

Abstract
Cyclin-dependent kinase 2 (CDK2) has been proposed to function as a master regulator of centrosome duplication. Using mouse embryonic fibroblasts (MEFs) in which Cdk2 has been genetically deleted, we show here that CDK2 is not required for normal centrosome duplication, maturation and bipolar mitotic spindle formation. By contrast, Cdk2 deficiency completely abrogates aberrant centrosome duplication induced by a viral oncogene. Mechanistically, centrosome overduplication in MEFs wild-type for Cdk2 involves the formation of supernumerary immature centrosomes. These results indicate that normal and abnormal centrosome duplication have significantly different requirements for CDK2 activity and point to a role of CDK2 in licensing centrosomes for aberrant duplication. Furthermore, our findings suggest that CDK2 may be a suitable therapeutic target to inhibit centrosome-mediated chromosomal instability in tumor cells.
Centrosomes serve as major microtubule-organizing center in animal and human cells (Bornens, 2002). The single centrosome of a cell duplicates precisely once prior to mitosis in synchrony with the cell division cycle (Hinchcliffe & Sluder, 2001; Delattre & Gonczy, 2004). Abnormal centrosome numbers are detected in virtually all human cancers where they can contribute to abnormal mitotic spindle polarity and chromosomal instability (Nigg, 2002; Salisbury et al., 1999; Brinkley, 2001; Doxsey, 2002; Duensing, 2005). The molecular mechanisms that govern normal and aberrant centrosome duplication are incompletely understood. CDK2 has been reported to control centrosome duplication in various model systems in which repeated centrosome reproduction was induced by a prolonged S phase arrest (Hinchcliffe et al., 1999; Lacey et al., 1999; Matsumoto et al., 1999; Meraldi et al., 1999). Several targets downstream of CDK2 relevant for the control of centrosome duplication have been identified including nucleophosmin/B23 (Okuda et al., 2000), Mps1 (Fisk & Winey, 2001) and CP110 (Chen et al., 2002). However, Cdk2-null mice are viable and develop essentially normal (Berthet et al., 2003; Ortega et al., 2003). Cells derived from these animals have no apparent proliferation defect in culture and re-enter the cell division cycle after serum deprivation without significant delay (Ortega et al., 2003). In addition, cancer cells were found to proliferate despite CDK2 inhibition (Tetsu & McCormick, 2003). Based on these results, we designed the present study to reassess the role of CDK2 in normal and aberrant centrosome duplication.
To determine the role of CDK2 in the regulation of centrosome duplication, we used mouse embryonic fibroblasts (MEFs) in which Cdk2 has been genetically deleted. Primary mouse embryonic fibroblasts (MEFs) derived from Cdk2−/− mice (Ortega et al., 2003) did not have any detectable CDK2 protein expression (data not shown). MEFs were analyzed for centrosome numbers using immunofluorescence microscopy for γ-tubulin, a commonly used marker for centrosomes. The γ-tubulin staining pattern in Cdk2−/− MEFs was normal (Fig. 1A) and indistinguishable from cells wild-type for Cdk2. Quantification of centrosome numbers per cell revealed that 14.1% of CDK2−/− MEFs had one centrosome compared to 37.3% in Cdk2+/+ cells (Fig. 1B). In Cdk2−/− MEFs, 70.1% of cells showed duplicated centrosomes (i.e., two per cell) compared to 54.9% in Cdk2+/+ cells. The percentage of cells with three, four or more than four centrosomes was 4.7%, 7.7% and 1.9% in Cdk2−/− cells compared to 3.4%, 3.8% and 0.6% in Cdk2+/+ MEFs. The increased fraction of Cdk2−/− MEFs with two centrosomes and the simultaneous decrease of cells with one centrosome is likely related to the fact that CDK2-deficient cells have the tendency to enter culture crisis earlier than Cdk2 wild-type cells (Ortega et al., 2003). Senescent cells have been reported to undergo growth arrest in a tetraploid state (Mason et al., 2004) and may therefore contain two centrosomes. The mitotic index, however, was not found to differ significantly between Cdk2−/− MEFs and wild-type MEFs (2.6% in both populations). Importantly, more than 90% of mitoses showed a normal bipolar spindle pole arrangement in both Cdk2−/− and Cdk2+/+ MEFs indicating that centrosomes functioned normally with respect to their role in spindle pole formation (Fig. 1A).
Figure 1. CDK2-independent centrosome duplication and maturation in Cdk2−/− MEFs.
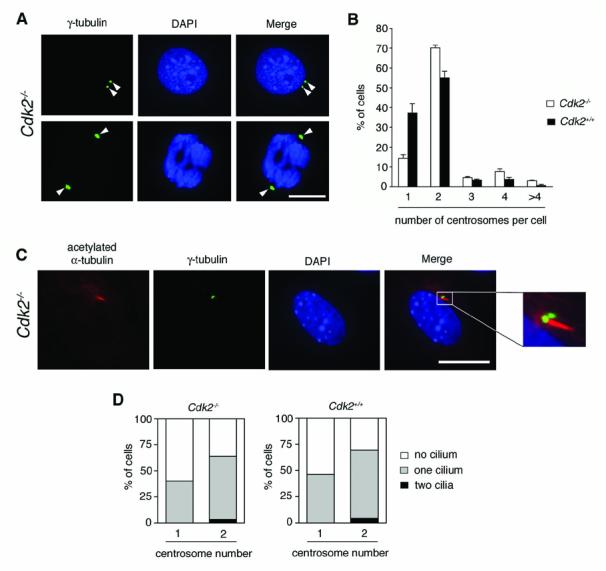
(A) Cdk2−/− MEFs were analyzed by immunofluorescence microscopy for γ-tubulin, a commonly used centrosome marker, to detect centrosomes (arrowheads) during interphase (top panels) and mitosis (bottom panels). Cells plated on coverslips were washed briefly in phosphate-buffered saline (PBS) and then fixed in 4% paraformaldehyde/PBS (pH 7.0) for 10 min at room temperature. Cells were permeabilized with 1% Triton-X 100 (Sigma) in PBS for 10 min at room temperature and non-specific staining was blocked with 10% normal donkey serum (Jackson Immunoresearch) in distilled water for 15 min at room temperature. Cells were then incubated with a primary mouse monoclonal anti-γ-tubulin antibody (Sigma) at a 1:1000 dilution in PBS overnight at 4°C followed by a FITC-conjugated donkey anti-mouse secondary antibody (Jackson Immunoresearch) at a 1:100 dilution in PBS for 2 h at 37°C. Cells were mounted with DAPI (Vector Laboratories) and analyzed using an Olympus AX70 epifluorescence microscope equipped with a SpotRT digital camera. Pictures were transferred to Adobe Photoshop for print-out. Scale bar equals 10 μm.
(B) Quantification of centrosome numbers following γ-tubulin staining in asynchronously growing Cdk2+/+ and Cdk2−/− MEFs. Each bar represents mean + standard error of three independent experiments and counts of at least one-hundred cells per experiment.
(C) Immunofluorescence microscopy for acetylated α-tubulin and γ-tubulin reveals the primary cilium-carrying centrosome in a Cdk2−/− MEF after serum-deprivation for 24 h. For detection of primary cilia, cells were first stained for γ-tubulin using a rabbit primary antibody (Santa Cruz) at a 1:100 dilution in PBS followed by a FITC-labeled donkey anti-rabbit secondary antibody (Jackson Immunoresearch) at a 1:200 dilution in PBS and then incubated with an anti-acetylated α-tubulin antibody (Zymed) at a 1:100 dilution in PBS followed by a Rhodamine Red-conjugated donkey anti-mouse secondary antibody (Jackson Immunoresearch) at a 1:500 in PBS. Cells were then processed as described above. Scale bar equals 10 μm.
(D) Quantification of primary cilium-carrying centrosomes in serum-starved Cdk2−/− and Cdk2+/+ MEFs in correlation to the total centrosome number per cell. Results from a representative experiment in which a total of n=186 cells were evaluated are shown.
Besides duplication, centrosomes undergo a process of maturation in order to become fully functional. Rodent fibroblasts grow a primary cilium specifically from the older, mature centriole of a centrosome under conditions of serum deprivation. A single, unduplicated centrosome therefore usually carries one primary cilium. Two primary cilia can be found at later stages of the G2 phase of the cell division cycle, when two centrosomes are present and each centrosome contains a centriole that has reached a certain level of maturation (Albrecht-Buehler & Bushnell, 1980; Alvarez-Salas et al., 1998; Wheatley et al., 1996). The analysis of primary cilium formation hence allows to evaluate the maturation status of centrosomes that are present in a cell (Guarguaglini et al., 2005). Primary cilia can be detected by immunostaining for acetylated α-tubulin. To analyze primary cilia in Cdk2−/− and Cdk2+/+ MEFs, cells were serum-deprived for 24 h to stimulate cilium formation followed by co-immunofluorescence analysis for γ-tubulin and acetylated α-tubulin (Fig. 1C). A single centrosome associated with one primary cilium was detected in 46.2% of Cdk2−/− and 40% of Cdk2+/+ MEFs, respectively (Fig. 1D). Duplicated centrosomes with one cilium-carrying centrosome were detected in 60.3% of Cdk2−/− cells and 65.2% of Cdk2+/+ MEFs, whereas two centrosomes each carrying a primary cilium were found in 3.9% of Cdk2−/− and 4.3% of Cdk2+/+ cells, respectively. Taken together, these results show that normal centrosome duplication and maturation are not affected by Cdk2 deficiency.
We next explored the possibility that ablation of Cdk2 may affect aberrant centrosome duplication. To stimulate centrosome overduplication, we used the E7 oncoprotein of human papillomavirus type 16 (HPV-16). HPV-16 E7 disrupts the retinoblastoma protein (pRB) tumor suppressor pathway through binding and degradation of pRB and the related p107 and p130 proteins as well as inactivation of the CDK inhibitors p21Cip1 and p27Kip1 (Munger & Howley, 2002). HPV-16 E7 rapidly stimulates aberrant centriole duplication and therefore predisposes cells to centrosome-related mitotic defects and chromosomal instability (Duensing et al., 2000). It has recently been shown that the HPV-16 E7 oncoprotein stimulates centriole overduplication through the increased formation of immature daughter centrioles (Guarguaglini et al., 2005).
Cdk2-deficient and Cdk2 wild-type MEFs transiently transfected with HPV-16 E7 did not differ significantly in the expression level of the E7 oncoprotein (Fig. 2A). To determine whether expression of HPV-16 E7 increases cyclin E- or cyclin A-associated kinase activities, an in vitro kinase assay was performed using a recombinant C-terminal fragment of pRB as a substrate. A modest increase of mainly cyclin E-associated kinase activity was detected in HPV-16 E7-transfected Cdk2+/+ cell populations but not in Cdk2-deficient MEFs (Fig. 2B). The relatively small changes in kinase activity are most likely related to the fact that experiments were performed after transient transfection of MEF populations that yielded transfection rates of approximately 5-10%.
Figure 2. CDK2 is required for centrosome overduplication induced by a viral oncogene.
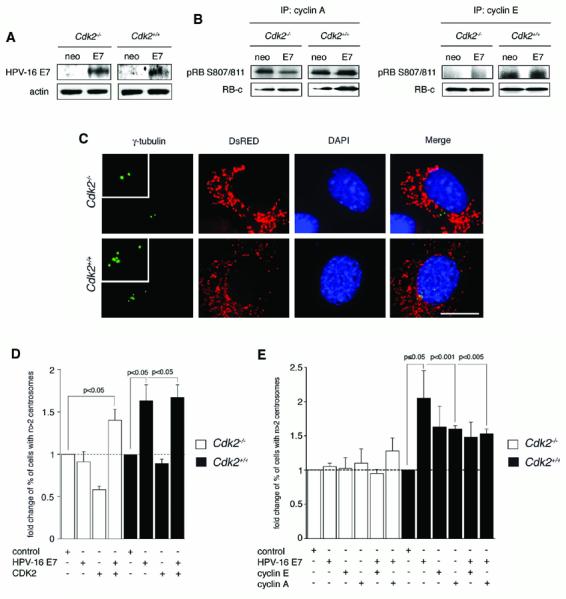
(A) Immunoblot analysis of HPV-16 E7 expression in Cdk2−/− and Cdk2+/+ MEFs compared to empty vector (neo) after transient transfection. Cells were transfected with 2 μg of plasmid DNA using Fugene 6 (Invitrogen) and processed for immunoblot analysis after 48 h. Whole cell lysates were prepared by scraping monolayer cells into ice-cold lysis buffer (1% NP-40, 50 mM Tris-Hcl pH 8.0, 100 mM sodium fluoride, 30 mM sodium pyrophosphate, 2 mM sodium molybdate, 5 mM EDTA, 2 mM sodium orthovanadate) containing 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM phenylmethylsulfonyl fluoride and additional sodium orthovanadate (2 mM). Lysates were incubated for 1 h with shaking at 4°C and then cleared by centrifugation for 30 min at 14,000 rpm at 4°C. SDS-gel electrophoresis and immunoblotting were performed using 4-12% Bis-Tris gels (Invitrogen) and a MiniTrans Blot Cell (Biorad Laboratories). Blot immunostains were detected by enhanced chemiluminescence (ECL, Amersham), exposed to X-ray film and developed in a Kodak X-OMAT 2000 film processor (Eastman Kodak). The pCDNA3-HPV-16 E7-HA construct was kindly provided by K. Munger (Harvard Medical School, Boston), empty pCDNA3-HA was kindly provided by Yuan Chang and Patrick Moore (University of Pittsburgh Cancer Institute, Pittsburgh).
(B) In vitro activity of cyclin A- (left panels) or cyclin E- (right panels) associated kinases following transient transfection of Cdk2−/− and Cdk2+/+ MEFs with HPV-16 E7 or empty vector (neo). Kinase reactions were performed with 375 μg of protein. Immunoprecipitations using agarose-conjugated cyclin A polyclonal antibodies (H-432, Santa Cruz) or cyclin E polyclonal antibodies (M2, Santa Cruz) were performed overnight at 4°C. Immunocomplexes were washed three times in lysis buffer (1% NP-40, 50 mM Tris-Hcl pH 8.0, 100 mM sodium fluoride, 30 mM sodium pyrophosphate, 2 mM sodium molybdate, 5 mM EDTA, 2 mM sodium orthovanadate) and three times in kinase buffer (10 mM MgCl2, 25 mM Tris HCl, 100 μM ATP, at pH 7.4). Samples were then resuspended in 20 μl of kinase buffer containing 0.5 μg of recombinant C-terminal fragment of the retinoblastoma protein (RB-c) comprising amino acids 773-928 (Upstate). Phosphorylation of RB-c was determined using a phospho-specific antibody against serine residues 807/811 (Cell Signaling). Total RB-c was measured using an antibody recognizing the C-terminus of pRB (4H1, Cell Signaling).
(C) Immunofluorescence analysis of Cdk−/− and Cdk2+/+ MEFs transiently transfected with HPV-16 E7. Cells were co-transfected with a plasmid encoding DsRED protein targeted to mitochondria (Clontech) at a 1:10 ratio to label transfected cells. Centrosomes were visualized by immunofluorescence staining for γ-tubulin using a mouse monoclonal antibody (see above). Nuclei were stained with DAPI. Scale bar equals 10 μm.
(D) Quantification of fold changes of the percentage of Cdk2−/− or Cdk2+/+ MEFs with abnormal centrosome numbers (i.e., more than two per cell) after transient transfection with HPV-16 E7 or CDK2 either alone or in combination. For cotransfection experiments, 1 μg of each plasmid DNA was used. The CDK2 expression construct was kindly provided by P. Hinds (Tufts University, Boston). Results were normalized to the mean percentage of cells with abnormal centrosome numbers in empty vector-transfected MEFs (control). Each bar indicates mean of fold changes and standard error obtained from at least three independent experiments and assessment of at least fifty DsRED-positive cells per experiment.
(D) Quantification of fold changes of the percentage of Cdk2−/− or Cdk2+/+ MEFs with abnormal centrosome numbers (i.e., more than two per cell) after transient transfection with cyclin E or cyclin A either alone or in combination with HPV-16 E7. For co-transfection experiments, 1 μg of each plasmid DNA was used. The cyclin E and cyclin A constructs were kindly provided by P. Hinds (Tufts University, Boston) Results were normalized to the mean percentage of cells with abnormal centrosome numbers in empty vector-transfected MEFs (control). Each bar indicates mean of fold changes and standard error obtained from at least three independent experiments and assessment of at least twenty-five DsRED-positive cells per experiment.
We next determined numerical centrosome aberrations in cells from both genotypes following transient transfection with HPV-16 E7 or empty vector control. This analysis was performed on a cell-per-cell basis using DsRED as marker for transfected cells (Fig. 2C). Fold-changes of the percentage of cells with abnormal centrosome numbers (i.e., more than two per cell) were calculated (Fig. 2D). HPV-16 E7-transfected Cdk2−/− MEFs showed no increase of abnormal centrosome numbers (0.9-fold) whereas Cdk2 wild-type MEFs showed a significant 1.6-fold increase of the proportion of cells with more than two centrosomes after transfection with HPV-16 E7 (p≤0.05, Student's t test for independent samples; Fig. 2D). Co-transfection of Cdk2−/− MEFs with HPV-16 E7 and a CDK2 expression plasmid compensated for Cdk2 deficiency and resulted in a significant 1.4-fold increase of the proportion of cells with numerical centrosome anomalies compared to controls (p≤0.05). This increase was similar to the increase detected in Cdk2+/+ MEFs transfected with HPV-16 E7 either alone or HPV-16 E7 in combination with CDK2 (1.7-fold; p≤0.05; Fig. 2D). Transient transfection of MEFs with CDK2 alone did not stimulate numerical centrosome aberrations in both Cdk2−/− or Cdk2+/+ cells (data not shown).
To determine whether cyclin E or cyclin A can rescue the HPV-16 E7-induced centrosome phenotype as CDK2 does, we transiently overexpressed cyclin E or cyclin A in Cdk2−/− and Cdk2+/+ MEFs either alone or in combination with HPV-16 E7 and analyzed centrosome numbers in transfected cells (Fig. 2E). Expression of cyclins was confirmed by immunoblotting (data not shown). Overexpression of cyclin E or cyclin A alone as well as co-expression of cyclin E or cyclin A with HPV-16 E7 did not lead to an increase of the proportion of cells with abnormal centrosome numbers in Cdk2−/− MEFs (Fig. 2E). A 1.3-fold increase detected in Cdk2−/− cells transfected with HPV-16 E7 and cyclin A did not reach statistical significance. In Cdk2 wild-type MEFs, cyclin A overexpression alone caused a statistically significant 1.6-fold increase of cells with abnormal centrosome numbers (p<0.001) and co-expression of cyclin A and HPV-16 E7 led to 1.5-fold increase of cells with abnormal centrosome numbers (p<0.005). Overexpression of cyclin E alone or in combination with HPV-16 E7 in Cdk2 wild-type MEFs stimulated a 1.6-fold and 1.5-fold increase of cells with abnormal centrosome numbers, respectively, that was in the range of cyclin A but did not reach statistical significance. Co-expression of cyclin A or cyclin E with HPV-16 E7 did not cause more centrosome abnormalities than overexpression of HPV-16 E7 alone (2.05-fold; p≤0.05). Collectively, these results show that overexpression of cyclin A or cyclin E cannot compensate for CDK2 loss with respect to HPV-16 E7-induced aberrant centrosome duplication.
To rule out the possibility that Cdk2−/− MEFs have adapted to the loss of CDK2 expression by complementing mutations, small-interfering RNA (siRNA) experiments were performed using Cdk2 wild-type MEFs and mouse-specific siRNA duplexes targeting CDK2 (Fig. 3). Depletion of CDK2 protein (Fig. 3A) was found to abolish the ability of HPV-16 E7 to induce abnormal centrosome numbers (Fig. 3B) without stimulating an increased cell death or growth arrest. These findings further support the notion that CDK2 is required for the induction of abnormal centrosome duplication.
Figure 3. Small-interfering RNA (siRNA)-mediated knock-down of CDK2 prevents HPV-16 E7-induced abnormal centrosome duplication.
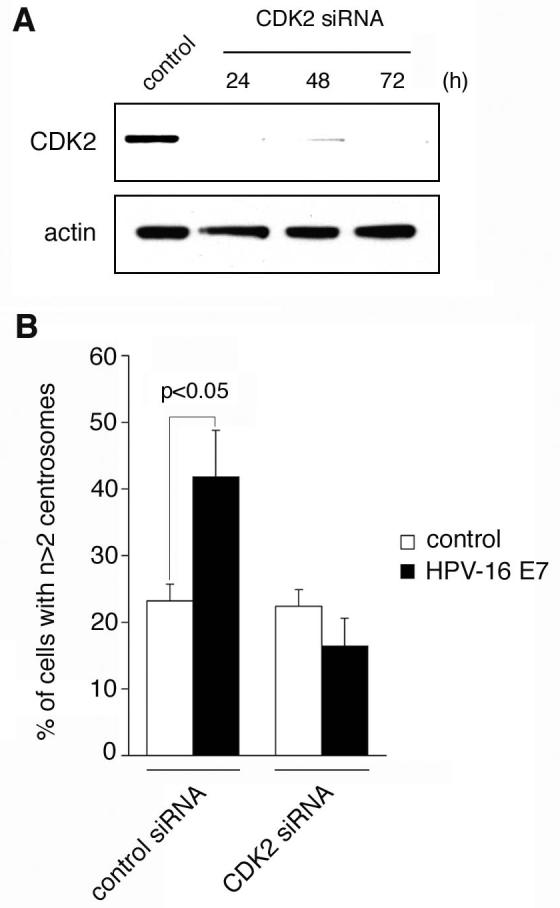
(A) Immunoblot analysis of wild-type MEFs for CDK2 expression following transfection with siRNA duplexes targeting CDK2 or control siRNAs. Two synthetic oligonucleotides targeting mouse CDK2 were used (5'-GGUGUACCCAGUACUGCCA-3' and 5'-GGAACUUAAUCACCCUAAU-3'; Silencer™, Ambion). Pre-designed control oligonucleotides were used as negative control (Silencer™ Negative, Ambion). Wild-type MEFs grown on coverslips in 60 mm plates with 2 ml DMEM free of antibiotics were transfected with 5 μl of 20 μM annealed duplexes using nucleofection (Amaxa). Whole cell lysates were prepared at the indicated time points and analyzed by immunoblotting as described above. Immunoblot for actin is shown to demonstrate loading of equal amounts of protein.
(B) Quantification of the percentage of MEFs with abnormal centrosome numbers following siRNA-directed knock-down of CDK2 protein or transfection with control duplexes and transient transfection with HPV-16 E7 or empty vector. Cells were transfected with siRNAs as described above and transfected with HPV-16 E7 or controls plasmids after 24 h. Cells were co-transfected with DsRED and processed for immunofluorescence microscopy for γ-tubulin after additional 48 h. Mean and standard errors of at least three independent experiments with at least 50 DsRED-positive cells counted per experiment are shown.
We next sought to determine whether aberrant centrosome numbers induced by HPV-16 E7 in Cdk2+/+ MEFs was caused by an increased generation of immature centrosomes. Cdk2+/+ MEFs transiently transfected with HPV-16 E7 were serum-deprived in order to stimulate the formation of primary cilia (Fig. 4). In a representative experiment, Cdk2+/+ MEFs with abnormal centrosome numbers after transient transfection with the HPV-16 E7 oncoprotein contained an average of 3.3 centrosomes in the presence of an average of 1.0 cilium-carrying centrosome per cell. In Cdk2+/+ MEFs without centrosome overduplication (one or two centrosomes per cell), an average of 0.8 cilium-carrying centrosomes was detected. These results underscore that HPV-16 E7 induces a bona fide centrosome overduplication in Cdk2+/+ MEFs as opposed to an accumulation of centrosomes, which would result in an increase of mature centrioles carrying primary cilia.
Figure 4. CDK2 mediates excessive generation of immature centrosomes induced by the HPV-16 E7 oncoprotein.
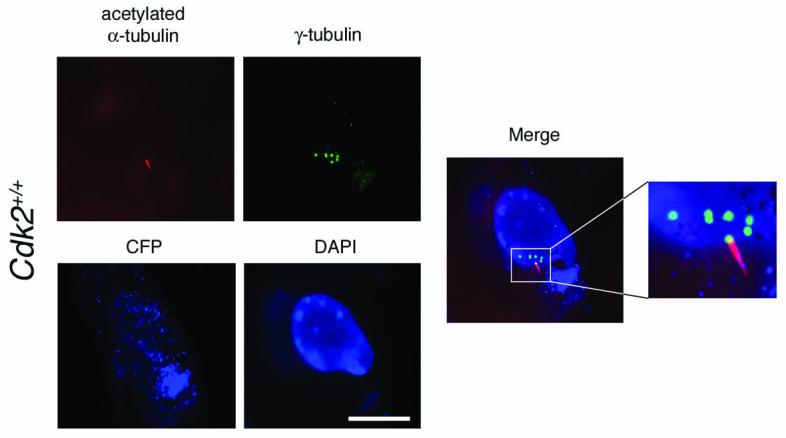
Immunofluorescence microscopy analysis for γ-tubulin and acetylated α-tubulin (see above) demonstrates multiple immature centrosomes in the presence of a single primary cilium-carrying centrosome in a HPV-16 E7-transfected cell. Cdk2+/+ MEFs were transiently transfected with HPV-16 E7 using cyan fluorescent protein (CFP) targeted to mitochondria (Clontech) at a 1:10 ratio as transfection marker. Pictures were taken from a representative experiment in which a total of n=53 transfected Cdk2+/+ MEFs were evaluated. Nucleus stained with DAPI. Scale bar equals 10 μm.
Collectively, our findings provide the first definitive demonstration of CDK2-independent centrosome duplication in somatic mammalian cells. Although CDK2 is not required for normal centrosome duplication, we demonstrate that CDK2 is a mediator of centrosome overduplication induced by the HPV-16 E7 oncoprotein.
In previous studies, small molecule CDK2 inhibitors were successfully used to interfere with centrosome duplication and it is important to mention that one inhibitor (Purvalanol A) was found to block normal centrosome duplication (Wong & Stearns, 2003) whereas another CDK inhibitor (Indirubin-3'-oxime) inhibited centrosome overduplication while leaving normal duplication unaffected (Duensing et al., 2004b). CDK inhibitors can differ significantly in both the spectrum of inhibited protein kinases and the inhibitory activity against individual kinases (Meijer et al., 1999). Since our results suggest that the activity of other kinases including cyclin A-associated CDK activity may compensate for the loss of CDK2 to allow normal centrosome duplication (Fig. 1), it is likely that differences in the activity spectrum of the small molecule inhibitors account for the observed discrepancies.
Cyclin A has previously been reported to be more effective than cyclin E in promoting aberrant centrosome duplication (Meraldi et al., 1999). We find a similar trend, although it is notable that cyclin E can induce abnormal centrosome numbers in Cdk2 wild-type cells to a similar extent as cyclin A, albeit without reaching statistical significance (Fig. 2E). Cyclin E is known to interact only with CDK2 and overexpression of cyclin E has been reported to stimulate normal centrosome duplication and aberrant centrosome numbers in p53-deficient cells (Mussman et al., 2000). We have previously shown that MEFs deficient of cyclins E1 and E2 maintain normal centrosome duplication (Duensing et al., 2004a) suggesting that CDK2-independent functions of cyclin E (Matsumoto & Maller, 2004) may play a role in the observed effects on the normal centrosome duplication cycle.
Centrosomes do not have unlimited capacity to reproduce during a normal duplication cycle. In fact, duplicated centrosomes are endowed with an intrinsic block to reduplicate even in a cellular milieu that normally supports their reproduction (Wong & Stearns, 2003). Therefore, our finding that CDK2 is necessary for centrosome overduplication may point to a role of CDK2 in licensing centrosomes for aberrant duplication. This is supported by the presence of multiple immature centrosomes in cells with HPV-16 E7-induced centrosome amplification (Fig. 4). Our study does not disagree with previous reports showing CDK2 to promote multiple rounds of centrosome duplication upon S phase arrest (Hinchcliffe et al., 1999; Lacey et al., 1999; Matsumoto et al., 1999; Meraldi et al., 1999) since it is conceivable that under such conditions the intrinsic block to reduplicate also needs to be relaxed. Whether and to what extent deregulated CDK2 activity drives centrosome overduplication and chromosome missegregation in human cancers remains to be determined. If such a function can be substantiated, our results suggest that CDK2 would be a suitable drug target to inhibit centrosome-mediated chromosomal instability in cancer patients.
Acknowledgments
We are grateful to K. Münger, P. Hinds, Y. Chang and P. Moore for sharing reagents. This work was supported by NIH/NCI grant CA112598 (to S.D.).
References
- Albrecht-Buehler G, Bushnell A. Exp Cell Res. 1980;126:427–37. doi: 10.1016/0014-4827(80)90282-7. [DOI] [PubMed] [Google Scholar]
- Alvarez-Salas LM, Cullinan AE, Siwkowski A, Hampel A, DiPaolo JA. Proc Natl Acad Sci U S A. 1998;95:1189–94. doi: 10.1073/pnas.95.3.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthet C, Aleem E, Coppola V, Tessarollo L, Kaldis P. Curr Biol. 2003;13:1775–85. doi: 10.1016/j.cub.2003.09.024. [DOI] [PubMed] [Google Scholar]
- Bornens M. Curr. Opin. Cell Biol. 2002;14:25–34. doi: 10.1016/s0955-0674(01)00290-3. [DOI] [PubMed] [Google Scholar]
- Brinkley BR. Trends Cell Biol. 2001;11:18–21. doi: 10.1016/s0962-8924(00)01872-9. [DOI] [PubMed] [Google Scholar]
- Chen Z, Indjeian VB, McManus M, Wang L, Dynlacht BD. Dev Cell. 2002;3:339–350. doi: 10.1016/s1534-5807(02)00258-7. [DOI] [PubMed] [Google Scholar]
- Delattre M, Gonczy P. J Cell Sci. 2004;117:1619–1630. doi: 10.1242/jcs.01128. [DOI] [PubMed] [Google Scholar]
- Doxsey S. Mol Cell. 2002;10:439–40. doi: 10.1016/s1097-2765(02)00654-8. [DOI] [PubMed] [Google Scholar]
- Duensing A, Geng Y, Sicinski P, Munger K, Duensing S. Proc Am Assoc Cancer Res. 2004a;45:2596. [Google Scholar]
- Duensing S. Cell Biol Int. 2005;29:352–359. doi: 10.1016/j.cellbi.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Duensing S, Duensing A, Lee DC, Edwards KM, Piboonniyom S, Manuel E, Skaltsounis L, Meijer L, Munger K. Oncogene. 2004b;23:8206–8215. doi: 10.1038/sj.onc.1208012. [DOI] [PubMed] [Google Scholar]
- Duensing S, Lee LY, Duensing A, Basile J, Piboonniyom S, Gonzalez S, Crum CP, Munger K. Proc Natl Acad Sci U S A. 2000;97:10002–7. doi: 10.1073/pnas.170093297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisk HA, Winey M. Cell. 2001;106:95–104. doi: 10.1016/s0092-8674(01)00411-1. [DOI] [PubMed] [Google Scholar]
- Guarguaglini G, Duncan PI, Stierhof YD, Holmstrom T, Duensing S, Nigg EA. Mol Biol Cell. 2005;16:1095–107. doi: 10.1091/mbc.E04-10-0939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinchcliffe EH, Li C, Thompson EA, Maller JL, Sluder G. Science. 1999;283:851–4. doi: 10.1126/science.283.5403.851. [DOI] [PubMed] [Google Scholar]
- Hinchcliffe EH, Sluder G. Genes Dev. 2001;15:1167–81. doi: 10.1101/gad.894001. [DOI] [PubMed] [Google Scholar]
- Lacey KR, Jackson PK, Stearns T. Proc Natl Acad Sci U S A. 1999;96:2817–22. doi: 10.1073/pnas.96.6.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason DX, Jackson TJ, Lin AW. Oncogene. 2004;23:9238–46. doi: 10.1038/sj.onc.1208172. [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Hayashi K, Nishida E. Curr Biol. 1999;9:429–32. doi: 10.1016/s0960-9822(99)80191-2. [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Maller JL. Science. 2004;306:885–8. doi: 10.1126/science.1103544. [DOI] [PubMed] [Google Scholar]
- Meijer L, Leclerc S, Leost M. Pharmacol Ther. 1999;82 doi: 10.1016/s0163-7258(98)00057-6. [DOI] [PubMed] [Google Scholar]
- Meraldi P, Lukas J, Fry AM, Bartek J, Nigg EA. Nat Cell Biol. 1999;1:88–93. doi: 10.1038/10054. [DOI] [PubMed] [Google Scholar]
- Munger K, Howley PM. Virus Res. 2002;89:213–228. doi: 10.1016/s0168-1702(02)00190-9. [DOI] [PubMed] [Google Scholar]
- Mussman JG, Horn HF, Carroll PE, Okuda M, Tarapore P, Donehower LA, Fukasawa K. Oncogene. 2000;19:1635–1646. doi: 10.1038/sj.onc.1203460. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Nature Rev Cancer. 2002;2:1–11. doi: 10.1038/nrc924. [DOI] [PubMed] [Google Scholar]
- Okuda M, Horn HF, Tarapore P, Tokuyama Y, Smulian AG, Chan PK, Knudsen ES, Hofmann IA, Snyder JD, Bove KE, Fukasawa K. Cell. 2000;103:127–40. doi: 10.1016/s0092-8674(00)00093-3. [DOI] [PubMed] [Google Scholar]
- Ortega S, Prieto I, Odajima J, Martin A, Dubus P, Sotillo R, Barbero JL, Malumbres M, Barbacid M. Nat Genet. 2003;35:25–31. doi: 10.1038/ng1232. [DOI] [PubMed] [Google Scholar]
- Salisbury JL, Whitehead CM, Lingle WL, Barrett SL. Biol Cell. 1999;91:451–60. [PubMed] [Google Scholar]
- Tetsu O, McCormick F. Cancer Cell. 2003;3:233–245. doi: 10.1016/s1535-6108(03)00053-9. [DOI] [PubMed] [Google Scholar]
- Wheatley DN, Wang AM, Strugnell GE. Cell Biol Int. 1996;20:73–81. doi: 10.1006/cbir.1996.0011. [DOI] [PubMed] [Google Scholar]
- Wong C, Stearns T. Nat Cell Biol. 2003;5:539–544. doi: 10.1038/ncb993. [DOI] [PubMed] [Google Scholar]