

Many laboratories study the movement of ions through single channel molecules every day using the dazzling techniques of molecular and membrane biology. Atomic details of a channel are changed and the correlations of structure and function are observed routinely, but the correlations have been difficult to understand without a quantitative physical theory that links structure to function. The inputs to such a theory are the structure of the channel, the physical properties of ions, and concentrations and electrical potentials in the bath. The output of the theory is the current observed in a single open channel for a range of transmembrane potentials and compositions and concentrations of the surrounding solutions.
Verbal Models and Direct Simulations
Physical laws are written as mathematical equations, and for good reason. Verbal formulations of physical laws are not specific and do not permit direct comparison with experiments (which usually have numbers as their output). Verbal formulations lead different people to different conclusions, depending on how they subjectively weigh different effects: verbal theories are rarely objective and often lead to interminable argument. Mathematical statements of physical laws are less subject to these human distortions, but they are hardly perfect either. Mathematical laws lead to mathematical complexities and computational problems quite independent of their physical meaning, and the necessary approximations are often illogical and can be distorted as well.
These generalities are easily replaced with specifics when theories of channels are considered. Verbal models of permeation cannot link structure and function because permeation usually involves competing effects. If one effect increases current and another decreases it, the net effect can only be determined quantitatively. It is natural then to turn to computational models of channels that promise to be complete and rigorous.
Simulations of molecular dynamics promise to directly compute properties of biological interest, starting with fundamental physical laws and decent representations of the properties of atoms. Unfortunately, the simulations of molecular dynamics cannot provide a quantitative theory of ions moving through a channel, driven by a gradient of concentration and electrical potential, even though the underlying equations of motion of ions and atoms in channels are reasonably well known. Direct simulations are not possible because the simulated system needs to be large enough and computed long enough to define the variables that are measured and controlled experimentally; e.g., current, transmembrane potential, and concentration. Direct simulation of atomic motions use a discrete time step of 10−16 s to resolve atomic vibrations. Calculations with (substantially) longer time steps than 10−16 s are no longer direct simulations, but depend on theory and assumptions to fill their gaps. A single ion takes some 100 ns to cross a channel (109 time steps), and calculations of thousands to billions of crossings must be made (Barcilon et al., 1993) to estimate measured currents. Direct integration of differential equations of molecular dynamics (i.e., the differential equations that are Newton's laws) is not reliable over times larger than several picoseconds because the calculated trajectories exponentially diverge (Ott, 1997) and are exquisitely sensitive to initial conditions and round-off error (Frenkel and Smit, 1996; Rapoport, 1997) as can easily be verified by running any of the codes widely available, some without cost.
It is not known mathematically (Frenkel and Smit, 1996) whether calculated trajectories (after a few picoseconds of calculation) are reliable, unbiased, or even useful estimates of the trajectories of real atoms (“unbiased estimate” here is defined as in probability theory and statistics). Simulated trajectories may not sample some of the phenomena of biological interest at all, particularly those phenomena that appear slowly (i.e., after microseconds). Trajectories of chaotic systems, starting from particular initial locations, are often confined to limited regions of “phase space” and do not explore other regions at all. Even within an accessible region of phase space, simulated trajectories may not be reliable or unbiased estimates of systems. After all, while the trajectories of real atoms are not subject to round off error, the computation is, and how the round off error influences the computation is not known (Beck and Schlögl, 1995).
Simulated systems of proteins and channels must also be large enough to define concentrations of ions in the bath, if those concentrations influence biological phenomena of interest (as they almost always do). Since errors tend to vary with the square root of the number of particles studied, something like 1,000 ions need be present in a simulation to define a concentration with 3% precision. Those thousand ions are solvated by a very much larger number of water molecules, since in biological systems the mole fraction of ions is small (ranging from, say, 0.150/55 for typical external sodium concentrations to 10−5/55 for typical internal calcium concentrations). Simulations need to involve some 3 × 105 molecules (nearly one million atoms) to define a typical extracellular Na+ concentration in the solutions surrounding a channel and 2 × 109 atoms to define a typical intracellular Ca2+ concentration.
Short duration simulations of small systems are often extrapolated to biological sizes and times in a sensible attempt to describe biological phenomena, but then they are indirect calculations, no better than the theory used to extrapolate them. Such indirect simulations are not a priori any more reliable than those made with a theory of lower resolution.
Finally, a technical difficulty is important when considering proteins like channels that act as devices in the engineering sense of the word. Devices and channels almost always function away from equilibrium, but simulations of molecular dynamics of channels are nearly always done at equilibrium and with treatments of the electric field that do not allow transmembrane potentials at all (Roux and Karplus, 1994). The mathematical difficulties involved in computing nonequilibrium biological and chemical systems with transmembrane potentials have not been overcome (Eisenberg, 1998), and the methods used by physicists to perform such calculations have not, to the best of our knowledge, been tried in channels, proteins, or ionic solutions. The best portal to these methods seems to be the Dacmocles website (www.research.ibm.com/0.1um/laux/dam.html).
Averaged Theories: Poisson-Nernst-Planck
Given the difficulties of direct simulations of molecular dynamics, it seems that some of their resolution must be abandoned if one wishes to actually fit biologically relevant data. The choice is only what to give up. The choice is made by using a series of simplified theories and picking the simplest that describes the experiments and behavior of interest, while retaining as much structural meaning as possible.
Here we will show that a simplified model that considers (mostly) the electrical properties of the open channel (the fixed charge on its walls and the mobile charge in its contents) does surprisingly well in understanding and predicting the currents that flow through several open channels, for a range of potential and concentrations of several ions. An electrical model seems a good place to begin a theory of channels, since the function of channels directly involves currents and transmembrane potentials and the structure of channels involves highly charged molecules lining tiny volumes. (One charge in a selectivity filter 7 × 10 Å is some 5 M.) It does not take much charge to produce huge forces (see first page of Feynman et al., 1963).
The model we will consider forms a working hypothesis, to be tested and then revised as it fails. In its simplest and original form, the working hypothesis was that a channel could be described as a one-dimensional distribution of fixed charge, corresponding in a crude way to the fixed charges on the atoms that line the wall of the channel's pore. In the crudest form, the effective charge would be the total charge on the wall of the channel in some cross-sectional slice. In more refined versions of the model, other estimates of the effective charge would be used. The only contribution of the channel protein to the permeation properties of mobile ions was supposed to be the electric field created by these charges, and the concomitant distribution of the probability of location (“concentration”) of the permeating ion. Single filing effects, and specific chemical interactions (not described by Coulomb's law) were purposely omitted from the first version of the model, so the entire force field on the permeating ions could be computed unambiguously (at this level of resolution) once the charges on the channel protein were specified (along with the bath concentrations, transmembrane potential, geometry, diffusion, and dielectric coefficients). The electric field in this model is computed by the Poisson equation, which is the differential form of Coulomb's law. Details are described in Eisenberg (1998), and the code that solves these equations is available in various forms at an anonymous ftp site (ftp.rush.edu in directory /users/Eisenberg). In the initial working hypothesis, the electric field is supposed to influence current only according to electrodiffusion as described by the Nernst-Planck equation. The model is specified by the Poisson-Nernst-Planck (PNP) equations, which, as it happens, are nearly identical to the drift diffusion equations that have been used for a very long time to describe the motion of charged quasi-particles like the holes, electrons, and superparticles of semiconductors (Lundstrom, 1992) or the hydrated ions of electrolyte solutions (Newman, 1991). We have suggested that the correlated motion of an ion, water, and atoms of the channel protein might act as a quasi-particle or super-particle, a permion (Elber et al., 1995), with the permion rather than the individual ions satisfying the conservation laws of PNP.
PNP depends on the same approximations and representations of underlying atomic variables as the Gouy-Chapman model of an electrified interface, the (nonlinear version of) the Debye-Hückel [DH] theory of ionic solutions), and the (nonlinear) Poisson-Boltzmann theory of proteins. Although the physics underlying PNP and these other models are similar, the actual behavior, mathematics, and computational properties of the systems are quite different because PNP includes flux in its every calculation, never requiring equilibrium (Eisenberg, 1998).
The Nernst-Planck equations have much more resolution than a crude continuum description of ionic concentration and movement. Theory and four types of simulations show that the Nernst-Planck equation with constrained potential describes the concentration and flux of discrete particles diffusing over barriers of arbitrary shape and size (Barcilon et al., 1993; Eisenberg et al., 1995) from a bath of one concentration and potential to another.
The Nernst-Planck equations use one parameter to describe frictional forces that limit the two components of flux, diffusion and drift, assuming that the Nernst-Einstein relation between diffusion coefficient and mobility is valid under all conditions of interest. Less elementary versions of PNP do not require that assumption. The diffusion coefficient of the ion in fact also depends on the properties of the channel wall, both because of steric effects and (probably much more importantly) because of dielectric friction produced by motions of the protein's atoms induced by the electric field of the permeating ion. But PNP does not explicity invoke this important physical mechanism (Wolynes, 1980).
The PNP model contains much less atomic detail than most models of channels or proteins. PNP neglects correlation effects of individual atoms. It contains only those effects that are mediated by the mean field. For example, PNP is rich with binding phenomena because it often predicts localized maxima in the concentration of permeating ions, but it does not predict the phenomena of single filing that depend on the correlated motion of ions. We have considered correlation effects to be important for a long time, and so it seemed highly unlikely to us (as we derived the model and as Duan Chen solved its equations) that PNP would actually fit data. Indeed, that is why we worked so hard on higher resolution models. However, the low resolution PNP model does fit a wide range of data, using a different distribution of fixed charge for each type of channel (corresponding presumably to the different structure of each type of channel) and different diffusion coefficients for each type of ion. Note that no parameters are changed as solutions or transmembrane potentials are changed other than the concentrations and potentials themselves.
PNP Fits Data
Thus far, the simple PNP model fits the highly rectifying (sublinear) I–V relations measured in a wide range of solutions, symmetrical and asymmetrical, from the synthetic cation-conducting leucine serine (LS) channel (Chen et al., 1997b). It fits most (but not all) of the highly rectifying, superlinear I–V relations in the neuronal background anion (NBAC) channel (Chen et al., 1995). It fits the rectifying sublinear I–V relations from porin (Tang et al., 1997), a channel of known structure, and its mutant, G119D, also of known structure. It fits I–V relations from both cardiac and skeletal forms of the calcium release channel (CRC) of sarcoplasmic reticulum (Chen et al., 1997a, 1999), all of which are quite linear; and it fits I–V relations from a number of other channels (most notably gramicidin, see below). So far, PNP has fit the I–V relations of every channel for which we have data, but we continually expect it to fail on the next attempt.
The simplest form of PNP fits most of the data but, in some cases, an extra parameter (a constant) is needed that describes chemical interaction, as described below. In most cases, PNP is the only theory that fits the entire data set, including asymmetrical solutions. The fact that PNP fits these data sets contradicts the general view, which we certainly shared until PNP showed us otherwise, that a theory of permeation must explicitly include much more than Coulomb's law. Specifically, it contradicts the view that a successful theory must have separate components and parameters that describe dehydration/ resolvation, obligatory single filing, or chemical interactions of the permeating ion with the channel protein.
Chemical Effects
Chemical interactions are seen in some cases. Binding must be included as an extra parameter in PNP when describing permeation of Li+ and Na+ in the cardiac CRC channel (Chen et al., 1999), or the anomalous mole fraction property of K+ channels (Nonner et al., 1998) or the interactions of Na+, Ca2+, and pH in the L-type calcium channel (Nonner and Eisenberg, 1998). It is enough (at the present level of experimental and structural resolution) to add a single constant for each ion that describes the excess chemical potential of that ion in the channel. That constant is the same for a given type of ion in a given type of channel and does not vary with transmembrane potential or concentration of any of the ions. This constant is the only representation of dehydration/resolvation, nonelectrostatic binding, and single filing needed to fit these data sets. Indeed, it is possible that this constant arises entirely from Coulomb's law, as a correction for effects of the three-dimensional field not correctly described in the one dimensional model. As we consider more complex channels and mixed solutions containing many different ionic species, it seems likely that more specific chemical effects will be seen. We propose to deal with these (in the first place) in the Mean Spherical Approximation (MSA) that has proven so successful in bulk solution, as discussed later in this paper.
PNP as a Structural Theory
If a theory is to serve as a link between structure and function of an open channel, it must do more than fit I–V relations. It must bear a well defined and close relation to the structure of the channel. The crudest form of PNP is a one-dimensional theory, and so the relation of its parameters and those of a three-dimensional structure are not immediately obvious even though the one-dimensional theory was derived by explicit mathematics; e.g., averaging (see Appendix of Chen and Eisenberg, 1993) of the structurally based three-dimensional (3-D) theory.
The question remains what is the meaning of the parameters estimated by fitting 1-D PNP to data? The answer to that question is not completely known, but we note that all of the parameter estimates are reasonable, and so it is possible that they may be meaningful and reliable estimates of the underlying physical properties, although that certainly has not been proven to be the case. For example, typical diffusion coefficients are some 10–50× smaller than in bulk solution; the fixed charge densities correspond to only one or two fixed charges in the “selectivity filter” (narrow part) of the channel, although those one or two charges induce a concentration of mobile charge in the channel of 5–10 M, since they attract one or two counter ions into the channel's pore, and the pore volume is very small. In fact, the fixed charge of the CRC channel turns out to be very close to −1 (Chen et al., 1997a, 1999), and this is an invariant of the curve fitting procedure (changing all sorts of parameters in the curve fitting does not change the estimate of the total fixed charge, although it changes the estimates of the diffusion coefficient), suggesting that the selectivity filter of this channel (in the form studied here) is dominated by a single fully ionized acidic amino acid, although of course the PNP results do not prove this.
The validity of 1-D PNP can be probed by studying the effects of mutations on its estimates of fixed charge. Tang et al. (1997) compared the estimates of charge in a wild-type porin and its aspartate-substituted mutant, G119D, which has one additional negative charge and known crystallographic structure. The estimated difference −0.93, found in those and many subsequent experiments, is surprisingly close to the value expected.
Three-Dimensional PNP
A three-dimensional version of the PNP theory has recently been developed and calculated (3DPNP), in which all atoms are assigned the charges used in a standard molecular dynamics program and all atoms are assigned positions known from nuclear magnetic resonance. Unlike 1DPNP, 3DPNP is a structural theory: the charges are estimated from the chemical literature, not from curve fitting. In 3DPNP, only the diffusion coefficient has to be specified by experimental measurements of I–V curves. Only the diffusion coefficient is left unspecified by a priori structural and physical data. In the first fairly low resolution finite difference calculation (Kurnikova et al., 1999), 3DPNP shows qualitative agreement between the theory and experimental data. A high resolution analysis (Hollerbach et al., 1999) using spectral elements shows quantitative agreement.
Why Does PNP Work?
It seems then that for a range of open channels, a simple electrostatic model is able to fit all the I–V data available in a range of solutions, sometimes with the addition of a single extra parameter to describe the excess chemical potential of a given ion in a given channel. There are physical reasons why a mean field theory of a tiny, highly charged, (nearly) one-dimensional system is a good approximation. Generally, one would expect low resolution–averaged theories to be a reasonable approximation because of the long duration of channel currents (on an atomic time scale). Very little temporal resolution is needed to describe single channel currents. One would also expect low resolution–spatially averaged theories to be a decent initial description of (tiny) channels because of the large number of atoms outside the channel involved in determining concentrations of ions, transmembrane potential, and the reaction field to the ions in the pore. The reaction field for a permeating ion is largely in the surrounding baths where a mean field theory is likely to be more than adequate. Specifically, it is known (and not just as a matter of speculation) that mean field terms dominate correlation terms when charge is distributed along a narrow cylinder (van den Brink and Sawatzky, 1998) or when systems are highly charged (Henderson et al., 1979).
Despite these arguments, a derivation of PNP is needed to understand its theoretical limitations. Deriving PNP requires a superior theory of higher resolution that describes single file motion of ions while still computing the electric field from the charges in and near the channel. Such a Langevin-Poisson theory is being worked on, by us and others, but is not yet available.
Traditional Barrier Models of Permeation
It seems idle to us to spend much further effort discussing models of permeation that do not fit data, when a simple model is available that does, particularly given our discussions in recent papers (e.g., see Appendices of Chen et al., 1997; Nonner and Eisenberg, 1998; see also Eisenberg, 1998; Nonner et al., 1998). Nonetheless, it is necessary, given the purpose and arena of this paper, to reiterate some of the things we have said previously about traditional barrier models of open channels that have been so widely used to study permeation.
It should be clearly understood that barrier models rarely are able to actually fit I–V curves measured over a range of transmembrane potentials and solutions, including asymmetrical solutions, even when using many adjustable parameters, including an incorrect or adjustable prefactor (see equation in Scheme I). As a rule of thumb, I–V relations observed in asymmetrical solutions are more linear, often much more linear, than predicted by barrier models.
Traditional barrier models (Heckmann, 1972; Hille, 1992) are not very useful for relating structure and function, because they involve states and transitions only vaguely related to the actual structure. The models do not contain spatial coordinates as variables. The positions of individual components like ions are not within the scope of the model and so the Heckmann level of description is a priori incapable of relating ion flow to the geometrical structure of a channel.
Traditional barrier models also contain two large errors, each factors ≈104, that act in opposite directions and so more or less balance each other in the limited sense that they allow the model to fit the current measured at one transmembrane potential in one symmetrical solution. Most glaring is the choice of prefactor in the expression for the current over a high barrier. For historical reasons, the prefactor used in nearly all barrier models of open channels is k B T/h, even though that prefactor leaves out friction altogether (i.e., it contains no diffusion coefficient or variable to describe friction). Friction is a dominant determinant of atomic movement in condensed phases (like ionic solutions, proteins, and channels) because condensed phases contain (almost) no empty space. Workers on channels pointed out this problem some time ago (Cooper et al., 1985, 1988a,b; Chiu and Jakobsson, 1989; Läuger, 1991; Roux and Karplus, 1991; Andersen and Koeppe, 1992; Barcilon et al., 1993; Crouzy et al., 1994; Eisenberg et al., 1995) and, indeed, Eyring clearly states that k B T/h is not to be used by itself as a prefactor in condensed phases (Wynne-Jones and Eyring, 1935). There is general agreement among workers on condensed phases that the expression k B T/h is inappropriate (Fleming and Hänggi, 1993; Hänggi et al., 1990, which cites some 700 papers relating to this matter). When reading these classic papers, it is important to be aware that the value of the transmission factor (Frauenfelder and Wolynes, 1985) is now known in condensed phases dominated by friction (Fleming and Hänggi, 1993; Pollak et al., 1994).
The correct expression for the rate constant k
j = J
f/ℓC
l in a condensed phase for one-dimensional (unidirectional) flux J
f from a solution of concentration C
l over a high barrier was apparently first published by Kramers, 1940. The rate constant and unidirectional flux can be written in a particularly neat form if the potential profile is a large symmetrical parabolic barrier spanning the whole length ℓ of the channel, with peak height φmax(x
max), at location x
max = ℓ/2, much larger than the transmembrane potential and k
B
T/e.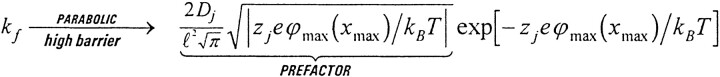
The prefactor can be viewed as a measure of the entropy of activation and thus a measure of the effective volume available for ions to diffuse in the channel compared with the volume available in the surrounding solution (Berry et al., 1980). It seems natural that the effective volume should involve the length and height of the potential barrier, and Hill discusses the role of diffusion velocity in the prefactor (Hill, 1976).
In this equation, D j is the diffusion coefficient in the channel of ion j of valence z j, e is the charge on the proton, k B is the Boltzmann constant, and T is the absolute temperature. Now, if the barrier is, say, k B T/e high and 1-nm long, and the diffusion coefficient is some 1.3 × 10−6 cm2 /s, the numerical value of the prefactor is ∼2.8 × 108 s−1. The numerical value of the usual prefactor in barrier theory is k B T/h, which is ∼2.2 × 104 times larger, ∼6.3 × 1012 s−1. As one might expect, ions hopping over barriers experience much less friction than ions diffusing over them, and the amount of the friction will depend on the identity of the ion. Numerical errors of this size have serious qualitative as well as quantitative consequences, as was pointed out some time ago (Cooper et al., 1988a).
The error in barrier models that more or less balances the error in the prefactor involves the potential barrier. The Heckmann model does not detail electrical interactions among its internal components. The spatial coordinates needed to specify an electric field are not present in the model as originally specified, but are put in by an artificial concept of electrical distance. The model postulates discrete sites when none need be present. It is not surprising that the Heckmann model does not predict the same electric field as Poisson's equation (see below).
In barrier models, and all other models of permeation familiar to us (that actually predict current), except PNP, the potential barrier is assumed, not computed. Potentials in channels arise, however, from the fixed charges on the protein, the mobile charges inside the channel's pores, and the charges in the solutions and electrodes outside the channel. These produce a potential profile that changes as conditions change; e.g., as transmembrane potential, bath concentration, or fixed charge on the channel protein changes. In general, the resulting potential profiles do not have large barriers, and so currents are much larger (indeed, exponentially larger) than they would be in otherwise similar theories with large barriers. The currents predicted are very different from those of traditional barrier models. Having been raised in the barrier tradition, we are often surprised by the electrostatic effects predicted by PNP, although they are easy to compute and to understand once they are computed.
An example may be useful. Nonner and Eisenberg (1998; see Appendix) compute the electric field of a barrier model of the L-type calcium channel using Poisson's equation instead of repulsion factors. The energy profile of Dang and McCleskey (1998) was included as a spatially varying excess chemical potential μ0 Ca(x) to describe the binding properties of the channel. Cl− was excluded from their calculation, so, to be fair and comparable, we also excluded Cl− by applying a large repulsive energy, specifically ∼12 k B T/e, just for Cl−, which we thought would reduce Cl− occupancy by ∼e12 ≈ 1.6 × 105. The repulsive potential did not act on the cations, and diffusion coefficients of all ions were set equal for illustrative purposes in this calculation.
Surprisingly, the PNP calculation using this profile predicted a reversal potential close to zero for external calcium concentrations <10 mM. That is, the “calcium” channel became a nonselective channel when Ca2+ was <10 mM. When Ca2+ in the external solution was 100 mM, the selectivity reversed (i.e., the reversal potential became −20 mV). The calcium channel had become a chloride channel, if we use common lab jargon. The change in selectivity of the channel was produced without invoking any specific chemical effects at all, it was produced by electrostatic repulsion, computed from PNP, just as the anomalous mole fraction effect (studied in Nonner et al., 1998) was a purely electrostatic effect. Neither single filing nor definite occupancy were involved. When placed in a 100 mM Ca2+ solution, 0.2 Ca ion was found in each pore in the calculation. This bound calcium produced a (small positive) local excess in net charge and that produced a large positive potential of nearly 110 mV (see Figure 12 of Nonner and Eisenberg, 1998). Of course, that potential was a severe barrier for cation movement. This potential barrier (produced by calcium binding) reduced cation movement (particularly divalent cation movement like calcium flux) so effectively that the residual conductance was dominated by chloride, even though chloride was subject to a repulsive potential of 12 k B T/e.
By explicitly using Poisson's equation, we had found that binding of calcium dramatically reduces the calcium current, not because of “interference” by single filing, or an effect on diffusion constant, but because of an electrostatic effect. Of course, the details of this effect depend on the size of the repulsive potential that we chose. If we had chosen a repulsive potential smaller in magnitude, the channel would have become a chloride channel at lower external calcium concentration. If we had chosen a repulsive potential larger in magnitude, the chloride “selectivity” would have not appeared in the 100 mM Ca2+ solution. But the electrostatic effects of the binding would have been profound in any case. Such effects are absent in traditional barrier models or other models that do not use Poisson's equation or Coulomb's law to go from charge to potential.
Binding invariably has a large effect on potential because of the accumulation of charge that binding necessarily entails (that is what the word “binding” means!). That charge changes potential, and the change in potential is large because the system is so small (i.e., its capacitance is tiny). Bound ions repel nearby ions and thus have large effects, creating, for example, depletion layers that can dominate channel properties. These electrostatic effects of binding will be seen no matter what the details of the calculation or choice of repulsion potential, and the existence of these effects is the main point of our discussion.
Barrier models miss the electrostatic effects of binding altogether. Rather than solving Poisson's equation, barrier models use fixed profiles of free energy to characterize the interactions of permeating ion and channel protein independent of charge; they use repulsion factors to characterize interactions between permeating ions as if the ions were always separated by a fixed distance, producing a fixed repulsive energy independent of the fixed charge of the protein and the screening of this fixed charge by nearby ions (in the pore and in the baths). Screening has been known to dominate the properties of electrolyte solutions and interfaces since the work of Debye-Hückel and Gouy and Chapman (Newman, 1991) and it seems unwise to ignore it in channels.
Using fixed profiles of free energy to characterize the interactions of permeating ion and channel protein is inaccurate because the potential profile inside the channel depends strongly on the concentration of ions in the bath, as is easily verified in a Poisson Boltzmann or 3DPNP calculation, because of the long range nature of the electric field: the ionic atmosphere of the fixed charge lining the channel's wall extends into the surrounding baths. Or, to put the same thing another way, the dielectric charge in the protein and lipid and the mobile charges in the channel's pore do not screen the fixed charge of the protein from the ions in the bath. Thus, the interactions of ions within the channel cannot be described by a theory that ignores the concentration of ions in the bath. The ions in the bath help determine the interaction between ions in the channel's pore.
Using fixed interionic distances in traditional barrier models is inaccurate because the distance between ions is in fact quite variable. The distance depends on screening that varies with the concentration of ions in the bath, the transmembrane potential, and the charges and shape of the channel protein itself. In fact, to maintain a fixed average distance between permeating ions (as conditions are changed), large amounts of energy would have to be injected directly into the channel's pore by a deus ex machina, always the theorists' most helping hand.
We suggest that the fundamentally flawed treatments of electrostatics in barrier models are likely to produce a qualitative misunderstanding of the role of occupancy and quantitative errors of at least one order of magnitude. Our calculation (Nonner et al., 1998) shows occupancy predicted by Poisson's equation is very different from that predicted with barrier models. Our calculations show that if a channel is to hold a certain number of mobile charges, it must have balancing structural charges, and the interactions of these mobile and fixed charges cannot be described by a fixed free energy. The wide variations in ionic occupancy that typically occur in barrier models are likely to be in severe conflict with the electrostatics of Poisson's equation customarily used to describe the electric field. That is to say, if such variations in occupancy actually occurred, the electrical potential would vary wildly because of the severe violations of local electrical neutrality.
If a channel is lined by a fixed structural charge, electrostatic effects tend to maintain a (nearly equal) number of mobile ions within the pore, thereby maintaining approximate electroneutrality. Wide variations in ionic occupancy are buffered by the need for approximate electrical neutrality; i.e., wide variations in occupancy can only be produced by large energies not typically available to channels. Even though ionic occupancy is buffered, screening depends on bath concentration because the electric field generated by the fixed charge lining the wall of channels reaches through the protein and lipid into the surrounding baths: the electric field is long range. Electroneutrality in the channel is approximate, not exact, and the residual (“unneutralized”) charge is large enough to have profound effects.
Conclusion
We conclude that traditional barrier models overestimate current because they neglect friction and underestimate current because they compute electrostatics incorrectly. These errors of course do not balance precisely and that is why barrier models fail to fit reasonably large data sets, particularly if the data sets include I–V relations measured in asymmetrical solutions. These errors are fundamental to the whole class of barrier models, and so we believe such models must be abandoned. When a model fails as badly as barrier models do, its qualitative features cannot be considered a reliable indicator of underlying mechanism. It is not useful for our main purpose.
To us, abandoning barrier models seems no great loss. Those models do not fit the data anyway (if the data is taken over a reasonable range of conditions). But abandoning barrier models is a great loss, in a more human sense, because so many gratifying insights into mechanism have been developed using them, often at great effort. Abandoning barrier models means calling these insights into question. It means these insights must be reexamined using theories that fit data and have some physical basis. It means that much more experimentation is needed to reexamine issues already thought to be settled. Reexamination of settled issues is bound to be unsettling.
Unsettled Questions
The outstanding problems in permeation are to understand the role of electrostatics, chemistry, and geometry in determining the movement of ions (in our view). So far, the role of electrostatics and geometry seems approachable by 3DPNP. We suspect, but have not proven, that the effective one-dimensional profile of charge we call P(x) will be approximated by derivatives of the solution Φ3DPNP(x,r,θ) of the three-dimensional PNP equation (specifically, the divergence of the three-dimensional electric field in the radial direction and equivalently its derivative along the path of permeation, with the concentration of permeating ions subtracted off). Further analysis will tell whether the excess free energy needed to fit some of our data sets is an expression of the three-dimensional field, of actual chemical interactions, or of some other effect.
Knowledge of permeation is limited by a surprising lack of published I–V curves in asymmetrical solutions. These are important because they are often much more linear than expected from traditional models. Knowledge is also limited because we have so little structural information, particularly of the eukaryotic channels of greatest anthropomorphic interest; e.g., the voltage-gated Na+ and K+ channels of nerve fibers. As we turn to these specialized channels, it seems likely that the simplest version of PNP will need to be supplemented by models containing more explicit chemistry; i.e., binding energies in binding regions. The question is how to introduce chemistry into a model without requiring analysis of uncomputable trajectories. We are following the chemists, taking a most successful theory of ions in bulk solution, namely the MSA and applying it inside a channel: it is comforting to work with a theory and people who have solved the problem of selectivity in bulk solution. The MSA has a long history (Blum et al., 1996), and recent versions have been remarkably successful at predicting the properties of solutions from infinite dilution to saturation, even molten salts (Simonin et al., 1999).
The MSA is a mean field theory that in essence reworks Debye-Hückel analysis, now treating ions as spheres. The distribution of point charges around a central sphere (as assumed in DH) is quite different from the distribution of spherical charges around a sphere (MSA), particularly at concentrations more than a millimole or so, because finite ion diameters create exclusion zones around ionic charges. The resulting charge distributions produce quite different electric fields, according to Poisson's equation, and this difference allows MSA to fit much data that DH cannot.
Fortunately, the MSA is hardly more complex than DH because both theories express thermodynamic functions in terms of one quantity, a characteristic length of screening κ−1 that is given by algebraic formulas, albeit more complex formulas in the MSA than in DH. MSA in its primitive form treats water as a continuous ideal dielectric, whereas “nonprimitive” versions include the solvent as a polar molecular species (up to octopolar, as is needed to model hydrogen bonding). More accurate theories are available (e.g., the hyper-netted chain [HNC]; Henderson, 1983), but they are more elaborate to compute and do not lead to algebraic expressions for activity coefficients.
Interestingly, most current theories of bulk solution and narrow spaces (DH, HNC, and MSA) are mean field theories; they do not try to follow or explicitly average individual ionic trajectories. These theories deal self-consistently with the average effects of excluded volume, including diameter-constrained electrostatic interactions. To be applied rigorously to channels, these theories need to be reexamined and rederived for the geometry of channels and the specific properties (e.g., excluded volume) of the amino acids that line the wall of the channel, taking note of its high surface charge density. Even better, density-functional theory (DFT, a generalization of HNC designed to describe inhomogeneous systems; Henderson, 1992) should be applied to channels and proteins. We are trying, thanks to Laura Frink of Sandia National Laboratory (Albuquerque, NM).
Even before these rigorous treatments are available, it is already clear that many of the distinguishing characteristics of K+, Na+, and Ca2+ channels can arise naturally from the two main (antagonistic) effects in MSA: electrostatic attraction between permeating cations and the groups forming the selectivity filter, and “chemical” repulsion arising from the effects of the finite volume of ions (Catacuzzeno et al., 1999). In the analysis of these three types of channels, no other specific chemical interactions are needed to describe selectivity (beyond those resulting from finite volume of ions as described by the MSA).
Single Filing
Finally, we address the issue of obligatory single filing, an important property of ionic channels that has concerned us for many years. Nonner et al. (1998) shows that one of the experimental phenomena (the anomalous mole fraction effect) thought to require an explanation involving obligatory single filing can in fact be explained without single filing. The effect appears (in a mean field theory without obligatory single filing) as a necessary consequence of (a small amount of) localized excess chemical potential; i.e., binding or repulsion.
Nonetheless, it is clear that PNP in its several forms does not predict the ratio of unidirectional fluxes observed in K+ channels, or expected in single file systems. (See citations in Nonner et al., 1998. Measurements are not available of flux ratios in most channels. Measurements of flux ratios independent of gating are not available at all, to the best of our knowledge.)
The paradoxical fact is that PNP predicts net fluxes (i.e., currents) in a wide range of channels and conditions, while it does not fit the ratio of the component unidirectional fluxes correctly, at least if we assume the flux ratio of all channels is rather like that of the K+ channel. Resolution of the paradox requires analysis or simulation of a system with both obligatory single filing and with an electric field computed from the charges present.
Wolfgang Nonner has constructed a mean field model of single filing, by extending PNP to include convection. This Navier-Stokes extension of PNP is clearly able to predict the appropriate ratios of unidirectional flux and I–V curves that PNP itself has not fit. We have also done much work (with Schuss and his students, available by anonymous FTP from ftp.rush.edu in directory /pub/Eisenberg/Schuss) to formulate a self-consistent model of Brownian motion; i.e., one in which ions move according to a Langevin equation in an electric field determined by all charges present, computed by a Poisson equation updated at each time step. The mean field arises naturally in the model because of the long range nature of the electric field. The fixed charge lining the wall of the channel is “neutralized” by ions in the bath (in large measure), and those ions can be described by a mean field theory under biological conditions. That is to say, the reaction field of the fixed charge on the wall of the channel is the mean field, even in a model constructed in atomic detail. It is clear from this work that a well-posed mathematical model can be constructed, and can predict flux ratios, but the model has not been solved in general.
Interestingly, the analysis with Schuss shows that nonindependent flux ratios can arise without changing net flux. In that analysis, a “single file term” is found in both the influx and efflux, and so the net flux is not changed by single filing, but the flux ratio is. Perhaps this is how PNP manages to fit net flux data so well in the K+ channel, while it gives ratios of unidirectional flux not expected in single file systems.
We, along with others, are also trying to simulate such a single file Langevin-Poisson system. Until this simulation is actually performed, it is wise to be prudent and not guess its outcome. Rather, we will trust the work, particularly the resulting numbers.
references
- Andersen OS, Koeppe RE. Molecular determinants of channel function. Physiol Rev. 1992;72:S89–S157. doi: 10.1152/physrev.1992.72.suppl_4.S89. [DOI] [PubMed] [Google Scholar]
- Barcilon V, Chen D, Eisenberg R, Ratner M. Barrier crossing with concentration boundary conditions in biological channels and chemical reactions. J Chem Phys. 1993;98:1193–1211. [Google Scholar]
- Beck, C., and F. Schlögl. 1995. Thermodynamics of Chaotic Systems. Cambridge University Press, New York.
- Berry, S.R., S.A. Rice, and J. Ross. 1980. Physical Chemistry. John Wiley & Sons Inc., New York. p. 1156.
- Blum L, Holovko MF, Protsykevych IA. A solution of the multiple-binding mean spherical approximation for ionic mixtures. J Stat Phys. 1996;84:191–203. [Google Scholar]
- Catacuzzeno L, Nonner W, Eisenberg B. PNP2 links crystallographic structure and conduction in K channels. Biophys J. 1999;76:A79. [Google Scholar]
- Chen D, Xu L, Tripathy A, Meissner G, Eisenberg B. Selectivity and permeation in calcium release channel of cardiac muscle: alkali metal ions. Biophys J. 1999;76:1346–1366. doi: 10.1016/S0006-3495(99)77297-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Xu L, Tripathy A, Meissner G, Eisenberg R. Permeation through the calcium release channel of cardiac muscle. Biophys J. 1997a;73:1337–1354. doi: 10.1016/S0006-3495(97)78167-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DP, Eisenberg RS. Charges, currents and potentials in ionic channels of one conformation. Biophys J. 1993;64:1405–1421. doi: 10.1016/S0006-3495(93)81507-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DP, Lear J, Eisenberg RS. Permeation through an open channel. Poisson-Nernst-Planck theory of a synthetic ionic channel. Biophys J. 1997b;72:97–116. doi: 10.1016/S0006-3495(97)78650-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DP, Nonner W, Eisenberg RS. PNP theory fits current–voltage (IV) relations of a neuronal anion channel in 13 solutions. Biophys J. 1995;68:A370. [Google Scholar]
- Chiu SW, Jakobsson E. Stochastic theory of singly occupied ion channels. II. Effects of access resistance and potential gradients extending into the bath. Biophys J. 1989;55:147–157. doi: 10.1016/S0006-3495(89)82786-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper K, Jakobsson E, Wolynes P. The theory of ion transport through membrane channels. Prog Biophys Mol Biol. 1985;46:51–96. doi: 10.1016/0079-6107(85)90012-4. [DOI] [PubMed] [Google Scholar]
- Cooper KE, Gates PY, Eisenberg RS. Diffusion theory and discrete rate constants in ion permeation. J Membr Biol. 1988a;109:95–105. doi: 10.1007/BF01871391. [DOI] [PubMed] [Google Scholar]
- Cooper KE, Gates PY, Eisenberg RS. Surmounting barriers in ionic channels. Q Rev Biophys. 1988b;21:331–364. doi: 10.1017/s0033583500004480. [DOI] [PubMed] [Google Scholar]
- Crouzy S, Woolf TB, Roux B. A molecular dynamics study of gating in dioxolane-linked gramicidin A channels. Biophys J. 1994;67:1370–1386. doi: 10.1016/S0006-3495(94)80618-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang TX, McCleskey EW. Ion channel selectivity through stepwise changes in binding affinity. J Gen Physiol. 1998;111:185–193. doi: 10.1085/jgp.111.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg B. Ionic channels in biological membranes. Electrostatic analysis of a natural nanotube. Contemp Phys. 1998;39:447–466. [Google Scholar]
- Eisenberg RS, Klosek MM, Schuss Z. Diffusion as a chemical reaction: stochastic trajectories between fixed concentrations. J Chem Phys. 1995;102:1767–1780. [Google Scholar]
- Elber R, Chen D, Rojewska D, Eisenberg RS. Sodium in gramicidin: an example of a permion. Biophys J. 1995;68:906–924. doi: 10.1016/S0006-3495(95)80267-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feynman, R.P., R.B. Leighton, and M. Sands. 1963. The Feynman: Lectures on Physics, Mainly Electromagnetism and Matter. Addison-Wesley Publishing Co., New York.
- Fleming, G., and P. Hänggi. 1993. Activated barrier crossing: applications in physics, chemistry and biology. World Scientific Publishing Co., Inc., River Edge, NJ. 327 pp.
- Frauenfelder H, Wolynes P. Rate theories and puzzles of hemeprotein kinetics. Science. 1985;229:337–345. doi: 10.1126/science.4012322. [DOI] [PubMed] [Google Scholar]
- Frenkel, D., and B. Smit. 1996. Understanding Molecular Simulation. Academic Press, Inc., New York. 62–63.
- Hänggi P, Talkner P, Borokovec M. Reaction-rate theory: fifty years after Kramers. Rev Mod Phys. 1990;62:251–341. [Google Scholar]
- Heckmann, K. 1972. Single-file diffusion. In Passive Permeability of Cell Membranes. F. Kreuzer and J.F.G. Slegers, editors. Plenum Publishing Corp., New York. p. 127.
- Henderson D. Recent progress in the theory of the electric double layer. Prog Surf Sci. 1983;13:197–224. [Google Scholar]
- Henderson, D. 1992. Fundamentals of Inhomogeneous Fluids. Marcel Dekker, New York.
- Henderson D, Blum L, Lebowitz JL. An exact formula for the contact value of the density profile of a system of charged hard spheres near a charged wall. The Journal of Electroanalytical Chemistry. 1979;102:315–319. [Google Scholar]
- Hill TL. Diffusion frequency factors in some simple examples of transition-state rate theory. Proc Natl Acad Sci USA. 1976;73:679–683. doi: 10.1073/pnas.73.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille, B. 1992. Ionic Channels of Excitable Membranes. Sinauer Associates Inc., Sunderland, MA. 607 pp.
- Hollerbach U, Chen D, Nonner W, Eisenberg B. Three-dimensional Poisson-Nernst-Planck Theory of Open Channels. Biophys J. 1999;76:A205. [Google Scholar]
- Kramers HA. Brownian motion in a field of force and the diffusion model of chemical reactions. Physica. 1940;7:284–304. [Google Scholar]
- Kurnikova MG, Coalson RD, Graf P, Nitzan A. A lattice relaxation algorithm for 3D Poisson-Nernst-Planck theory with application to ion transport through the gramicidin A channel. Biophys J. 1999;76:642–656. doi: 10.1016/S0006-3495(99)77232-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Läuger, P. 1991. Electrogenic Ion Pump. Sinauer Associates Inc., Sunderland, MA.
- Lundstrom, M. 1992. Fundamentals of Carrier Transport. Addison-Wesley Publishing Co., New York. 308 pp.
- Newman, J.S. 1991. Electrochemical Systems. 2nd ed. Prentice Hall, Englewood Cliffs, NJ. 560 pp.
- Nonner W, Chen DP, Eisenberg B. Anomalous mole fraction effect, electrostatics, and binding in ionic channels. Biophys J. 1998;74:2327–2334. doi: 10.1016/S0006-3495(98)77942-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonner W, Eisenberg B. Ion permeation and glutamate residues linked by Poisson-Nernst-Planck theory in L-type calcium channels. Biophys J. 1998;75:1287–1305. doi: 10.1016/S0006-3495(98)74048-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott, E. 1997. Chaos in Dynamical Systems. Cambridge University Press, New York. 385 pp.
- Pollak E, Berezhkovskii A, Schuss Z. Activated rate processes: a relation between Hamiltonian and stochastic theories. J Chem Phys. 1994;100:334–339. [Google Scholar]
- Rapoport, D.C. 1997. The Art of Molecular Dynamics Simulations. Cambridge University Press, New York. p. 70.
- Roux B, Karplus M. Ion transport in a gramicidin-like channel: dynamics and mobility. J Phys Chem. 1991;95:4856–4868. [Google Scholar]
- Roux B, Karplus M. Molecular dynamics simulations of the gramicidin channel. Annu Rev Biophys Biomol Struct. 1994;23:731–761. doi: 10.1146/annurev.bb.23.060194.003503. [DOI] [PubMed] [Google Scholar]
- Simonin J-P, Bernard O, Blum L. Ionic solutions in the binding mean spherical approximation. Thermodynamic properties of mixtures of associating electrolytes. J Phys Chem B. 1999;103:699–704. [Google Scholar]
- Tang J, Chen D, Saint N, Rosenbusch J, Eisenberg R. Permeation through porin and its mutant G119D. Biophys J. 1997;72:A108. [Google Scholar]
- van den Brink, J., and G.A. Sawatzky. 1998. Non-conventional screening of the Coulomb interaction. In Electronic Properties of Novel Materials: Progress in Molecular Nanostructures. H. Kuzmany, editor. American Institute of Physics, New York. 152 pp.
- Wolynes P. Dynamics of electrolyte solutions. Annu Rev Phys Chem. 1980;31:345–376. [Google Scholar]
- Wynne-Jones WFK, Eyring H. The absolute rate of reactions in condensed phases. J Chem Phys. 1935;3:492–502. [Google Scholar]