at rest [30] as well as during dobutamine-induced beta-adrenergic stimulation or exercise [31]. Interestingly, the benefits of allopurinol and ascorbate in dogs with heart failure could be prevented by NOS inhibition, suggesting that XO-derived superoxide may interfere with NO regulation of myocardial energetics [31]. More recently, Khan and colleagues [34] have demonstrated that the neuronal nitric oxide synthase (nNOS) and XO are in physical proximity in the sarcoplasmic reticulum of the cardiac myocytes of mice. Deficiency of nNOS but not eNOS was associated with profound increases in XO-mediated super-oxide production, which in turn depressed myocardial excitation-contraction coupling in a manner reversible by XO inhibition with allopurinol [8]. These data suggested that nNOS not only regulates the SR Ca2+ cycle [3,34], but also represents an important antioxidant system inhibiting XO activity [34,35]. Similarly to allopurinol, oxypurinol also improved cardiac contractility and mechanoenergetic coupling in rat model of heart failure [36]. In a mouse model of heart failure, allopurinol doubled the survival, decreased pathologically elevated XO activity, improved contractility and response to isoproterenol both in vivo and in isolated cardiac muscle [37]. Consistently with these results, in another recent murine heart failure study [38], investigators have demonstrated reduction of ROS production and decreased myocardial dysfunction following allopurinol treatment. Importantly, in addition to the beneficial effect of the drug on the left ventricular contractile function, allopurinol treatment also attenuated the left ventricular cavity dilation and reduced the myocardial hypertrophy and the intestinal fibrosis [38].
In CHF patients, elevated circulating uric acid levels strongly correlate with inflammatory markers (ICAM-1, TNFα, soluble TNF receptor 2 and E-selectin) and with the increased mortality [39–42]. In patients with idiopathic dilated cardiomyopathy, intracoronary administration of allopurinol resulted in an acute, significant improvement in myocardial efficiency, by diminishing the oxygen consumption in the presence of standard supportive therapy [40]. Acute intravenous infusion of allopurinol or chronic treatment with the drug for 1 month improved endothelial function in patients with CHF [42,43]. In a retrospective study, high-dose of allopurinol was found to beneficially affect survival [44]. On the other hand, the degree of autonomic dysfunction was unaffected by allopurinol [45]. Long-term prospective evaluation of the possible benefits of allopurinol treatment in CHF is currently lacking. These and the above mentioned preclinical studies have encouraged the clinical testing of oxypurinol for CHF. Such large-scale clinical studies are currently on-going and the interim analysis has produced some encouraging results [46].
Preliminary evidence suggests that NAD(P)H oxidase-derived superoxide production also contributes to the endothelial and myocardial dysfunction in CHF both in animal models and humans [47,48]. Further studies are definitely needed to elucidate the link between NAD(P)H oxidase activation, myocardial inflammation and contractile dysfunction during the development of heart failure.
EVIDENCE FOR INCREASED NITROSATIVE STRESS IN HEART FAILURE
NO produced by Ca2+-dependent NOS in endothelium, in cardiomyocytes and in cardiac nerves serves a number of important roles in the regulation of cardiac function including coronary vasodilation, inhibiting platelet and neutrophil actions, antioxidant effects, modulation of cardiac contractile function, and inhibiting cardiac oxygen consumption [3, 7, 50–67]. In excess concentrations, NO could be potentially toxic. It is now generally accepted that many of the toxic actions of NO are not directly due to NO itself, but are mediated via the production of highly reactive oxidant peroxynitrite, the reaction product of NO and superoxide [7,68].
Increased expression and activity of inducible isoform of nitric oxide synthase (iNOS) was demonstrated in the myocardium and vasculature of animals and patients with heart failure [69–79]. One study in mice found correlation between the chronic overexpression of iNOS and peroxynitrite generation with cardiac enlargement, conduction defects, sudden cardiac death and, less commonly, heart failure in mice [79], while an other study came to the conclusion that cardiac-specific overexpression of iNOS was not associated with severe cardiac dysfunction [80]. Increased myocardial iNOS activity was found in rats with volume-overload HF, which contributed to depressed myocardial contractility and beta-adrenergic hyporesponsiveness [81]. Although the above-mentioned evidence exists, the role of iNOS in the development and progression of heart failure is a subject of recent debate. For example, a recent study has demonstrated that deficiency of iNOS does not attenuate severe congestive heart failure in mice [82]. Neuronal nitric oxide synthase may represent an additional source of increased NO production in the failing human heart [83]. Additionally, upregulation of the endothelial isoform of NOS was also demonstrated in banding-induced chronic heart failure model [84].
As mentioned above, the combination of superoxide and NO yields peroxynitrite. Peroxynitrite generation has been demonstrated in various forms of acute and chronic heart failures both in experimental animals and biopsies obtained from human subjects, and this species has been shown to impair cardiac function via multiple mechanisms [7,85].
The reaction of peroxynitrite with different enzymes, macromolecules and lipids, has been shown to influence several cellular functions. For instance, tyrosine nitration may lead to dysfunction of nitrated proteins, as has been shown or suggested in the case of superoxide dismutase, cytoskeletal actin, neuronal tyrosine hydroxylase, cytochrome P450 and prostacyclin synthase [reviewed in 86,87]. Oxidation of the critical sulfhydryl groups is responsible for the inhibition of mitochondrial and cytosolic aconitase and other critical enzymes in the mitochondrial respiratory chain [88]. Peroxynitrite can also cause covalent modification of an active site thiol of glyceraldehyde-3-phosphate dehydrogenase and in creatine kinase [86,87]. Peroxynitrite-mediated nitration of myofibrillar creatine kinase activity may lead to contractile dysfunction of the heart [69]. Peroxynitrite-modified cellular proteins are subject to accelerated degradation via the proteosome. Peroxynitrite has been shown to inhibit a variety of ion pumps including calcium pumps, calcium-activated potassium channels and also the membrane Na+/K+ ATP-ase activity [86,87].
The reaction of lipids with peroxynitrite leads to peroxidation (malondialdehyde and conjugated diene formation) and formation of nitrito-, nitro-, nitrosoperoxo- and/or nitrated lipid oxidation adducts. Peroxynitrite also potently oxidizes various biomolecules including tetrahydrobiopterin (BH4) to quinonoid 5,6-dihydrobiopterin. A large proportion of the quinonoid isomer readily loses its side chain to form 7,8-dihydropterin, which is not a co-factor for NO synthase. Thus, in endothelial cells and other cell types, pathophysiologically low levels of BH4 can promote a cycle of its own destruction mediated by NO synthase-dependent formation of peroxynitrite. This mechanism might also contribute to vascular endothelial dysfunction induced by oxidative stress in heart failure. Peroxynitrite generation also decreases the nitric oxide available for G-protein stimulation and vasodilation, thus contributing to endothelial dysfunction. It is important to note that peroxynitrite can inhibit superoxide dismutase, glutaredoxin and other antioxidant molecules and systems. Peroxynitrite-mediated depletion of one of the key cellular antioxidants, glutathione can lead to positive feedback cycles of intracellular oxidant generation and exacerbation of the oxidative cellular injury [reviewed in 86,87].
Tissue injury caused by oxidative stress may be a consequence of activation of the precursors of matrix metalloproteinase (proMMPs). Recent work suggests that the activation of proMMPs is triggered by peroxynitrite generation, via an extensive S-glutathiolation reaction [reviewed in Pacher et al. 2005].
Peroxynitrite can also induce DNA single strand breakage and activation of the nuclear enzyme PARP, which can trigger a cellular suicide pathway [reviewed in 7, 89 and see also below].
In contrast to high concentrations of peroxynitrite which leads to rapid cell death, associated with rapid energetic derangements and PARP activation, lower concentrations of peroxynitrite, after several hours, can result in cytochrome c release from mitochondria and caspase 3-, 2-, 8- and 9- dependent apoptotic cell death.
In addition to being a terminal mediator of cell injury, peroxynitrite also enhances and triggers a variety of pro-inflammatory processes. For example, peroxynitrite enhances the expression of P-selectin and ICAM-1 in human endothelial cells, and it mediates the cytokine-induced IL-8 expression in human leukocytes. In human neutrophils, peroxynitrite triggers the down regulation of L-selectin expression, and up regulation of CD11b/CD18 expression. These effects are likely to be mediated, at least in part, by the ability of peroxynitrite to trigger and enhance nuclear factor kappa B (NF-κB) mediated pro-inflammatory signal trans-duction pathways [reviewed in 89]. These alterations can culminate in a global dysregulation of cellular signal transduction pathways.
As mentioned above, there is circumstantial evidence that oxidative/nitrosative stress importantly contributes to the pathogenesis of various forms of myocardial injury and heart failure. It appears that peroxynitrite decomposition catalysts improve cardiac function and overall outcome in these models. For instance, a novel metalloporphyrin peroxynitrite decomposition catalyst, FP15, reduced myocardial necrosis in a rat model of acute myocardial infarction as well as in a recent porcine study [90,91]. Furthermore, FP15 significantly improved cardiac function in a diabetic cardiomyopathy model [92,93]. In addition, a recent study provided evidence that the treatment with a novel potent peroxynitrite decomposition catalyst attenuated the development of cardiac dysfunction and increased the survival in doxorubicin-induced cardiomyopathy model [90]. The mechanism by which peroxynitrite neutralization protects hearts from dysfunction may involve protection against vascular and myocardial tyrosine nitration, PARP activation, lipid peroxidation and multiple other mechanisms, as all these mechanisms have previously been linked to peroxynitrite-induced cardiac injury. Additional mechanisms of peroxynitrite-mediated diabetic cardiac dysfunction may include inhibition of myofibrillar creatine kinase and of succinyl-CoA:3-oxoacid CoA-transferase or activation of metalloproteinases [reviewed in 7,85]. These observations support the concept that peroxynitrite is a major mediator of myocardial injury in various pathophysiological conditions, and its effective neutralization can be of significant therapeutic benefit.
EVIDENCE FOR INCREASED POLY(ADP-RIBOSE) POLYMERASE (PARP) ACTIVATION IN HEART FAILURE
Poly(ADP-ribose) polymerase-1 (PARP-1) is a nuclear enzyme with multiple regulatory functions. PARP-1 cleaves NAD+ to nicotinamide and ADP-ribose to form long branched (ADP-ribose) polymers on glutamic acid residues of a number of target proteins including histones and PARP-1 (automodification domain) itself. Poly(ADP-ribosyl)ation is involved in the regulation of many cellular processes such as DNA repair, gene transcription, cell cycle progression, cell death, chromatin function and genomic stability. When PARP is activated by oxygen- and nitrogen-derived radical-induced DNA single-strand breaks, it catalyzes the cleavage of NAD+ into nicotinamide and ADP-ribose, and uses the latter to synthesize branched nucleic acid-like polymers poly(ADP-ribose) covalently attached to nuclear acceptor proteins. The branched polymer, the size of which varies from a few to 200 ADP-ribose units, may facilitate recruitment of DNA repair enzymes to the sites of DNA injury [7, 89; Fig. 1]. In vivo, the most abundantly poly-ADP-ribosylated protein is PARP-1 itself and auto-poly(ADP-ribosylation) represents a major regulatory mechanism for PARP-1 resulting in the downregulation of the enzyme activity. The polymer is degraded by poly(ADP-ribose) glycohydrolase (PARG) and ADP-ribosyl protein lyase with the latter enzyme removing the protein proximal ADP-ribose residue [89; Fig. 1]. The concerted action of PARP-1 and PARG maintains a highly accelerated ADP-ribose turnover in peroxynitrite exposed cells. As a result, NAD becomes depleted in the cells leading to malfunctioning glycolysis, Krebs cycle, mitochondrial electron transport and eventually to ATP depletion. Moreover, shortage on ATP is exaggerated by attempts of the cells to resynthesize NAD from ATP and nicotinamide. The net result of this pathway is a dramatic drop in cellular ATP [7,89; Fig. 1].
Fig. 1. Proposed role of oxidative/nitrosative stress and downstream pathway in heart failure.
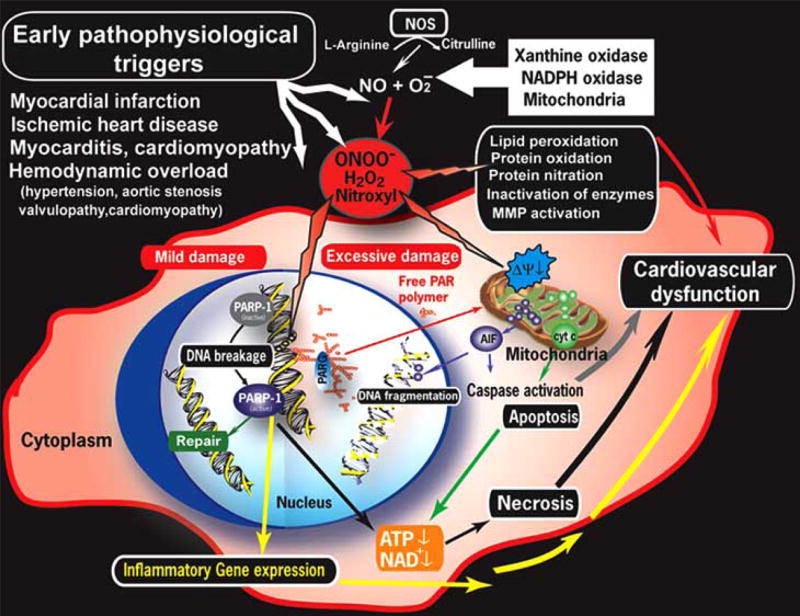
Peroxynitrite, formed from superoxide and NO, causes cell injury via lipid peroxidation, inactivation of enzymes and other proteins by oxidation and nitration and also activation of matrix metalloproteinases (MMPs) just to mention a few. Peroxynitrite also triggers the release of proapoptotic factors such as cytochrome c and apoptosis inducing factor (AIF) from the mitochondria, which mediate caspase dependent and independent apoptotic death pathways. Peroxynitrite together with other oxidants, induces stand breaks in deoxyribonucleic acid (DNA), which in turn activates PARP. Mild damage of DNA activates the DNA repair machinery, but once excessive oxidative/nitrosative stress-induced damage occurs (like in various forms of myocardial reperfusion injury and heart failure), overactivated PARP initiates an energy-consuming cycle by transferring adenosine diphosphateribose units from nicotinamide adenine dinucleotide (NAD+) to nuclear proteins. This process will lead to depletion of the intracellular NAD+ and adenosine triphosphate (ATP) pools, slowing the rate of glycolysis and mitochondrial respiration, eventually culminating to cardiovascular dysfunction or death. Poly (ADP-ribose) glycohydrolase (PARG) degrades poly (ADP-ribose) (PAR) polymers, generating free PAR polymer and ADP-ribose. PARP also regulates the expression of a variety of inflammatory mediators, which may facilitate the progression of heart failure.
PARP-1 also regulates the expression of various proteins at transcriptional level. Of special importance is the regulation by PARP-1 of the production of inflammatory mediators such as inducible nitric oxide synthase (iNOS), intercellular adhesion molecule-1 (ICAM-1) and major histocompatibility complex class II (MHC Class II) [89]. NF-κB is a key transcription factor in the regulation of this set of proteins, and PARP has been shown to act as a co-activator in the NF-κB-mediated transcription. Poly(ADP-ribosylation) can loosen up the chromatin structure, thereby making genes more accessible for the transcriptional machinery [89].
The PARP-mediated pathway of cell necrosis and the PARP-mediated pathway of inflammatory signal transduction and gene expression may be interrelated in pathophysiological conditions. Oxidant stress can generate DNA single-strand breaks. DNA strand breaks then activate PARP, which in turn potentiates NF-κB activation and AP-1 expression, resulting in greater expression of the AP-1 and NF-κB dependent genes, such as the gene for ICAM-1, as well as chemokines such as MIP-1alpha and cytokines such as TNF-alpha. Chemokine generation, in combination with increased endothelial expression of ICAM-1, recruits more activated leukocytes to inflammatory foci, producing greater oxidant stress. It is possible that a low-level, localized inflammatory response may be beneficial in recruiting mononuclear cells to an inflammatory site. However, in many pathophysiological states, the above described feedback cycles amplify themselves beyond control.
A recently characterized mechanism of myocardial dysfunction involves the activation of the nuclear enzyme PARP by DNA single strand breaks generated in response to increased oxidative and nitrosative stress. It is well-established that this pathway plays a pivotal role in various models of myocardial ischemia-reperfusion injury (a condition in which oxidative and nitrosative stress plays a key pathogenetic role) [reviewed in 7,94]. Currently, there is also emerging evidence that PARP pathway also plays pathogenetic role in the development of various forms of cardiomyopathy and heart failure [7]. As a first example, in diabetic cardiomyopathy models that spontaneously develop in the non-obese diabetic mice and in the streptozotocin-induced models of diabetes, the marked depression of myocardial contractile function is associated with a significant increase in poly(ADP-ribosyl)ation in cardiac myocytes and endothelium [93, 95,96]. Increased nitrosative stress and poly(ADP-ribosyl)ation were demonstrated in CHF following coronary artery ligation and associated with advanced aging both in the cardiac myocytes and endothelial cells. In these studies pharmacological inhibition of PARP improved cardiac contractility and endothelial function [97–101]. The role of increased oxidative-nitrosative stress-induced PARP activation in the development of cardiac dysfunction induced by doxorubicin [90,102–104] or bacterial endotoxin was also demonstrated [105].
Chronic mechanical overloading of the heart leads to ventricular hypertrophy, which, at later stages, is also associated with a progressive decline of myocardial contractility. Recent studies suggest that heart failure in mice with chronic aortic banding is associated with PARP activation, and PARP activation was also present in myocardial samples of patients with CHF [106,107]. Importantly, both pharmacological inhibition of PARP and genetic PARP deficiency prevented the pressure-overload induced decrease in cardiac contractile function [106,107] and attenuated hypertrophy. Whether PARP inhibition exerts its beneficial effect in this model by affecting NAD/NADH levels or by other mechanisms (by reducing myocardial apoptosis or by limiting inflammatory processes) is not yet understood. Previous studies showed that cardiac NAD/NADH supply may change in hearts with mechanical overload, which was, in part, attributed to altered cellular metabolism (for example, oxidative utilization of glucose is slightly depressed in volume-overloaded hearts [108]. Yet, the role of NAD utilization by PARP in reducing NADH supplies remains to be explored. We have recently showed that in aortic banded rats with cardiac hypertrophy, [109] in which overt heart failure is not yet present, cardiac NADH levels do not decline substantially (unpublished data).
In contrast, using a pacing-induced heart failure model in dogs [51,56,110], we have demonstrated that development of overt heart failure is associated with a significant decrease in NADH supply (Fig. 2). Previous studies demonstrated a metabolic shift in severe heart failure, including a depressed free fatty acid (FFA) oxidation (likely due to reduced activity of mitochondrial fatty acid oxidation enzymes) and a downregulation of some of the key enzymes of the carbohydrate metabolism [56; 110]. We propose that the observed metabolic changes may aggravate the effects of PARP activation (e.g. by depleting NAD/NADH stores). Thus, future studies should investigate the potential beneficial effects of PARP inhibition in this large animal model, which shows many similarities to human heart failure.
Fig. 2. Cardiac NADH concentration in dogs with pacing-induced heart failure.
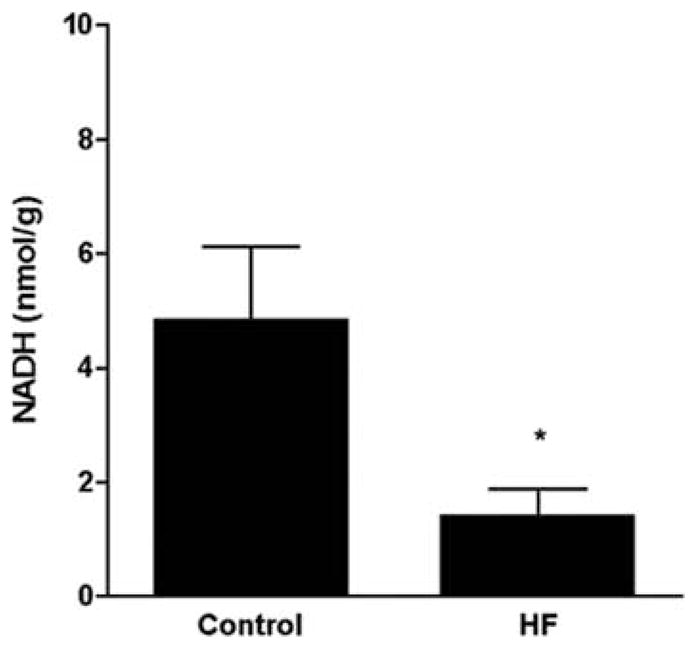
The heart was paced at 210 bpm for 3 weeks, and then the pacing rate was increased to 240 bpm until LV end-diastolic pressure reached 25 mm Hg and clinical signs of severe decompensation were observed (for details of the method see refs. 56,110). NADH content was measured by HPLC as we described [119,120]. Data are mean±S.E.M. (n=5 in each group). * p<0.05
Several reports indicate the importance of PARP activation in the development of mitochondrial dysfunction under conditions of oxidative stress [111, 112]. Even though the major isoform of the PARP family, PARP-1, is widely considered as a nuclear enzyme, there may also be mitochondrial isoforms [112], and there is a nuclear-to mitochondrial signaling process, which initiates early mitochondrial alterations, as demonstrated in neurons [111], in cardiac myocytes in vitro [113] and in myocardial infarction in vivo [114]. Importantly, pharmacological inhibition of PARP or genetic PARP-1 deficiency reverse, mitochondrial-to nuclear translocation of AIF in banding-induced murine heart failure model [107], consistently with earlier studies demonstrating that PARP-1 regulates the translocation of this cell death factor.
An additional factor to be considered in the context of PARP activation and the pathogenesis of endothelial dysfunction in heart failure is angiotensin II, which is a well-known secondary mediator in the pathogenesis of CHF. Recent studies have demonstrated that angiotensin II can induce intraendothelial peroxynitrite formation [115,116], as well as PARP activation [117]. PARP may also regulate the expression of endothelin, which is also an important secondary mediator in CHF [118].
The protective effect of pharmacological inhibition of PARP or in preventing cardiovascular dysfunction has been demonstrated in experimental models of reperfusion injury and chronic heart failure. The mode of PARP inhibitors’ cardioprotective action involves conservation of myocardial energetics, as well as prevention of the upregulation of various pro-inflammatory pathways (cytokines, adhesion receptors, mononuclear cell infiltration) triggered by ischemia and reperfusion [reviewed in 7]. Similar to acute myocardial ischemia and reperfusion, both oxidant-mediated myocardial injury, as well as upregulation of various proinflammatory processes, plays important roles in CHF. It is conceivable that PARP inhibition exerts beneficial effects in CHF by affecting both above referenced pathways of injury, and also by suppressing positive feedback cycles initiated by them.
Although the exact molecular triggers of PARP activation in chronic heart failure are not known, the above mentioned studies support the view that PARP activation importantly contributes to the pathogenesis of cardiovascular dysfunction in experimental models of heart failure.
FUTURE PROSPECTS
Based on the evidence reviewed herein, we conclude that oxidative-nitrosative stress-PARP pathway plays very important regulatory roles in the pathogenesis of cardiac and endothelial dysfunction in pathophysiological conditions associated with oxidative stress, including various forms of heart failure. It remains to be studied whether various clinical therapeutic or experimental therapeutic interventions, which are known to have some cardiac and vascular protective effects in chronic heart failure (antioxidant therapies, statins, β-blockers, ACE-inhibitors, etc.), are able to suppress the activation of PARP in the cardiovascular system. A multitude of novel PARP inhibitors are in various stages of preclinical or clinical development, many with potency that greatly exceeds the prototypic agents successfully used in earlier animal studies. The remarkable efficiency of new PARP inhibitors against various forms of myocardial injury in animal models strongly suggests that these agents hold considerable promise and hope in the treatment of cardiovascular disorders in humans.
References
- 1.Ferrari R, Guardigli G, Mele D, Percoco GF, Ceconi C, Curello S. Oxidative stress during myocardial ischaemia and heart failure. Curr Pharm Des. 2004;10(14):1699–711. doi: 10.2174/1381612043384718. [DOI] [PubMed] [Google Scholar]
- 2.Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005;115(3):500–8. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hare JM, Stamler JS. NO/redox disequilibrium in the failing heart and cardiovascular system. J Clin Invest. 2005;115(3):509–17. doi: 10.1172/JCI200524459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huss JM, Kelly DP. Mitochondrial energy metabolism in heart failure: a question of balance. J Clin Invest. 2005;115(3):547–55. doi: 10.1172/JCI200524405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McKinsey TA, Olson EN. Toward transcriptional therapies for the failing heart: chemical screens to modulate genes. J Clin Invest. 2005;115(3):538–46. doi: 10.1172/JCI24144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morita H, Seidman J, Seidman CE. Genetic causes of human heart failure. J Clin Invest. 2005;115(3):518–26. doi: 10.1172/JCI200524351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pacher P, Schulz R, Liaudet L, Szabó C. Nitrosative stress and pharmacological modulation of heart failure. In press: Trends in Pharmacological Sciences. 2005 doi: 10.1016/j.tips.2005.04.003. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berry CE, Hare JM. Xanthine oxidoreductase and cardiovascular disease: molecular mechanisms and pathophysiological implications. J Physiol. 2004;555(Pt 3):589–606. doi: 10.1113/jphysiol.2003.055913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drexler H, Hornig B. Importance of endothelial function in chronic heart failure. J Cardiovasc Pharmacol. 1996;27(Suppl 2):S9–12. doi: 10.1097/00005344-199600002-00003. [DOI] [PubMed] [Google Scholar]
- 10.Sun D, Huang A, Zhao G, Bernstein R, Forfia P, Xu X, et al. Reduced NO-dependent arteriolar dilation during the development of cardiomyopathy. Am J Physiol Heart Circ Physiol. 2000;278(2):H461–8. doi: 10.1152/ajpheart.2000.278.2.H461. [DOI] [PubMed] [Google Scholar]
- 11.Landmesser U, Spiekermann S, Dikalov S, Tatge H, Wilke R, Kohler C, et al. Vascular oxidative stress and endothelial dysfunction in patients with chronic heart failure: role of xanthine-oxidase and extracellular superoxide dismutase. Circulation. 2002;106(24):3073–8. doi: 10.1161/01.cir.0000041431.57222.af. [DOI] [PubMed] [Google Scholar]
- 12.Colombo PC, Banchs JE, Celaj S, Talreja A, Lachmann J, Malla S, et al. Endothelial cell activation in patients with decompensated heart failure. Circulation. 2005;111(1):58–62. doi: 10.1161/01.CIR.0000151611.89232.3B. [DOI] [PubMed] [Google Scholar]
- 13.Li JM, Shah AM. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol. 2004;287(5):R1014–30. doi: 10.1152/ajpregu.00124.2004. [DOI] [PubMed] [Google Scholar]
- 14.Fischer D, Rossa S, Landmesser U, Spiekermann S, Engberding N, Hornig B, et al. Endothelial dysfunction in patients with chronic heart failure is independently associated with increased incidence of hospitalization, cardiac transplantation, or death. Eur Heart J. 2005;26(1):65–9. doi: 10.1093/eurheartj/ehi001. [DOI] [PubMed] [Google Scholar]
- 15.Cesselli D, Jakoniuk I, Barlucchi L, Beltrami AP, Hintze TH, Nadal-Ginard B, et al. Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ Res. 2001;89(3):279–86. doi: 10.1161/hh1501.094115. [DOI] [PubMed] [Google Scholar]
- 16.Polidori MC, Savino K, Alunni G, Freddio M, Senin U, Sies H, et al. Plasma lipophilic antioxidants and malondialdehyde in congestive heart failure patients: relationship to disease severity. Free Radic Biol Med. 2002;32(2):148–52. doi: 10.1016/s0891-5849(01)00782-1. [DOI] [PubMed] [Google Scholar]
- 17.Polidori MC, Pratico D, Savino K, Rokach J, Stahl W, Mecocci P. Increased F2 isoprostane plasma levels in patients with congestive heart failure are correlated with antioxidant status and disease severity. J Card Fail. 2004;10(4):334–8. doi: 10.1016/j.cardfail.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 18.Nakamura K, Kusano KF, Matsubara H, Nakamura Y, Miura A, Nishii N, et al. Relationship between oxidative stress and systolic dysfunction in patients with hypertrophic cardiomyopathy. J Card Fail. 2005;11(2):117–23. doi: 10.1016/j.cardfail.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 19.Granger DN, Rutili G, McCord JM. Superoxide radicals in feline intestinal ischemia. Gastroenterology. 1981;81(1):22–9. [PubMed] [Google Scholar]
- 20.McCord JM. Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med. 1985;312(3):159–63. doi: 10.1056/NEJM198501173120305. [DOI] [PubMed] [Google Scholar]
- 21.MacGowan SW, Regan MC, Malone C, Sharkey O, Young L, Gorey TF, et al. Superoxide radical and xanthine oxidoreductase activity in the human heart during cardiac operations. Ann Thorac Surg. 1995;60(5):1289–93. doi: 10.1016/0003-4975(95)00616-S. [DOI] [PubMed] [Google Scholar]
- 22.Xia Y, Zweier JL. Substrate control of free radical generation from xanthine oxidase in the postischemic heart. J Biol Chem. 1995;270(32):18797–803. doi: 10.1074/jbc.270.32.18797. [DOI] [PubMed] [Google Scholar]
- 23.Bergsland J, LoBalsamo L, Lajos P, Feldman MJ, Mookerjee B. Allopurinol in prevention of reperfusion injury of hypoxically stored rat hearts. J Heart Transplant. 1987;6(3):137–40. [PubMed] [Google Scholar]
- 24.Vinten-Johansen J, Chiantella V, Faust KB, Johnston WE, McCain BL, Hartman M, et al. Myocardial protection with blood cardioplegia in ischemically injured hearts: reduction of reoxygenation injury with allopurinol. Ann Thorac Surg. 1988;45(3):319–26. doi: 10.1016/s0003-4975(10)62472-1. [DOI] [PubMed] [Google Scholar]
- 25.Coghlan JG, Flitter WD, Clutton SM, Panda R, Daly R, Wright G, et al. Allopurinol pretreatment improves postoperative recovery and reduces lipid peroxidation in patients undergoing coronary artery bypass grafting. J Thorac Cardiovasc Surg. 1994;107(1):248–56. [PubMed] [Google Scholar]
- 26.Castelli P, Condemi AM, Brambillasca C, Fundaro P, Botta M, Lemma M, et al. Improvement of cardiac function by allopurinol in patients undergoing cardiac surgery. J Cardiovasc Pharmacol. 1995;25(1):119–25. doi: 10.1097/00005344-199501000-00019. [DOI] [PubMed] [Google Scholar]
- 27.Akizuki S, Yoshida S, Chambers DE, Eddy LJ, Parmley LF, Yellon DM, et al. Infarct size limitation by the xanthine oxidase inhibitor, allopurinol, in closed-chest dogs with small infarcts. Cardiovasc Res. 1985;19(11):686–92. doi: 10.1093/cvr/19.11.686. [DOI] [PubMed] [Google Scholar]
- 28.Ferdinandy P, Panas D, Schulz R. Peroxynitrite contributes to spontaneous loss of cardiac efficiency in isolated working rat hearts. Am J Physiol. 1999;276(6 Pt 2):H1861–7. doi: 10.1152/ajpheart.1999.276.6.H1861. [DOI] [PubMed] [Google Scholar]
- 29.Ferdinandy P, Danial H, Ambrus I, Rothery RA, Schulz R. Peroxynitrite is a major contributor to cytokine-induced myocardial contractile failure. Circ Res. 2000;87(3):241–7. doi: 10.1161/01.res.87.3.241. [DOI] [PubMed] [Google Scholar]
- 30.Ekelund UE, Harrison RW, Shokek O, Thakkar RN, Tunin RS, Senzaki H, et al. Intravenous allopurinol decreases myocardial oxygen consumption and increases mechanical efficiency in dogs with pacing-induced heart failure. Circ Res. 1999;85(5):437–45. doi: 10.1161/01.res.85.5.437. [DOI] [PubMed] [Google Scholar]
- 31.Saavedra WF, Paolocci N, St John ME, Skaf MW, Stewart GC, Xie JS, et al. Imbalance between xanthine oxidase and nitric oxide synthase signaling pathways underlies mechanoenergetic uncoupling in the failing heart. Circ Res. 2002;90(3):297–304. doi: 10.1161/hh0302.104531. [DOI] [PubMed] [Google Scholar]
- 32.de Jong JW, Schoemaker RG, de Jonge R, Bernocchi P, Keijzer E, Harrison R, et al. Enhanced expression and activity of xanthine oxidoreductase in the failing heart. J Mol Cell Cardiol. 2000;32(11):2083–9. doi: 10.1006/jmcc.2000.1240. [DOI] [PubMed] [Google Scholar]
- 33.Ukai T, Cheng CP, Tachibana H, Igawa A, Zhang ZS, Cheng HJ, et al. Allopurinol enhances the contractile response to dobutamine and exercise in dogs with pacing-induced heart failure. Circulation. 2001;103(5):750–5. doi: 10.1161/01.cir.103.5.750. [DOI] [PubMed] [Google Scholar]
- 34.Khan SA, Lee K, Minhas KM, Gonzalez DR, Raju SV, Tejani AD, et al. Neuronal nitric oxide synthase negatively regulates xanthine oxidoreductase inhibition of cardiac excitation-contraction coupling. Proc Natl Acad Sci U S A. 2004;101(45):15944–8. doi: 10.1073/pnas.0404136101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bonaventura J, Gow A. NO and superoxide: opposite ends of the seesaw in cardiac contractility. Proc Natl Acad Sci USA. 2004;101(47):16403–4. doi: 10.1073/pnas.0405859101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kogler H, Fraser H, McCune S, Altschuld R, Marban E. Disproportionate enhancement of myocardial contractility by the xanthine oxidase inhibitor oxypurinol in failing rat myocardium. Cardiovasc Res. 2003;59(3):582–92. doi: 10.1016/s0008-6363(03)00512-1. [DOI] [PubMed] [Google Scholar]
- 37.Stull LB, Leppo MK, Szweda L, Gao WD, Marban E. Chronic treatment with allopurinol boosts survival and cardiac contractility in murine postischemic cardiomyopathy. Circ Res. 2004;95(10):1005–11. doi: 10.1161/01.RES.0000148635.73331.c5. [DOI] [PubMed] [Google Scholar]
- 38.Engberding N, Spiekermann S, Schaefer A, Heineke A, Wiencke A, Muller M, et al. Allopurinol attenuates left ventricular remodeling and dysfunction after experimental myocardial infarction: a new action for an old drug? Circulation. 2004;110(15):2175–9. doi: 10.1161/01.CIR.0000144303.24894.1C. [DOI] [PubMed] [Google Scholar]
- 39.Leyva F, Anker SD, Godsland IF, Teixeira M, Hellewell PG, Kox WJ, et al. Uric acid in chronic heart failure: a marker of chronic inflammation. Eur Heart J. 1998;19(12):1814–22. doi: 10.1053/euhj.1998.1188. [DOI] [PubMed] [Google Scholar]
- 40.Cappola TP, Kass DA, Nelson GS, Berger RD, Rosas GO, Kobeissi ZA, et al. Allopurinol improves myocardial efficiency in patients with idiopathic dilated cardiomyopathy. Circulation. 2001;104(20):2407–11. doi: 10.1161/hc4501.098928. [DOI] [PubMed] [Google Scholar]
- 41.Anker SD, Doehner W, Rauchhaus M, Sharma R, Francis D, Knosalla C, et al. Uric acid and survival in chronic heart failure: validation and application in metabolic, functional, and hemodynamic staging. Circulation. 2003;107(15):1991–7. doi: 10.1161/01.CIR.0000065637.10517.A0. [DOI] [PubMed] [Google Scholar]
- 42.Doehner W, Anker SD. Uric acid in chronic heart failure. Semin Nephrol. 2005;25(1):61–6. doi: 10.1016/j.semnephrol.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 43.Farquharson CA, Butler R, Hill A, Belch JJ, Struthers AD. Allopurinol improves endothelial dysfunction in chronic heart failure. Circulation. 2002;106(2):221–6. doi: 10.1161/01.cir.0000022140.61460.1d. [DOI] [PubMed] [Google Scholar]
- 44.Struthers AD, Donnan PT, Lindsay P, McNaughton D, Broomhall J, MacDonald TM. Effect of allopurinol on mortality and hospitalisations in chronic heart failure: a retrospective cohort study. Heart. 2002;87(3):229–34. doi: 10.1136/heart.87.3.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shehab AM, Butler R, MacFadyen RJ, Struthers AD. A placebo-controlled study examining the effect of allopurinol on heart rate variability and dysrhythmia counts in chronic heart failure. Br J Clin Pharmacol. 2001;51(4):329–34. doi: 10.1046/j.1365-2125.2001.01361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Freudenberger RS, Schwarz RP, Jr, Brown J, Moore A, Mann D, Givertz MM, et al. Rationale, design and organisation of an efficacy and safety study of oxypurinol added to standard therapy in patients with NYHA class III – IV congestive heart failure. Expert Opin Investig Drugs. 2004;13(11):1509–16. doi: 10.1517/13543784.13.11.1509. [DOI] [PubMed] [Google Scholar]
- 47.Li JM, Gall NP, Grieve DJ, Chen M, Shah AM. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension. 2002;40(4):477–84. doi: 10.1161/01.hyp.0000032031.30374.32. [DOI] [PubMed] [Google Scholar]
- 48.Takayama T, Wada A, Tsutamoto T, Ohnishi M, Fujii M, Isono T, et al. Contribution of vascular NAD(P)H oxidase to endothelial dysfunction in heart failure and the therapeutic effects of HMG-CoA reductase inhibitor. Circ J. 2004;68(11):1067–75. doi: 10.1253/circj.68.1067. [DOI] [PubMed] [Google Scholar]
- 49.Heymes C, Bendall JK, Ratajczak P, Cave AC, Samuel JL, Hasenfuss G, et al. Increased myocardial NADPH oxidase activity in human heart failure. J Am Coll Cardiol. 2003;41(12):2164–71. doi: 10.1016/s0735-1097(03)00471-6. [DOI] [PubMed] [Google Scholar]
- 50.Linke A, Recchia F, Zhang X, Hintze TH. Acute and chronic endothelial dysfunction: implications for the development of heart failure. Heart Fail Rev. 2003;8(1):87–97. doi: 10.1023/a:1022151106019. [DOI] [PubMed] [Google Scholar]
- 51.Trochu JN, Mital S, Zhang X, Xu X, Ochoa M, Liao JK, et al. Preservation of NO production by statins in the treatment of heart failure. Cardiovasc Res. 2003;60(2):250–8. doi: 10.1016/j.cardiores.2003.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mital S, Barbone A, Addonizio LJ, Quaegebeur JM, Mosca RJ, Oz MC, et al. Endogenous endothelium-derived nitric oxide inhibits myocardial caspase activity: implications for treatment of end-stage heart failure. J Heart Lung Transplant. 2002;21(5):576–85. doi: 10.1016/s1053-2498(01)00404-1. [DOI] [PubMed] [Google Scholar]
- 53.Adler S, Huang H, Trochu JN, Xu X, Gupta S, Hintze TH. Simvastatin reverses impaired regulation of renal oxygen consumption in congestive heart failure. Am J Physiol Renal Physiol. 2001 Nov;281(5):F802–9. doi: 10.1152/ajprenal.2001.281.5.F802. [DOI] [PubMed] [Google Scholar]
- 54.Recchia FA. Role of nitric oxide in the regulation of substrate metabolism in heart failure. Heart Fail Rev. 2002 Apr;7(2):141–8. doi: 10.1023/a:1015324508556. [DOI] [PubMed] [Google Scholar]
- 55.Tada H, Thompson CI, Recchia FA, Loke KE, Ochoa M, Smith CJ, et al. Myocardial glucose uptake is regulated by nitric oxide via endothelial nitric oxide synthase in Langendorff mouse heart. Circ Res. 2000;86(3):270–4. doi: 10.1161/01.res.86.3.270. [DOI] [PubMed] [Google Scholar]
- 56.Recchia FA, McConnell PI, Bernstein RD, Vogel TR, Xu X, Hintze TH. Reduced nitric oxide production and altered myocardial metabolism during the decompensation of pacing-induced heart failure in the conscious dog. Circ Res. 1998;83(10):969–79. doi: 10.1161/01.res.83.10.969. [DOI] [PubMed] [Google Scholar]
- 57.Kinugawa S, Huang H, Wang Z, Kaminski PM, Wolin MS, Hintze TH. A defect of neuronal nitric oxide synthase increases xanthine oxidase-derived superoxide anion and attenuates the control of myocardial oxygen consumption by nitric oxide derived from endothelial nitric oxide synthase. Circ Res. 2005;96(3):355–62. doi: 10.1161/01.RES.0000155331.09458.A7. [DOI] [PubMed] [Google Scholar]
- 58.Li W, Mital S, Ojaimi C, Csiszar A, Kaley G, Hintze TH. Premature death and age-related cardiac dysfunction in male eNOS-knockout mice. J Mol Cell Cardiol. 2004;37(3):671–80. doi: 10.1016/j.yjmcc.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 59.Walsh EK, Huang H, Wang Z, Williams J, de Crom R, van Haperen R, et al. Control of myocardial oxygen consumption in transgenic mice overexpressing vascular eNOS. Am J Physiol Heart Circ Physiol. 2004;287(5):H2115–21. doi: 10.1152/ajpheart.00267.2004. [DOI] [PubMed] [Google Scholar]
- 60.Adler A, Huang H, Wang Z, Conetta J, Levee E, Zhang X, et al. Endocardial endothelium in the avascular frog heart: role for diffusion of NO in control of cardiac O2 consumption. Am J Physiol Heart Circ Physiol. 2004;287(1):H14–21. doi: 10.1152/ajpheart.01235.2003. [DOI] [PubMed] [Google Scholar]
- 61.Hintze TH. Prologue: Nitric oxidehormones, metabolism, and function. Am J Physiol Heart Circ Physiol. 2001;281(6):H2253–5. doi: 10.1152/ajpheart.2001.281.6.H2253. [DOI] [PubMed] [Google Scholar]
- 62.Trochu JN, Bouhour JB, Kaley G, Hintze TH. Role of endothelium-derived nitric oxide in the regulation of cardiac oxygen metabolism: implications in health and disease. Circ Res. 2000;87(12):1108–17. doi: 10.1161/01.res.87.12.1108. [DOI] [PubMed] [Google Scholar]
- 63.Zhao G, Bernstein RD, Hintze TH. Nitric oxide and oxygen utilization: exercise, heart failure and diabetes. Coron Artery Dis. 1999;10(5):315–20. doi: 10.1097/00019501-199907000-00007. [DOI] [PubMed] [Google Scholar]
- 64.Wolin MS, Xie YW, Hintze TH. Nitric oxide as a regulator of tissue oxygen consumption. Curr Opin Nephrol Hypertens. 1999;8(1):97–103. doi: 10.1097/00041552-199901000-00015. [DOI] [PubMed] [Google Scholar]
- 65.Xie YW, Shen W, Zhao G, Xu X, Wolin MS, Hintze TH. Role of endothelium-derived nitric oxide in the modulation of canine myocardial mitochondrial respiration in vitro Implications for the development of heart failure. Circ Res. 1996;79(3):381–7. doi: 10.1161/01.res.79.3.381. [DOI] [PubMed] [Google Scholar]
- 66.Massion PB, Feron O, Dessy C, Balligand JL. Nitric oxide and cardiac function: ten years after, and continuing. Circ Res. 2003;93(5):388–98. doi: 10.1161/01.RES.0000088351.58510.21. [DOI] [PubMed] [Google Scholar]
- 67.Paulus WJ, Bronzwaer JG. Nitric oxide's role in the heart: control of beating or breathing? Am J Physiol Heart Circ Physiol. 2004;287(1):H8–13. doi: 10.1152/ajpheart.01147.2003. [DOI] [PubMed] [Google Scholar]
- 68.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271(5 Pt 1):C1424–37. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 69.Mihm MJ, Coyle CM, Schanbacher BL, Weinstein DM, Bauer JA. Peroxynitrite induced nitration and inactivation of myofibrillar creatine kinase in experimental heart failure. Cardiovasc Res. 2001;49(4):798–807. doi: 10.1016/s0008-6363(00)00307-2. [DOI] [PubMed] [Google Scholar]
- 70.Feng Q, Lu X, Jones DL, Shen J, Arnold JM. Increased inducible nitric oxide synthase expression contributes to myocardial dysfunction and higher mortality after myocardial infarction in mice. Circulation. 2001;104(6):700–4. doi: 10.1161/hc3201.092284. [DOI] [PubMed] [Google Scholar]
- 71.Chen Y, Traverse JH, Du R, Hou M, Bache RJ. Nitric oxide modulates myocardial oxygen consumption in the failing heart. Circulation. 2002;106(2):273–9. doi: 10.1161/01.cir.0000021120.90970.b9. [DOI] [PubMed] [Google Scholar]
- 72.Funakoshi H, Kubota T, Kawamura N, Machida Y, Feldman AM, Tsutsui H, et al. Disruption of inducible nitric oxide synthase improves beta-adrenergic inotropic responsiveness but not the survival of mice with cytokine-induced cardiomyopathy. Circ Res. 2002;90(9):959–65. doi: 10.1161/01.res.0000017632.83720.68. [DOI] [PubMed] [Google Scholar]
- 73.Kobayashi N, Horinaka S, Mita S, Yoshida K, Honda T, Kobayashi T, et al. Aminoguanidine inhibits mitogen-activated protein kinase and improves cardiac performance and cardiovascular remodeling in failing hearts of salt-sensitive hypertensive rats. J Hypertens. 2002;20(12):2475–85. doi: 10.1097/00004872-200212000-00028. [DOI] [PubMed] [Google Scholar]
- 74.Mungrue IN, Stewart DJ, Husain M. The Janus faces of iNOS. Circ Res. 2003;93(7):e74. doi: 10.1161/01.RES.0000095452.77140.7A. [DOI] [PubMed] [Google Scholar]
- 75.Vanderheyden M, Bartunek J, Knaapen M, Kockx M, De Bruyne B, Goethals M. Hemodynamic effects of inducible nitric oxide synthase and nitrotyrosine generation in heart failure. J Heart Lung Transplant. 2004;23(6):723–8. doi: 10.1016/j.healun.2003.07.015. [DOI] [PubMed] [Google Scholar]
- 76.Ferreiro CR, Chagas AC, Carvalho MH, Dantas AP, Scavone C, Souza LC, et al. Expression of inducible nitric oxide synthase is increased in patients with heart failure due to ischemic disease. Braz J Med Biol Res. 2004;37(9):1313–20. doi: 10.1590/s0100-879x2004000900005. [DOI] [PubMed] [Google Scholar]
- 77.Yang B, Larson DF, Watson RR. Modulation of iNOS activity in age-related cardiac dysfunction. Life Sci. 2004;75(6):655–67. doi: 10.1016/j.lfs.2003.09.076. [DOI] [PubMed] [Google Scholar]
- 78.Colombo PC, Banchs JE, Celaj S, Talreja A, Lachmann J, Malla S, et al. Endothelial cell activation in patients with decompensated heart failure. Circulation. 2005;111(1):58–62. doi: 10.1161/01.CIR.0000151611.89232.3B. [DOI] [PubMed] [Google Scholar]
- 79.Mungrue IN, Gros R, You X, Pirani A, Azad A, Csont T, et al. Cardiomyocyte overexpression of iNOS in mice results in peroxynitrite generation, heart block, and sudden death. J Clin Invest. 2002;109(6):735–43. doi: 10.1172/JCI13265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Heger J, Godecke A, Flogel U, Merx MW, Molojavyi A, Kuhn-Velten WN, et al. Cardiac-specific overexpression of inducible nitric oxide synthase does not result in severe cardiac dysfunction. Circ Res. 2002;90(1):93–9. doi: 10.1161/hh0102.102757. [DOI] [PubMed] [Google Scholar]
- 81.Gealekman O, Abassi Z, Rubinstein I, Winaver J, Binah O. Role of myocardial inducible nitric oxide synthase in contractile dysfunction and beta-adrenergic hyporesponsiveness in rats with experimental volume-overload heart failure. Circulation. 2002;105(2):236–43. doi: 10.1161/hc0202.102015. [DOI] [PubMed] [Google Scholar]
- 82.Jones SP, Greer JJ, Ware PD, Yang J, Walsh K, Lefer DJ. Deficiency of iNOS does not attenuate severe congestive heart failure in mice. Am J Physiol Heart Circ Physiol. 2005;288(1):H365–70. doi: 10.1152/ajpheart.00245.2004. [DOI] [PubMed] [Google Scholar]
- 83.Damy T, Ratajczak P, Shah AM, Camors E, Marty I, Hasenfuss G, et al. Increased neuronal nitric oxide synthase-derived NO production in the failing human heart. Lancet. 2004;363(9418):1365–7. doi: 10.1016/S0140-6736(04)16048-0. [DOI] [PubMed] [Google Scholar]
- 84.Dai ZK, Tan MS, Chai CY, Yeh JL, Chou SH, Chiu CC, et al. Upregulation of endothelial nitric oxide synthase and endothelin-1 in pulmonary hypertension secondary to heart failure in aorta-banded rats. Pediatr Pulmonol. 2004;37(3):249–56. doi: 10.1002/ppul.10413. [DOI] [PubMed] [Google Scholar]
- 85.Turko IV, Murad F. Protein nitration in cardiovascular diseases. Pharmacol Rev. 2002;54(4):619–34. doi: 10.1124/pr.54.4.619. [DOI] [PubMed] [Google Scholar]
- 86.Szabo C. Multiple pathways of peroxynitrite cytotoxicity. Toxicol Lett. 2003;140–141:105–12. doi: 10.1016/s0378-4274(02)00507-6. [DOI] [PubMed] [Google Scholar]
- 87.Virag L, Szabo E, Gergely P, Szabo C. Peroxynitrite-induced cytotoxicity: mechanism and opportunities for intervention. Toxicol Lett. 2003;140–141:113–24. doi: 10.1016/s0378-4274(02)00508-8. [DOI] [PubMed] [Google Scholar]
- 88.Hausladen A, Fridovich I. Superoxide and peroxynitrite inactivate aconitases, but nitric oxide does not. J Biol Chem. 1994;269(47):29405–8. [PubMed] [Google Scholar]
- 89.Virag L, Szabo C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54(3):375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- 90.Pacher P, Liaudet L, Bai P, Mabley JG, Kaminski PM, Virag L, et al. Potent metalloporphyrin peroxynitrite decomposition catalyst protects against the development of doxorubicin-induced cardiac dysfunction. Circulation. 2003;107(6):896–904. doi: 10.1161/01.cir.0000048192.52098.dd. [DOI] [PubMed] [Google Scholar]
- 91.Bianchi C, Wakiyama H, Faro R, Khan T, McCully JD, Levitsky S, Szabo C, Sellke FW. A novel peroxynitrite decomposer catalyst (FP-15) reduces myocardial infarct size in an in vivo peroxynitrite decomposer and acute ischemia-reperfusion in pigs. Ann Thorac Surg. 2002;74(4):1201–7. doi: 10.1016/s0003-4975(02)03953-x. [DOI] [PubMed] [Google Scholar]
- 92.Szabo C, Mabley JG, Moeller SM, Shimanovich R, Pacher P, Virag L, et al. Part I: pathogenetic role of peroxynitrite in the development of diabetes and diabetic vascular complications: studies with FP15, a novel potent peroxynitrite decomposition catalyst. Mol Med. 2002;8(10):571–80. [PMC free article] [PubMed] [Google Scholar]
- 93.Pacher P, Obrosova IG, Mabley JG, Szabo C. Role of nitrosative stress and peroxynitrite in the pathogenesis of diabetic complications. Emerging new therapeutical strategies. Curr Med Chem. 2005;12(3):267–75. doi: 10.2174/0929867053363207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Szabo G, Liaudet L, Hagl S, Szabo C. Poly(ADP-ribose) polymerase activation in the reperfused myocardium. Cardiovasc Res. 2004;61(3):471–80. doi: 10.1016/j.cardiores.2003.09.029. [DOI] [PubMed] [Google Scholar]
- 95.Soriano FG, Pacher P, Mabley J, Liaudet L, Szabo C. Rapid reversal of the diabetic endothelial dysfunction by pharmacological inhibition of poly(ADP-ribose) polymerase. Circ Res. 2001;89(8):684–91. doi: 10.1161/hh2001.097797. [DOI] [PubMed] [Google Scholar]
- 96.Pacher P, Liaudet L, Soriano FG, Mabley JG, Szabo E, Szabo C. The role of poly(ADP-ribose) polymerase activation in the development of myocardial and endothelial dysfunction in diabetes. Diabetes. 2002;51(2):514–21. doi: 10.2337/diabetes.51.2.514. [DOI] [PubMed] [Google Scholar]
- 97.Pacher P, Liaudet L, Mabley J, Komjati K, Szabo C. Pharmacologic inhibition of poly(adenosine diphosphateribose) polymerase may represent a novel therapeutic approach in chronic heart failure. J Am Coll Cardiol. 2002;40(5):1006–16. doi: 10.1016/s0735-1097(02)02062-4. [DOI] [PubMed] [Google Scholar]
- 98.Pacher P, Mabley JG, Soriano FG, Liaudet L, Szabo C. Activation of poly(ADP-ribose) polymerase contributes to the endothelial dysfunction associated with hypertension and aging. Int J Mol Med. 2002;9(6):659–64. [PubMed] [Google Scholar]
- 99.Pacher P, Mabley JG, Soriano FG, Liaudet L, Komjati K, Szabo C. Endothelial dysfunction in aging animals: the role of poly(ADP-ribose) polymerase activation. Br J Pharmacol. 2002;135(6):1347–50. doi: 10.1038/sj.bjp.0704627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pacher P, Mabley JG, Liaudet L, Evgenov OV, Marton A, Hasko G, et al. Left ventricular pressure-volume relationship in a rat model of advanced aging-associated heart failure. Am J Physiol Heart Circ Physiol. 2004;287(5):H2132–7. doi: 10.1152/ajpheart.00405.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pacher P, Vaslin A, Benko R, Mabley JG, Liaudet L, Hasko G, et al. A new, potent poly(ADP-ribose) polymerase inhibitor improves cardiac and vascular dysfunction associated with advanced aging. J Pharmacol Exp Ther. 2004;311(2):485–91. doi: 10.1124/jpet.104.069658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pacher P, Liaudet L, Bai P, Virag L, Mabley JG, Hasko G, et al. Activation of poly(ADP-ribose) polymerase contributes to development of doxorubicin-induced heart failure. J Pharmacol Exp Ther. 2002;300(3):862–7. doi: 10.1124/jpet.300.3.862. [DOI] [PubMed] [Google Scholar]
- 103.Bai P, Mabley JG, Liaudet L, Virag L, Szabo C, Pacher P. Matrix metalloproteinase activation is an early event in doxorubicin-induced cardiotoxicity. Oncol Rep. 2004;11(2):505–8. [PubMed] [Google Scholar]
- 104.Szenczi O, Kemecsei P, Holthuijsen MF, van Riel NA, van der Vusse GJ, Pacher P, et al. Poly(ADP-ribose) polymerase regulates myocardial calcium handling in doxorubicin-induced heart failure. Biochem Pharmacol. 2005;69(5):725–32. doi: 10.1016/j.bcp.2004.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pacher P, Cziraki A, Mabley JG, Liaudet L, Papp L, Szabo C. Role of poly(ADP-ribose) polymerase activation in endotoxin-induced cardiac collapse in rodents. Biochem Pharmacol. 2002;64(12):1785–91. doi: 10.1016/s0006-2952(02)01421-1. [DOI] [PubMed] [Google Scholar]
- 106.Pillai JB, Russell HM, Raman J, Jeevanandam V, Gupta MP. Increased expression of poly(ADP-ribose) polymerase-1 contributes to caspase-independent myocyte cell death during heart failure. Am J Physiol Heart Circ Physiol. 2005;288(2):H486–96. doi: 10.1152/ajpheart.00437.2004. [DOI] [PubMed] [Google Scholar]
- 107.Xiao CY, Chen M, Zsengeller Z, Li H, Kiss L, Kollai M, et al. Poly(ADP-Ribose) polymerase promotes cardiac remodeling, contractile failure, and translocation of apoptosis-inducing factor in a murine experimental model of aortic banding and heart failure. J Pharmacol Exp Ther. 2005;312(3):891–8. doi: 10.1124/jpet.104.077164. [DOI] [PubMed] [Google Scholar]
- 108.El Alaoui-Talibi Z, Guendouz A, Moravec M, Moravec J. Control of oxidative metabolism in volume-overloaded rat hearts: effect of propionyl-L-carnitine. Am J Physiol. 1997;272(4 Pt 2):H1615–24. doi: 10.1152/ajpheart.1997.272.4.H1615. [DOI] [PubMed] [Google Scholar]
- 109.Ungvari Z, Csiszar A, Kaminski PM, Wolin MS, Koller A. Chronic high pressure-induced arterial oxidative stress: involvement of protein kinase C-dependent NAD(P)H oxidase and local renin-angiotensin system. Am J Pathol. 2004;165(1):219–26. doi: 10.1016/S0002-9440(10)63290-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lei B, Lionetti V, Young ME, Chandler MP, d'Agostino C, Kang E, et al. aradoxical downregulation of the glucose oxidation pathway despite enhanced flux in severe heart failure. J Mol Cell Cardiol. 2004;36(4):567–76. doi: 10.1016/j.yjmcc.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 111.Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297(5579):259–63. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- 112.Du L, Zhang X, Han YY, Burke NA, Kochanek PM, Watkins SC, et al. Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J Biol Chem. 2003;278(20):18426–33. doi: 10.1074/jbc.M301295200. [DOI] [PubMed] [Google Scholar]
- 113.Chen M, Zsengeller Z, Xiao CY, Szabo C. Mitochondrial-to-nuclear translocation of apoptosis-inducing factor in cardiac myocytes during oxidant stress: potential role of poly(ADP-ribose) polymerase-1. Cardiovasc Res. 2004;63(4):682–8. doi: 10.1016/j.cardiores.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 114.Xiao CY, Chen M, Zsengeller Z, Szabo C. Poly(ADP-ribose) polymerase contributes to the development of myocardial infarction in diabetic rats and regulates the nuclear translocation of apoptosis-inducing factor. J Pharmacol Exp Ther. 2004;310(2):498–504. doi: 10.1124/jpet.104.066803. [DOI] [PubMed] [Google Scholar]
- 115.Wattanapitayakul SK, Weinstein DM, Holycross BJ, Bauer JA. Endothelial dysfunction and peroxynitrite formation are early events in angiotensin-induced cardiovascular disorders. FASEB J. 2000;14(2):271–8. doi: 10.1096/fasebj.14.2.271. [DOI] [PubMed] [Google Scholar]
- 116.Mihm MJ, Wattanapitayakul SK, Piao SF, Hoyt DG, Bauer JA. Effects of angiotensin II on vascular endothelial cells: formation of receptor-mediated reactive nitrogen species. Biochem Pharmacol. 2003;65(7):1189–97. doi: 10.1016/s0006-2952(03)00012-1. [DOI] [PubMed] [Google Scholar]
- 117.Szabo C, Pacher P, Zsengeller Z, Vaslin A, Komjati K, Benko R, Chen M, Mabley JG, Kollai M. Angiotensin II-mediated endothelial dysfunction: role of poly(ADP-ribose) polymerase activation. Mol Med. 2004;10(1–6):28–35. doi: 10.2119/2004-00001.szabo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Minchenko AG, Stevens MJ, White L, Abatan OI, Komjati K, Pacher P, et al. Diabetes-induced overexpression of endothelin-1 and endothelin receptors in the rat renal cortex is mediated via poly(ADP-ribose) polymerase activation. FASEB J. 2003;17(11):1514–6. doi: 10.1096/fj.03-0013fje. [DOI] [PubMed] [Google Scholar]
- 119.Gupte SA, Arshad M, Viola S, Kaminski PM, Ungvari Z, Rabbani G, et al. Pentose phosphate pathway coordinates multiple redox-controlled relaxing mechanisms in bovine coronary arteries. Am J Physiol Heart Circ Physiol. 2003;285(6):H2316–26. doi: 10.1152/ajpheart.00229.2003. [DOI] [PubMed] [Google Scholar]
- 120.Gupte SA, Kaminski PM, Floyd B, Agarwal R, Ali N, Ahmad M, et al. Cytosolic NADPH may regulate differences in basal Nox oxidase-derived superoxide generation in bovine coronary and pulmonary arteries. Am J Physiol Heart Circ Physiol. 2005;288(1):H13–21. doi: 10.1152/ajpheart.00629.2004. [DOI] [PubMed] [Google Scholar]