

Modeling of biological ion channels has a long history, going back more then 100 yr (Hille 1984). In the old, premolecular biology era, interpretation of these simple models provided the primary source of information about channel structure. As molecular biology and, now, x-ray diffraction have provided direct information about structure, modeling has been reduced to playing more of a secondary role in channel biochemistry. The early channel models were always understood to be highly simplified approximations that, hopefully, captured the “essential” features of the channel. The combination of atomic resolution channel structures plus highly sophisticated computer routines raises the possibility of obtaining “exact” (molecular dynamic) solutions for channel flux—something that would have seemed a wild dream only a few years ago. Such solutions have the purpose of interpreting and relating the channel structure to its function. As will be discussed below, although this exact solution is still beyond our reach, a combination of currently available approaches now make it possible to obtain solutions that approach this ideal. In this review I will attempt to illustrate the various approaches and approximations that are currently being used to model ion channels by starting with this exact solution, and then describing the series of assumptions that are made as one progresses to simpler and simpler models. No attempt is made to provide a comprehensive review of each of the approximations and the few specific examples that are referred to were chosen simply to provide illustrations of the different approaches (see Jakobsson, 1998, for a recent review).
Exact Molecular Dynamic Solution
This approach is made possible by the availability of channel structures that are known to atomic resolution. Until recently, the only channel that met this condition was the relatively simple gramicidin channel. However, with structures now available for the porin channel (Kreusch and Schulz, 1994), potassium channel (Doyle et al., 1998), iron transporting channel (Ferguson et al., 1998), and a mechanosensitive channel (Chang et al., 1998) (all from bacteria), this approach now has the potential to be applied to these biologically interesting channels. The basic idea is quite simple. First, one assembles an initial atomic model of the channel protein, the channel water, a nearby region of the channel lipid membrane, and samples of the bulk water at both ends of the channel. One then places some ions in the water, sets the temperature, and applies a voltage or concentration gradient and directly measures the ion flux as a function of time as the exact atomic dynamics of the model are simulated on the computer. At each time step in the simulation, all the forces on each atom are calculated (covalent, fixed charges, dipolar, etc.) and the atom moves under this force for a time period short enough that the forces should remain constant.
Unfortunately (or, fortunately, if you want to justify the use of other models), this direct approach is still beyond our current computational limits. The simulation must be run for a time period that is at least long enough to observe an ion crossing the channel. In fact, since the ion can take a large variety of paths through the channel and since the specific interactions with the channel and the other ions will vary randomly during the crossing, one should sample a minimum of 10 such crossings to estimate the channel conductance. For a channel with a conductance of 50 pS and an applied voltage of 100 mV, 10 crossings would take about 0.4 μs. For comparison, a recent simulation of the porin trimer channel OmpF (consisting of 1,020 amino acid residues, 300 phosphatidyl choline molecules, 12,992 water molecules, and 27 sodium atoms) required 2 h of computer time for 1 ps of real time (Tieleman and Berendsen, 1998). Thus, the 0.4 μs of real time needed to simulate the 10 ion crossings would require ∼100 yr of computer time. This calculation is for the ideal case where the entire channel and part of the lipid are included in the simulation. Computer times could be reduced by simulating only the protein residues lining the channel and replacing the rest of the protein and lipid by some continuum approximation. For example, in the molecular dynamic (MD) simulation of the acetylcholine channel (Sansom et al., 1998), only the M2 helix bundles were directly simulated and the computer time was reduced to ∼1 yr for a single ion crossing. Because porin has such a high conductance, Suenaga et al. (1998) were able to observe a single Na+ ion crossing the channel during a 1.3-ns MD simulation of a reduced porin model. Given the projected improvements in computational speed, such direct MD simulations should become possible within a few years.
These numerical calculations also have some other limitations, beyond that of the computer time. The most serious problem is a fundamental limitation in the accuracy of the atomic force constants. For example, if one wants to estimate an ion channel conductance accurate to within a factor of 7, one needs to be able to determine the interaction energy of the ion and channel to within a factor of ∼2 kT. This is less than half the energy in a single hydrogen bond! Second and third order effects (polarization, etc.) that are usually neglected in molecular dynamic calculations can easily lead to errors many times this size. In addition, although the models of the water molecules that are used in these calculations have been carefully refined and tested, they still are not likely to be accurate enough for the purposes of calculating ion channel flux. These fundamental limitations are clearly illustrated by the recent MD studies of gramicidin. Because of its small size and known atomic structure, gramicidin has, historically, been the proving ground for ion channel models. It is the channel for which any new MD approach or extension is first tested. It presents a particularly rigorous test of these models because the small uniform channel radius (≈3 Å) means that the ion directly interacts with the channel wall along its entire length (≈30 Å). Most importantly, there is direct experimental NMR information about the specific localized binding sites of ions in the channel. Woolf and Roux (1997) attempted to predict these NMR results using MD simulations. They used a fully solvated gramicidin-dimyristoyl phosphatidylcholine model and modified the standard CHARMM (Brooks et al., 1983) force field to include first and second order polarization effects of the ion on the peptide. Despite the fact that this MD model was optimized to fit gramicidin, the theoretical predictions of the ion binding sites still differed significantly from the experimental results. Accurate predictions of the ion binding locations require that the ion free energy as a function of position be calculated to an accuracy of a few tenths of a kilocalorie per mole (Woolf and Roux, 1997), and this is beyond the limits of current MD calculations.
Despite these current limitations, MD simulations still have an important function in modeling ion channels. From a cursory survey of the current literature, they are now the most popular approach to the theoretical investigation of ion channels. Simulations for short times (10 ps) can be used to obtain information about the local potential energy and diffusion coefficient of the ion as a function of position in the channel (Smith and Sansom, 1998; Tieleman and Berendsen, 1998; Zhong et al., 1998). This information can then be used in combination with Brownian dynamics or Poisson-Nernst-Planck theory (see below) to estimate the channel flux. Molecular dynamics is becoming a relatively routine procedure thanks to the availability of extremely sophisticated software (AMBER, GROMOS, CHARMM, XPLOR). Although MD calculations of flux in ion channels now require some special modifications of these routines, it is likely that some user-friendly interface will soon be developed to bring these calculations to the masses.
Three-Dimensional Brownian Dynamics Approach
The above discussion makes it clear that some simplifying assumptions are required. In the approach described in this section, it is assumed that the protein structure is held fixed and the water molecules are replaced by a continuum. With these assumptions, the three-dimensional (3-D) movement of ion i can be described by the following simple equation:
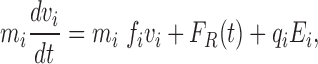 |
1 |
where m i, v i, q i and f i are the mass, velocity, charge, and frictional coefficient on the ith ion, respectively. F R is a random thermal force representing the effects of collisions with the water and channel wall. E i is the total electrical field on the ion, including the partial charges in the protein, all the other ions in the system, and the induced charges from the variation in the dielectric constant at the boundaries between the protein, water, and lipid. It is assumed in Eq. 1 that the only significant forces on the ion are long range electrical forces plus some kind of short range scattering condition when the ion contacts the hard sphere radius of the channel wall. Although one could always add other short-range specific force terms, this would, in effect, be adding an empirical term that did not arise directly from the known protein structure. The solution for this approach proceeds as in the above molecular dynamics method. The channel boundaries are defined, all the ions in the channel and attached bulk reservoirs are positioned, and then, for each ion i, Eq. 1 is integrated in discrete time steps (Brownian dynamics). Because the dynamics of the water and protein are no longer included and relatively long time steps can be taken for the ion motion, this approach is many orders of magnitude faster than the exact molecular dynamics approach (see below for a specific example).
The ability to accurately account for the interaction between ions in the channel system is one of the most difficult and critical aspects of modeling ion channels. In the absence of such interactions, the channel conductance will vary linearly with the ion concentration. Since almost all ion channels show some nonlinearity (e.g., saturation) in the physiological concentration range, it is essential that this interaction be accurately modeled. A major advantage of this Brownian dynamic approach is that it allows a direct simulation of this ion–ion interaction. At each step in the dynamics, the position of all the ions in the channel system are determined and their interaction energy (E i) is calculated for the next time step. One difficulty with this approach is that, because of the induced charges at the membrane and channel water interface, the calculation of the electrostatic energy (E i) at each step requires an involved, time-consuming calculation. S.H. Chung's group has recently described two three-dimensional Brownian dynamics (BD) simulations that use exact 3-D electrostatic potentials (Chung et al., 1998; Li et al., 1998). In the first calculation, Li et al. (1998) used an idealized, unrealistic channel model geometry that allowed an analytical solution for E i. It provides an interesting analysis of the effects of allowing the ion to wander over the entire mouth and pore region of the channel area, rather then be constrained to the channel axis as in the standard 1-D solutions. The second calculation was for an idealized acetylcholine receptor channel (ACHR) (Chung et al., 1998). For this case, it was necessary to first use a numerical procedure to solve for the electric field and potential on a grid. This data was then stored in a lookup table that was used during the BD simulation of a channel that was in contact with reservoirs large enough to hold 52 ions. A 1-μs real time simulation of this ACHR channel required only 18 h of computer time, making this approach four to five orders of magnitude faster than exact MD simulations.
It is now generally recognized that a combination of MD and BD provides the best available approach to modeling the ion conductance of channels whose atomic structure is known. In this combination approach, local MD simulations are carried out for different positions of the ion. These calculations are then used to determine the local diffusion coefficient and the perturbation of the local channel structure (e.g., main chain and carbonyl shifts) induced by the ion. This local value for the diffusion coefficient is then used to determine the frictional coefficient (f i, Eq. 1) and the perturbed structure is used for the fixed channel structure and both are varied as a function of the position of the ion in the BD calculations.
Three-Dimensional Poisson-Nernst-Planck Approach
The next simplifying approximation is to keep Eq. 1, but replace the exact expression for E i by a mean field approximation Ei that represents a sort of average over all the possible positions of the other ions in the system. This E is calculated using Poisson's equation. This combination of random thermal motion of the ion combined with a Poisson solution for E is referred to as the Poisson-Nernst-Plank solution (PNP). Although most PNP solutions have been for the 1-D case (see below), a general 3-D PNP solver has recently been described (Kurnikova et al., 1999). The 3-D steady state Nernst-Planck equation is given by:
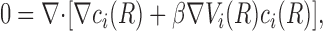 |
2 |
where R is the 3-D position vector, c i is the concentration of the ith ion, β = 1/kT and Vi is total potential energy of the ith ion and is described by:
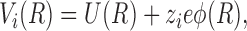 |
3 |
where U(R) is the potential due to nonelectrostatic forces, φ is the electrostatic potential, z i is the valence of the ith ion, and e is the electron charge. The value of φ is then determined from the solution to the 3-D Poisson equation. Given the concentration and potential on the boundaries in the bulk solution, these equations have a unique interior solution for c i from which the channel flux of ion i can be obtained. PNP is much faster than BD because it replaces the simulation of the ion movement at a sequence of time steps by a global numerical solution of a differential equation. A unique feature of the approach of Kurnikova et al. (1999) is that the solution to Eqs. 2 and 3 is combined with the standard Poisson-Boltzmann Equation solver Delphi (Nicholls et al., 1990), which is routinely used to solve for the electrostatic potentials in proteins.
This solver was tested using the gramicidin channel (Kurnikova et al., 1999). A long standing question about gramicidin has been the origin of its cation selectivity. Since the channel is uncharged, this selectivity presumably arises from partial dipolar charges in the channel. This is now a classical problem that has been studied by a large number of investigators and it has been a challenge to obtain theoretical results that are in good agreement with experiment without imposing some arbitrary adjustable parameters. For example, in the molecular dynamics study of Roux et al. (1995) and the reduced dynamic model of Dorman et al. (1996), special care and adjustments had to be made to get the models to agree with experimental results. Both of these models are significantly more complicated than the PNP model of Kurnikova et al. (1999), which was applied to the gramicidin channel without any modifications. (Kurnikova et al., 1999, did arbitrarily assume a channel diffusion coefficient of 1.27 × 10−6 cm2/s to fit the data). In general, the results of this 3-D PNP solution for gramicidin were in remarkably good agreement with the experimental results except, possibly, for the location of one of the high affinity sites in the channel. This PNP result, which uses just the fixed, ion-free gramicidin structure, is surprising because the molecular dynamic calculations indicated that it was essential to include local ion-induced peptide (carbonyl) perturbations in channel structure to explain the qualitative features of the gramicidin channel. In any case, this PNP result is very impressive considering that it represents a direct application of this general “off the shelf” model.
There are clearly some limitations to this 3-D PNP approach. The use of a mean field approximation (Poisson equation) is clearly a major simplification. It reduces the specific ion–ion interaction to an interaction between the ion and this mean field. It is difficult to quantitate the accuracy of this approximation, and its range of validity is the subject of considerable debate (Jakobsson, 1998). Mean field theories have a habit of working better than one might expect (e.g., the Debye-Huckel theory), and the only way to test them is by comparison with theoretical models of higher accuracy; e.g., BD. Despite its obvious limitations, the 3-D PNP solver of Kurnikova et al. (1999) has the potential to become the routine, quick first approach for estimating the conductance of a channel whose atomic structure is known or can be estimated. All the user of this software would need to do is to input the atom coordinates, the bulk concentrations and applied potential, and the program would then output the fluxes of the different ions.
One-Dimensional Brownian Dynamic and Poisson-Nernst-Plank Approaches
The next major simplification is to replace the 3-D concentration function used above by a 1-D concentration averaged over the radial cross section of the channel. As with the use of the mean field approximation, it is difficult to quantitate this 1-D assumption, and its range of validity is not known. In general, with each additional assumption that has been described here, the model results become more empirical and less related to the detailed knowledge of the channel structure. However, if the channel structure is not known to atomic resolution, which is still the usual case, then the error introduced by these approximations is probably of the same order as the uncertainty in the structure, and there is no advantage in using more complicated models.
As with the 3-D case, the 1-D BD approach has the major advantage that it allows for accurate modeling of ion–ion interactions. Despite this apparent theoretical advantage, there are not many examples of the use of this approach. One of the most detailed is the simulation of a multiply occupied cation channel by Bek and Jakobsson (1994). These authors were only interested in the qualitative features of the solution, and did not try to model the real channel electrostatic potential terms. One problem that limits the accuracy of this approach is the difficulty of simulating the 3-D bulk solutions by an equivalent 1-D model.
The most complete application of the 1-D PNP model to ion channels is the modeling of the acetylcholine receptor channel by Levitt (1991a,b). In this solution, realistic channel geometry and electrostatics were used and the cation and anion flux as a function of varying channel partial charges was obtained. This solution is appealing because just the fundamental structural features of the channel are used, and there is a minimum of adjustable parameters. The resulting ion flux was in good agreement with the experimental data.
Recently, Nonner and Eisenberg (1998) have used this approach to model L-type calcium channels. Their solution does not attempt to rigorously model the channel geometry or electrostatics, but rather is used to illustrate the general features that are required to fit the flux data for these calcium channels. It had previously been assumed that calcium channel kinetics required direct interactions between two or more ion binding sites. The results of Nonner and Eisenberg (1998) demonstrated that a simple PNP model could qualitatively reproduce many of the experimental calcium channel properties. An essential feature of this solution is the addition of a local, nonelectrostatic ion– channel interaction (Nonner and Eisenberg, 1998). For example, to fit the calcium channel data, the channel was assumed to have a relatively high affinity site for calcium relative to sodium that could not be explained in terms of the partial charges in the channel. This is a semi-empirical correction factor and has the disadvantage that the channel behavior can no longer be understood in terms of its well understood long range electrostatic properties. Of course, it is possible that some nonelectrostatic ion–channel interactions are important in determining channel behavior. One potential problem in using a large local attractive potential is that there is nothing in the standard PNP theory to prevent the local ion concentration from reaching the unphysical concentration of more than one ion per hard sphere ion volume. In the approach of Levitt (1991), a simple modification of the PNP theory was introduced to correct for this problem.
As with the 3-D PNP approach, the 1-D PNP solution uses an approximation to the direct ion–ion interaction whose validity is difficult to evaluate. It certainly can reproduce some aspects of this interaction, as is evidenced by its ability to mimic the concentration dependence of the conductance for the calcium (Nonner and Eisenberg, 1998) and acetylcholine receptor channels (Levitt, 1991). Levitt (1987) has described a modification of the PNP solution to allow for direct interaction between two ions. Although the approach seems to provide a more accurate way to treat ion–ion interaction, it does so at the cost of a large increase in mathematical complexity.
Reaction-Rate Approach
In this final simplification, it is assumed that the ions are localized in specific regions of the channel, and the kinetics are represented by the rate constants for jumping between these regions and between these regions and the bulk solution. This approximation obviously represents a gross simplification that reduces the channel kinetics to its bare fundamentals. The reaction-rate (RR) model should be classified as a simplification of BD rather than PNP because it does not use the mean field assumption. In fact, the RR approximation is ideally suited for modeling strong ion–ion interactions. There has been a long-standing debate (Levitt, 1986) centered on the issue of reaction-rate “versus” continuum theory (where continuum theory refers either to 1-D NP or PNP approximations). The answer depends on what one is trying to model. If one wants to interpret the channel conductance in terms of the actual channel geometry and electrostatics, then some sort of continuum model is essential since it provides a first order approximation to the actual channel structure. On the other hand, if one is simply trying to parameterize the channel or one knows that strong ion–ion interactions are important, then the RR model may be preferable (for example, see Dang and McCleskey, 1998).
The problem with the RR model occurs when the investigator over-interprets the results and attempts to relate the rate constants to real energy barriers (Levitt, 1986). For example, the rate of going from the bulk solution to a binding site in the channel may simply be limited by the ion diffusion rate and there may not be any physical energy barrier. The problems become particularly severe if one tries to explain the experimental current–voltage curves using the voltage dependence of the RR energy barriers. For example, most biological ion channels have relatively linear current–voltage relations, while the voltage dependence of a single RR energy barrier is highly nonlinear (Levitt, 1986). To fit a linear I–V curve with the RR model, it is necessary to add multiple energy barriers that probably have no relation to the actual physical energy barriers in the channel.
Summary and Conclusions
As illustrated in this brief review, there is a hierarchy of approaches to modeling ion channels ranging from the exact molecular dynamics simulation down to reaction-rate theory. Each new simplification introduces a new limitation or uncertainty in the result. In the past, there was little reason to use the most exact solutions because of the computational limitations and, more importantly, without atomic resolution channel structures, these solutions were basically empty exercises. That situation is clearly about to change and we are at the beginning of a new era in ion channel modeling. The obvious first candidate for this new approach is the Streptomyces lividans potassium channel, whose structure has been recently solved by x-ray diffraction (Doyle et al., 1998). This channel should become a major testing ground for checking and calibrating this new generation of ion channel models. There is no question that strong ion– ion interactions are important for this channel since at least two ions are directly observed near the selectivity filter. Thus, an accurate simulation of this ion–ion simulation should require the use of the 3-D Brownian dynamic approach and rules out some sort of Poisson mean field approximation. One will probably need to use molecular dynamics to estimate the cation diffusion coefficient and potential function in the region of the narrow selectivity filter. A stringent test of the competing models will be provided by their ability to predict the conductance changes that occur for specific channel mutations.
For most channels, the structure is not known, and the old fashioned use of channel models to guide the interpretation of flux measurements in terms of structure is still relevant. This is illustrated by the current debate about the selectivity mechanism of the calcium channel (Dang and McCleskey, 1998; Nonner and Eisenberg, 1998). Since the two protagonists in this debate are represented by separate articles in this issue, no more will be added here. Suffice it to say, the excitement that is generated by this issue is the best illustration that these old fashioned, semi-empirical models still have a role to play in our understanding of ion channel function.
references
- Bek S, Jakobsson E. Brownian dynamics study of a multiply-occupied cation channel: application to understanding permeation in potassium channels. Biophys J. 1994;66:1028–1038. doi: 10.1016/S0006-3495(94)80884-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: a program for macromolecular energy minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- Chang G, Spencer RH, Lee AT, Barclay MT, Rees DC. Structure of the MscL homolog from Mycobacterium tuberculosis: a gated mechanosensitive ion channel. Science. 1998;282:2220–2226. doi: 10.1126/science.282.5397.2220. [DOI] [PubMed] [Google Scholar]
- Chung SH, Hoyles M, Allen T, Kuyucak S. Study of ionic currents across a model membrane channel using Brownian dynamics. Biophys J. 1998;75:793–809. doi: 10.1016/S0006-3495(98)77569-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang TX, McCleskey EW. Ion channel selectivity through stepwise changes in binding affinity. J Gen Physiol. 1998;111:185–193. doi: 10.1085/jgp.111.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorman V, Partenskii MB, Jordan PC. A semi-microscopic Monte Carlo study of permeation energetics in a gramicidin-like channel: the origin of cation selectivity. Biophys J. 1996;70:121–134. doi: 10.1016/S0006-3495(96)79554-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Ferguson AD, Hofmann E, Coulton JW, Diederichs K, Welte W. Siderophore-mediated iron transport: crystal structure of FhuA with bound lipopolysaccharide. Science. 1998;282:2215–2220. doi: 10.1126/science.282.5397.2215. [DOI] [PubMed] [Google Scholar]
- Hille, B. 1984. Ionic Channels of Excitable Membranes. Sinauer Associates, Inc., Sunderland, MA.
- Jakobsson E. Using theory and simulation to understand permeation and selectivity in ion channels. Methods (Orlando) 1998;14:342–351. doi: 10.1006/meth.1998.0589. [DOI] [PubMed] [Google Scholar]
- Kreusch A, Schulz GE. Refined structure of the porin from Rhodopseudomonas blastica. Comparison with the porin from Rhodobacter capsulatus. . J Mol Biol. 1994;243:891–905. doi: 10.1006/jmbi.1994.1690. [DOI] [PubMed] [Google Scholar]
- Kurnikova MG, Coalson RD, Graf P, Nitzan A. A lattice relaxation algorithm for three-dimensional Poisson-Nernst-Planck theory with application to ion transport through the gramicidin A channel. Biophys J. 1999;76:642–656. doi: 10.1016/S0006-3495(99)77232-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt DG. Interpretation of biological ion channel flux data--reaction-rate versus continuum theory. Annu Rev Biophys Biophys Chem. 1986;15:29–57. doi: 10.1146/annurev.bb.15.060186.000333. [DOI] [PubMed] [Google Scholar]
- Levitt DG. Exact continuum solution for a channel that can be occupied by two ions. Biophys J. 1987;52:455–466. doi: 10.1016/S0006-3495(87)83234-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt DG. General continuum theory for multiion channel. I. Theory. Biophys J. 1991a;59:271–277. doi: 10.1016/S0006-3495(91)82220-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt DG. General continuum theory for multiion channel. II. Application to acetylcholine channel. Biophys J. 1991b;59:278–288. doi: 10.1016/S0006-3495(91)82221-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SC, Hoyles M, Kuyucak S, Chung SH. Brownian dynamics study of ion transport in the vestibule of membrane channels. Biophys J. 1998;74:37–47. doi: 10.1016/S0006-3495(98)77764-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls, A., K.A. Shap, and B. Honig. 1990. DelPhi V3.0. Columbia University Press, New York.
- Nonner W, Eisenberg B. Ion permeation and glutamate residues linked by Poisson-Nernst-Planck theory in L-type calcium channels. Biophys J. 1998;75:1287–1305. doi: 10.1016/S0006-3495(98)74048-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux B, Prod'hom B, Karplus M. Ion transport in the gramicidin channel: molecular dynamics study of single and double occupancy. Biophys J. 1995;68:876–892. doi: 10.1016/S0006-3495(95)80264-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom MS, Adcock C, Smith GR. Modelling and simulation of ion channels: applications to the nicotinic acetylcholine receptor. J Struct Biol. 1998;121:246–262. doi: 10.1006/jsbi.1997.3950. [DOI] [PubMed] [Google Scholar]
- Smith GR, Sansom MSP. Dynamic properties of Na+ions in models of ion channels: a molecular dynamics study. Biophys J. 1998;75:2767–2782. doi: 10.1016/S0006-3495(98)77720-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suenaga A, Komeiji Y, Uebayasi M, Meguro T, Saito M, Yamato I. Computational observation of an ion permeation through a channel protein. Biosci Rep. 1998;18:39–48. doi: 10.1023/a:1022292801256. [DOI] [PubMed] [Google Scholar]
- Tieleman DP, Berendsen HJ. A molecular dynamics study of the pores formed by Escherichia coliOmpF porin in a fully hydrated palmitoyloleoylphosphatidylcholine bilayer. Biophys J. 1998;74:2786–2801. doi: 10.1016/S0006-3495(98)77986-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf TB, Roux B. The binding site of sodium in the gramicidin A channel: comparison of molecular dynamics with solid-state NMR data. Biophys J. 1997;72:1930–1945. doi: 10.1016/S0006-3495(97)78839-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Q, Jiang Q, Moore PB, Newns DM, Klein ML. Molecular dynamics simulation of a synthetic ion channel. Biophys J. 1998;74:3–10. doi: 10.1016/S0006-3495(98)77761-6. [DOI] [PMC free article] [PubMed] [Google Scholar]