

Summary
The N-aryl carbamate URB602 (biphenyl-3-ylcarbamic acid cyclohexyl ester) is an inhibitor of monoacylglycerol lipase (MGL), a serine hydrolase involved in the biological deactivation of the endocannabinoid 2-arachidonoyl-sn-glycerol (2-AG). Here, we investigated the mechanism by which URB602 inhibits purified recombinant rat MGL using a combination of biochemical and structure-activity relationship (SAR) approaches. We found that URB602 weakly inhibits recombinant MGL (IC50 = 223±63 μM) through a rapid and noncompetitive mechanism. Dialysis experiments and SAR analyses suggest that URB602 acts through a partially reversible mechanism rather than by irreversible carbamoylation of MGL. Finally, URB602 (100 μM) elevates 2-AG levels in hippocampal slice cultures without affecting levels of other endocannabinoid-related substances. Thus, URB602 may provide a useful tool to investigate the physiological roles of 2-AG and explore the potential interest of MGL as a therapeutic target.
Introduction
2-Arachidonoyl-sn-glycerol (2-AG) is a major monoacylglycerol (MAG) species present in mammalian tissues and, along with anandamide, an endogenous ligand for cannabinoid (CB) receptors [1, 2]. These lipid messengers, called endocannabinoids, are produced on demand through cleavage of membrane phospholipid precursors and are implicated in a variety of short-range signaling processes [2, 3]. In the central nervous system (CNS), the endocannabinoids activate CB1 receptors on presynaptic terminals to regulate calcium and potassium channel activity, as well as neurotransmitter release [4]. These neuromodulatory effects have a wide range of functional consequences. In particular, their proposed involvement in the regulation of pain, appetite, and mood has significant therapeutic potential and highlights the importance of understanding the mechanisms by which endocannabinoids are produced and eliminated [5].
The biological deactivation of 2-AG and anandamide is thought to require a two-step process involving carrier-mediated transport into cells and subsequent intracellular hydrolysis [6-8]. 2-AG is hydrolyzed by two or more monoacylglycerol lipase (MGL) enzymes [9, 10], one of which has been molecularly cloned [11, 12], while anandamide is hydrolyzed by fatty-acid amide hydrolase (FAAH) [13] and N-acylethanolamine acid amidase (NAAA) [14].
The cloned MGL is a 303 amino-acid serine hydrolase that resides in the cell cytosol and cleaves MAGs into fatty acid and glycerol [11, 12]. Three lines of evidence suggest that this enzyme plays a key role in the physiological deactivation of 2-AG in the CNS. First, MGL is expressed at high levels in the rodent brain [12], where it is predominantly found in presynaptic nerve terminals of both excitatory and inhibitory neurons [15]. Second, virally induced overexpression of MGL dampens N-methyl-D-aspartate (NMDA)-dependent accumulation of 2-AG in rat cortical neurons [12], while RNAi-mediated silencing of constitutive MGL expression increases 2-AG levels in HeLa cells [16]. Finally, pharmacological blockade of intracellular MGL activity by two structurally unrelated inhibitors, URB602 (biphenyl-3-ylcarbamic acid cyclohexyl ester) and methylarachidonylfluorophosphonate (MAFP), elevates 2-AG levels and enhances 2-AG-mediated signaling in neurons [17, 18]. For example, microinjections of URB602 or MAFP in the periaqueductal grey matter of the midbrain selectively increases 2-AG levels, but not anandamide levels, and enhance CB1 receptor-mediated stress-induced analgesia in rats [17]. Further, administration of URB602 or MAFP prolongs endocannabinoid-mediated changes in synaptic strength in acutely dissected rat hippocampal slices [18]. Even further, administration of URB602 in rodents has been shown to produce antiinflammatory and antinociceptive effects that are selectively blocked by CB2 receptor antagonists [19, 20]. Thus, URB602 and MAFP may be used to investigate the physiological roles of 2-AG and the pharmacological potential of MGL inhibition, provided that appropriate precautions are taken to control for the limitations of these agents. In particular, questions that remain to be addressed concern (i) the molecular identity of the MAG-hydrolyzing enzyme(s) targeted by URB602 and MAFP; and (ii) the mechanisms by which URB602 and MAFP inhibit these enzyme(s). Answering these questions may help resolve current inconsistencies in the literature, such as a recent study reporting that URB602 exerts little or no inhibitory effect on 2-AG hydrolysis in broken-cell preparations of brain tissue [21]. Therefore, in the present study we have sought to (i) reexamine the inhibitory effects of URB602 on MGL activity, using both a purified recombinant MGL and intact brain neurons; and (ii) investigate the mechanism by which URB602 inhibits cloned MGL, using an approach that combined kinetic, dialysis, and structure-activity relationship (SAR) analyses.
Results and Discussion
Expression of Recombinant MGL
We overexpressed recombinant rat MGL in E.coli and purified it by affinity chromatography. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analyses of the purified protein, followed by Coomassie-blue staining or immunoblotting, revealed a single band that closely matched the calculated molecular weight of histidine-tagged cloned rat brain MGL (35.7 kD) (Figure 1A). As expected, recombinant MGL hydrolyzed the MAG substrate 2-oleoylglycerol (2-OG) in a time-dependent and protein concentration-dependent manner (Figure 1B; data not shown).
Figure 1. Expression of Recombinant MGL.

(A) Coomassie staining (left) and Western blot (right) analyses of purified recombinant MGL expressed in E.coli.
(B) Time-course of oleic acid (OA) production from 2-oleoyl-sn-glycerol (2-OG) in the presence of purified MGL (10 ng). Results are the average of two experiments, each performed in duplicate.
Inhibition of Recombinant MGL
Figure 2A shows the concentration-dependent inhibition of purified recombinant MGL by URB602 (IC50 = 223±63 μM, n=4) and by the broad-spectrum lipase inhibitor MAFP (35±13 nM, n=3) under standard conditions (10 min, 37°C) [22, 23, 24]. In some experiments, we measured the ability of URB602 to inhibit MGL activity in assays of 2.5-min duration, obtaining similar results (IC50 = 185±13 μM, n=3). Figure 2A also illustrates the effect of the MGL inhibitor N-arachidonylmaleimide (NAM) [25] (IC50 = 46±7 nM, n=3). Figure 2B depicts the effects of URB602, MAFP, and NAM on recombinant MGL overexpressed in HeLa cells (IC50 values: URB602, 81±13 μM; MAFP, 23±0.1 nM; NAM, 41±19 nM; n=3-4). Notably, URB602 was significantly more potent at inhibiting non-purified MGL expressed in HeLa than purified MGL expressed in E.coli (p<0.05, unpaired Student's t-test), suggesting that post-translational modifications or differences in enzyme assay environment may alter the inhibitory potency of this compound. An alternate interpretation is that HeLa cells express another 2-OG-hydrolyzing enzyme that has higher sensitivity to URB602 than the cloned MGL.
Figure 2. Inhibition of MGL Activity by Various Agents.

(A, B) Effects of URB602 (circles, n=4), MAFP (triangles, n=3), or NAM (squares, n=3 on either purified MGL expressed in E.coli (A) or MGL expressed in HeLa cells (B). Results are expressed as mean ± SEM, each assay performed in duplicate.
(C) Inhibition of MGL activity in cerebellar membranes by URB602, MAFP, or NAM. Results are expressed as mean ± SEM (n=3), each assay performed in triplicate. *p<0.05, **p<0.01, ANOVA, followed Dunnett's test; #p<0.05, unpaired Student's t-test.
Inhibition of Cerebellar MGL
The ability of URB602, MAFP, and NAM to inhibit MGL activity was further investigated using cerebellar membranes. A previous study using this preparation reported no effect of URB602 on 2-AG hydrolysis [21]. We found, however, that both URB602 and NAM inhibited MAG hydrolysis in cerebellar membranes in a concentration dependent manner (Figure 2C). Interestingly, neither agent achieved complete inhibition, even when tested at concentrations that were maximally effective on cloned MGL. By contrast, MAFP virtually eliminated 2-OG hydrolysis, suggesting that cerebellar membranes contain multiple MGL activities, and that URB602 and NAM only affect a subset of such activities.
Kinetic Analyses of MGL Inhibition
To explore the mechanism by which URB602 inhibits MGL, we measured recombinant MGL activity at various substrate concentrations, in the absence or presence of URB602 (0.3 and 1 mM) (Figure 3A). Incubation with URB602 resulted in an increase in the Michaelis-Menten constant (Km) and a decrease in the maximal velocity (Vmax) of the MGL reaction (Table 1). This result suggests that URB602 inhibits MGL activity in a noncompetitive manner. In particular, the reciprocal effect of URB602 on Km and Vmax indicates that the inhibitor may bind with greater affinity to the free enzyme than the enzyme-substrate complex. [26]
Figure 3. Characterization of the Mechanism of MGL Inhibition by URB602.

(A) Michaelis-Menten analysis of the MGL reaction in the presence of vehicle (DMSO, 1%) (circles, n=4), 0.3 mM URB602 (triangles, n=3), or 1 mM URB602 (squares, n=3). Results are expressed as mean ± SEM.
(B) Time-course of the MGL reaction in the presence of 1% DMSO (circles) or 0.3 mM URB602 (triangles). Results are expressed as mean ± SEM (n=5).
(C) Changes in MGL inhibition over time were replotted as percent of maximal inhibition (closed squares); also shown are changes in URB602 concentration over time, plotted as percent of initial concentration (open triangles). Results are expressed as mean ± SEM (n=3).
(D) Effects of dialysis (24 h, 0-4°C) on MGL activity by URB602. Results are expressed as mean ± SEM (n=6). **p<0.01, ***p<0.001, unpaired Student's t-test.
Table 1.
Michaelis-Menten Constant (Km), and Maximal Rate of Reaction (Vmax) for Purified Recombinant MGL Expressed in E.coli
| URB602 (mM) | 0 | 0.3 | 1 |
|---|---|---|---|
| Km (μM) | 16.92 ± 3.188 | 27.05 ± 2.045 | 32.11± 1.083 |
| Vmax (pmol/min/ng) | 120.6 ± 12.44 | 106.1 ± 2.750 | 82.68 ± 6.273 |
Data are represented as mean ± SEM (average of 3 experiments performed in triplicate)
Time-Course of MGL Inhibition
We next examined the time dependence of MGL inhibition by URB602 (0.3 mM). Maximal percent inhibition occurred within 2 min of URB602 addition, and this level remained unchanged for up to 1 h (Figure 3B,C). To determine whether URB602 itself may be hydrolyzed by MGL during the incubation period, we measured URB602 concentrations by liquid chromatography/mass spectrometry (LC/MS). Although a small decrease in URB602 levels was seen immediately after initiating the incubation, possibly due to absorption to glassware, no further change occurred for the rest of the experiment (Figure 3C). The results indicate that URB602 (i) inhibits MGL in a time-independent manner, and (ii) does not serve as a substrate for MGL.
Reversibility of MGL Inhibition
The O-aryl carbamate URB597 (cyclohexylcarbamic acid 3'-carbamoylbiphenyl-3-yl ester) inhibits FAAH activity by an irreversible mechanism that likely involves carbamoylation of an active-site serine residue of the enzyme [27, 28]. To test whether URB602, an N-aryl carbamate, also inhibits MGL irreversibly, we incubated the purified enzyme in the presence of URB602 (30, 100 μM) and then subjected the incubation mixture to extensive dialysis (24 h, 0-4°C). Dialysis partially reversed the inhibition of MGL activity by URB602 (Figure 3D), suggesting that this agent inhibits MGL through a mechanism that is at least partially reversible.
Structure-activity relationship studies
We also investigated the chemical requirements for MGL inhibition by URB602 (Table 2). A series of modifications were made to the two substituents on the carbamate moiety, focusing on the importance of substituent aromaticity (3a), ester moiety (3j, 3l), N-alkylation (3o), shape of the N-biphenyl-3-yl group (3b, 3d-g, 3k), role of the O-cyclohexyl group (3c), and potential of the O-substituent to serve as a leaving group by mimicking the glycerol fragment of 2-AG (3h, 3i, 3m, 3n). Finally, as our kinetic and dialysis results challenged the role of the carbamate group in MGL inhibition, we replaced this group with various isosteric moieties (4a, 5, 6). Only one compound in this focused series, the non-carbamate derivative 4a, was found to be more effective than URB602 at inhibiting MGL activity (IC50 = 115 μM) (Table 2). As compound 4a retains a high steric similarity with URB602, but has much lower tendency to give a covalent bond with a nucleophilic serine, this result supports the possibility that URB602 and 4a are not active site-directed MGL inhibitors.
Table 2.
IC50 Values for Inhibition of Purified Recombinant MGL by Various URB602 Derivatives
| Compound | Structure | IC50 (μM) |
|---|---|---|
| URB602 | 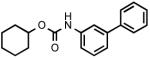 |
223 ± 63 |
| 3a | 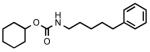 |
> 300 |
| 3b | > 300 | |
| 3c | > 300 | |
| 3d | 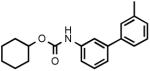 |
> 300 |
| 3e | 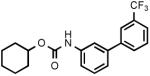 |
> 300 |
| 3f | > 300 | |
| 3g | 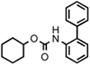 |
> 300 |
| 3h | > 300 | |
| 3i | 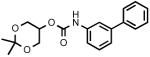 |
> 300 |
| 3j | > 300 | |
| 3k | 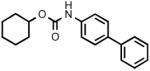 |
> 300 |
| 3l | 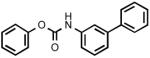 |
243 (n=2) |
| 3m | 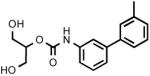 |
> 300 |
| 3n | 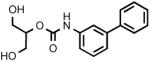 |
> 300 |
| 3o | 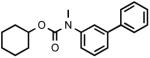 |
> 300 |
| 4a | 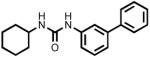 |
115 (n=2) |
| 5 | 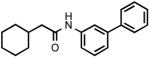 |
> 300 |
| 6 | 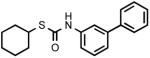 |
> 300 |
Data expressed as mean ± SEM (n=3)
Recently, a carbamate, SPB 01403 [n-butylcarbamic acid 4-(4,5-dihydrothiazol-2-yl)phenyl ester], that inhibits MAG-hydrolyzing activity in rat cerebellar membranes with an IC50 value of 31 μM has been reported [29] (Figure 4). Although belonging to the same chemical class as URB602, this compound has a carbamate group with a different electronic environment, due to the presence of an aromatic O-substituent with an electron-withdrawing group in a conjugated position. While the mechanism of this inhibitor has not been elucidated, it may differ from that which is reported here for URB602, given the large difference in reactivity between their carbamate fragments.
Figure 4.
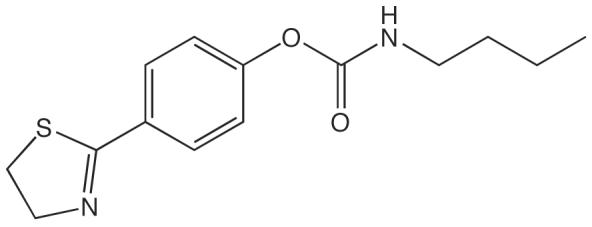
Structure of MGL inhibitor SPB 01403
Effect of URB602 on 2-AG and Anandamide Levels in Neurons
Previous studies indicate that URB602 protects endogenous 2-AG from degradation in intact brain tissue [17, 18]. Thus, as an additional test of the ability of URB602 to selectively inhibit MGL, we examined the effects of this agent on 2-AG content in rat hippocampal slices in primary cultures. As shown in Figure 5A, treatment with URB602 (100 μM, 25 min) produced a significant elevation in 2-AG levels compared to vehicle (0.1% DMSO in Dulbecco-modified Eagle's medium) (n=5). In the same slices, the levels of the FAAH substrates anandamide (Figure 5B) and palmitoylethanolamide (PEA) (Figure 5C) were not affected. These findings suggest that URB602 inhibits the degradation of 2-AG without affecting that of other endocannabinoid-related substances.
Figure 5. Effects of URB602 on Endocannabinoid Levels in Rat Brain Slices.

Effects of URB602 (100 μM) or vehicle (0.1% DMSO in DMEM) on (A) 2-AG levels; (B) anandamide levels, and (C) PEA levels in rat organotypic hippocampal slices in culture. Results are expressed as mean ± SEM (n=4-5). **p<0.01, ns = not significant, unpaired Student's t-test.
Significance
In the present study, we show that the N-aryl carbamate URB602 inhibits purified recombinant rat brain MGL and increases 2-AG levels in intact brain neurons. We further show that MGL inhibition by URB602 occurs through a rapid, noncompetitive and partially reversible mechanism, suggesting that URB602 – unlike O-aryl carbamate FAAH inhibitors such as URB597 [27] – does not covalently modify MGL. SAR studies confirm this possibility, pointing to substituted ureas as a starting point for the discovery of novel MGL inhibitors. Such agents are important for three reasons. First, they may help uncover specific functions served by 2-AG in endocannabinoid signaling. Second, they may provide an experimental tool to validate MGL as a therapeutic target. Lastly, they may serve as chemical scaffolds for the development of drug-like MGL inhibitors with potential applications in the treatment of stress-related disorders [17], pain [19], and inflammation [20]. Thus, despite its limited potency, URB602 remains a useful tool to investigate the roles of 2-AG and validate MGL as a pharmacological target.
Experimental Procedures
Chemicals
MAFP and NAM were purchased from Cayman Chemical (Ann Arbor, MI), 2-OG from Sigma-Aldrich (St. Louis, MO), and 2-[2H8]-AG from Cayman Chemical; [2H4]-anandamide and [2H4]-palmitoylethanolamide were synthesized in the lab [30].
Chemistry
All chemicals were purchased from Sigma-Aldrich in the highest quality commercially available. Solvents were RP grade, unless otherwise indicated. Chromatographic separations were performed on open silica gel columns by flash chromatography (Kieselgel 60, 0.040-0.063 mm, Merck). The weight ratio between the material loaded in the column and the stationary phase was about 1:50. Thin-layer chromatography (TLC) analyses were performed on precoated silica gel sheets (Kieselgel 60 F254, Merck). Melting points were determined on a Büchi SMP-510 capillary melting point apparatus. EI-MS spectra (70 eV) were recorded with a Fisons Trio 1000 spectrometer; only molecular ions (M+) and base peaks are given. 1H NMR spectra were recorded on an AVANCE Bruker 200 spectrometer; chemical shifts are reported in δ scale and were measured by using the central peak of the solvent. Infrared spectra (IR) were obtained on a Shimadzu FT-8300, or a Nicolet Avatar, spectrometer; absorbances are reported in ν (cm-1).
Synthetic procedures
URB602 was synthesized as previously reported [17]. Carbamates 3a-l were prepared by addition to carbonyldiimidazole (CDI) of an opportune amine (1) and an alcohol (2), all commercially available except 5-hydroxy-2,2-dimethyl-1,3-dioxane (2a) prepared according to literature [31], biphenyl-3-ylamine (1a) [32], 5-phenyl-1-pentylamine (1c) [33], and substituted anilines 1d-f. 1d,e were obtained by a Suzuki coupling of 3-bromoaniline and the opportune boronic acid. 1f resulted from coupling of potassium 3-phenylphenolate and 3-bromoaniline at high temperature in the presence of copper powder. N-Methylcarbamate 3o was obtained by alkylation of URB602 in basic conditions. Acid hydrolysis of acetal 3i afforded 3n. Urea 4a was synthesized by addition of 1c and cyclohexylamine to CDI. Amide 5 was prepared from the corresponding acid via acyl chloride; thiocarbamate 6 by addition of 1c and cyclohexanethiol to CDI.
N-Aryl-O-alkylcarbamates 3a-l
To a stirred solution of the suitable amine (1) (2 mmol) in dry CH3CN (22 ml), CDI (0.648 g, 4 mmol; 3e: 0.324 g, 2 mmol) and triethylamine (TEA) (0.71 ml, 5 mmol) were added. After the reactants were refluxed for 4 h (2 h: 3h,i,k,l; 8 h: 3g; 20 h: 3b,j), the appropriate alcohol 2 (2 mmol) was added, and the mixture reacted for the opportune time (3f: 1 h; 3c-e,j: 2 h; 3h,i,k: 4 h; 3g: 8 h; 3l: 9 h; 3a,b: 20 h). In some cases a further amount of alcohol (3d,e: 1 mmol; 3a: 6 mmol) and heating (3d,e: 0.5 h; 3a: 24 h) were necessary. The mixture was cooled and concentrated. Purification of the residue by column chromatography (cyclohexane/EtOAc 9:1: 3a,d,f,j; 85:15: 3c; 8:2: 3b,e,g,l; 7:3 then EtOAc/MeOH 98:2: 3h; petroleum ether/EtOAc 75:25: 3i) and, in the case of solids, recrystallization gave the desired compounds.
(5-Phenylpentyl)carbamic acid cyclohexyl ester (3a)
Colorless oil. Yield: 26% (0.150 g). MS (EI): 289 (M+); 207 (100). 1H NMR (CDCl3): 1.28-1.93 (m, 16H); 2.62 (t, 2H); 3.16 (t, 2H); 4.64 (br s, 2H); 7.16-7.32 (m, 5H) ppm. IR (Neat): 3344, 3083, 3064, 3021, 2932, 2847, 1689.
Phenylcarbamic acid cyclohexyl ester (3b)
White crystals. Yield: 91% (0.399 g). Mp: 81-83°C (petroleum ether; sealed capillary tube) (lit. 81-82°C). MS (EI): 219 (M+); 93 (100). 1H NMR and IR are according to literature [34].
Biphenyl-3-ylcarbamic acid 5-phenylpentyl ester (3c)
White tufts. Yield: 96% (0.689 g). Mp: 92-93°C (EtOH; sealed capillary tube). MS (EI): 359 (M+); 91 (100). 1H NMR (CDCl3): 1.45-1.80 (m, 6H); 2.65 (t, 2H); 4.19 (t, 2H); 6.66 (br s, 1H); 7.17-7.66 (m, 14H) ppm. IR (Nujol): 3316, 1703.
(3'-Methylbiphenyl-3-yl)carbamic acid cyclohexyl ester (3d)
Yellow oil. Yield: 35% (0.216 g). MS (EI): 309 (M+); 183 (100). 1H NMR (CDCl3): 1.26-1.94 (m, 10H); 2.42 (s, 3H); 4.78 (m, 1H); 6.60 (d, 1H); 7.15-7.67 (m, 8H) ppm. IR (Neat): 3321, 3055, 3022, 2925, 2849, 1730, 1698.
(3'-Trifluoromethylbiphenyl-3-yl)carbamic acid cyclohexyl ester (3e)
White needles. Yield: 56% (0.406 g). Mp: 72-74°C (petroleum ether). MS (EI): 363 (M+); 281 (100). 1H NMR (CDCl3): 1.26-1.99 (m, 10H); 4.78 (m, 1H); 6.67 (br s, 1H); 7.30-7.83 (m, 8H) ppm. IR (Nujol): 3321, 1692.
[3-(Biphenyl-3-yloxy)phenyl]carbamic acid cyclohexyl ester (3f)
Pale yellow oil. Yield: 13% (0.101 g). MS (EI): 387 (M+); 287 (100). 1H NMR (CDCl3): 1.27-1.90 (m, 10H); 4.74 (m, 1H); 6.56 (br s, 1H); 6.73-7.59 (m, 13H) ppm. IR (Neat): 3397, 3316, 1714.
Biphenyl-2-ylcarbamic acid cyclohexyl ester (3g)
Brown oil. Yield: 97% (0.572 g). MS (EI): 295 (M+); 169 (100). 1H NMR (CDCl3): 1.27-1.91 (m, 10H); 4.73 (m, 1H); 6.59 (s, 1H); 7.08-7.55 (m, 8H); 8.14 (d, 1H) ppm. IR (Nujol): 3421, 1728.
Biphenyl-3-ylcarbamic acid 2,3-dihydroxypropyl ester (3h)
White solid. Yield: 21% (0.121 g). Mp: 130-132°C (EtOAc/petroleum ether; sealed capillary tube). MS (EI): 287 (M+); 195 (100). 1H NMR (d6-acetone): 3.58 (d, 2H); 3.88 (m, 2H); 4.20 (m, 3H); 7.29-7.66 (m, 8H); 7.91 (s, 1H); 8.80 (s, 1H) ppm. IR (Neat): 3362, 1790, 1714.
Biphenyl-3-ylcarbamic acid 2,2-dimethyl[1,3]dioxan-5-yl ester (3i)
White crystals. Yield: 64% (0.419 g). MS (EI): 327 (M+); 195 (100). Mp: 116-117°C [(iPr)2O]. 1H NMR (CDCl3): 1.49 (d, 6H); 3.92-4.24 (m, 4H); 4.74 (m, 1H); 6.95 (s, 1H); 7.29-7.65 (m, 9H) ppm. IR (Nujol): 3348, 1725.
Biphenyl-3-ylcarbamic acid biphenyl-3-yl ester (3j)
Pearly crystals. Yield: 18% (0.131 g). Mp: 166-169°C (EtOH; sealed capillary tube). MS (EI): 365 (M+); 115 (100). 1H NMR (CDCl3): 7.07 (br s, 1H); 7.18-7.24 (m, 1H); 7.33-7.51 (m, 12H); 7.59-7.64 (m, 4H); 7.78 (br s, 1H) ppm. IR (Nujol): 3337, 1704.
Biphenyl-4-ylcarbamic acid cyclohexyl ester (3k)
White crystals. Yield: 37% (0.218 g). Mp: 142-143°C (EtOH). MS (EI): 295 (M+), 213 (100). 1H NMR (CDCl3): 1.26-1.95 (m, 10H); 4.78 (m, 1H); 6.65 (s, 1H); 7.32-7.60 (m, 9H) ppm. IR (Nujol): 3376, 3338, 1698.
Biphenyl-3-ylcarbamic acid phenyl ester (3l)
Pearly crystals. Yield: 15% (0.088 g). Mp: 84-85°C (EtOH). MS (EI): 289 (M+); 94 (100). 1H-NMR (CDCl3): 7.08 (br s, 1H); 7.25 (m, 3H); 7.42 (m, 8H); 7.62 (m, 2H); 7.77 (br s, 1H) ppm. IR (Nujol): 3293, 1710.
(3'-Methylbiphenyl-3-yl)carbamic acid 2-hydroxy-1-hydroxymethylethyl ester (3m)
To a solution of 3'-methylbiphenyl-3-ylamine (1d) (1.03 g, 5.63 mmol) in dry CH3CN (80 ml) CDI (1.754 g, 10.83 mmol) and TEA (2.5 ml, 34.5 mmol) were added. The mixture was refluxed for 2 h, then 2a was added (0.740 g, 5.61 mmol) and again refluxed (4 h). The solvent was evaporated, and purification of the residue by column chromatography (cyclohexane/EtOAc 8:2) gave a sample (0.800 g) of (3'-methylbiphenyl-3-yl)carbamic acid 2,2-dimethyl[1,3]dioxan-5-yl ester [MS (EI): 341 (M+), 209 (100)] and a product whose Rf corresponds to that of 1d (approximate ratio 1:1 by TLC). This mixture was added to a 1:50 mixture of 37% HCl and THF (20 ml) and allowed to react under stirring at room temperature overnight. The solvent was evaporated, and purification of the residue by column chromatography (EtOAc) gave 3m as a colorless, fruit-smelling oil which solidified in the freezer and, after recrystallization, turned into a white solid. Yield: 27% (0.450 g). Mp: 100-103°C (EtOAc/petroleum ether; sealed capillary tube). MS (EI): 301 (M+); 209 (100). 1H NMR (d6-acetone): 2.40 (s, 3H); 3.76 (m, 4H); 3.95 (m, 2H); 4.87 (m, 1H); 7.57 (m, 1H); 7.16-7.46 (m, 6H); 7.93 (t, 1H); 8.70 (s, 1H) ppm. IR (Neat): 3342, 2926, 1710.1-(3'-methylbiphenyl-3-yl)-3-(3'-methylbiphenyl-4-yl)urea (4b) [0.230 g; Mp: 214-217°C (EtOH); MS (EI): 392 (M+), 209 (100); 1H NMR (CDCl3): 2.09 (s, 6H); 7.17-7.99 (m, 14H); 7.89 (m, 2H); 8.28 (s, 2H). IR (Nujol): 3287, 1710] was also isolated as a side-product of the reaction.
Biphenyl-3-ylcarbamic acid 2-hydroxy-1-hydroxymethylethyl ester (3n)
To a 1:50 mixture of 37% HCl and THF (8 ml) 3i (0.170 g; 0.52 mmol) was added and the solution reacted under stirring at room temperature for 3 h. The mixture was quenched with 2N NaHCO3 and extracted with EtOAc. The organic phase was dried and concentrated. Purification of the residue by column chromatography (EtOAc/MeOH 200:5) and recrystallization gave 3n as colorless crystals. Yield: 40% (0.060 g). Mp: 100-102°C (EtOAc/petroleum ether; sealed capillary tube). MS (EI): 287 (M+); 195 (100). 1H NMR (d6-acetone): 3.75 (m, 4H); 3.97 (br s, 2H); 4.86 (m, 1H); 7.29-7.67 (m, 8H); 7.94 (s, 1H); 8.72 (s, 1H) ppm. IR (Neat): 3289, 1779, 1692.
Biphenyl-3-ylmethylcarbamic acid cyclohexyl ester (3o)
To a solution of URB602 (0.127 g, 0.43 mmol) in dry DMF (5 ml) Cs2CO3 (0.420 g, 1.29 mmol) and tetrabutylammonium iodide (0.480 g, 1.29 mmol) were added. The mixture was stirred at room temperature for 30 min, added of CH3I (0.183 g, 0.8 ml, 1.29 mmol) and allowed to react further (8 h). H2O was then added and the mixture extracted with EtOAc. The organic phase was washed with brine, dried and concentrated. Purification of the residue by column chromatography (cyclohexane/EtOAc 7:3) afforded 3o as a colorless oil. Yield: 49% (0.065 g). MS (EI): 309 (M+); 152 (100). 1H NMR (CDCl3): 1.26-1.84 (m, 10H); 3.36 (s, 3H); 4.77 (m, 1H); 7.32-7.60 (m, 9H) ppm. IR (Neat): 3061, 3033, 2930, 2849, 1698.
1-(Biphenyl-3-yl)-3-cyclohexyl urea (4a)
To a solution of 1c (0.169 g, 2 mmol) and CDI (0.324 g, 2 mmol) in dry CH3CN (11 ml) TEA (0.253 g, 0.35 ml, 2.5 mmol) was added, and the mixture stirred under reflux for 2 h. Then cyclohexylamine (0.099 g, 2.5 mmol, 0.105 ml) was added, the mixture stirred under reflux for 4 h and concentrated. Purification of the residue by column chromatography (cyclohexane/EtOAc 8:2) and recrystallization gave 4a as a white solid. Yield: 51% (0.300 g). Mp: 179-183°C (EtOH). MS (EI): 294 (M+); 169 (100). 1H NMR (d6-acetone): 0.90-1.72 (m, 10H); 3.38 (m, 1H); 6.29 (m, 1H); 7.03-7.36 (m, 8H); 7.55 (s, 1H); 7.86 (br s, 1H) ppm. IR (Nujol): 3332, 1714.
N-Biphenyl-3-yl-2-cyclohexylacetamide (5)
To a cooled (0°C) solution of cyclohexylacetic acid (0.142 g, 0.14 ml, 1 mmol) in dry THF (3.3 ml) oxalyl chloride (0.508 g, 0.35 ml, 4 mmol) was added under nitrogen. The mixture was stirred at room temperature for 2 h, concentrated, and the residue dissolved in CH2Cl2 (3.3 ml). TEA (0.283 g, 0.39 ml, 2.8 mmol) and 1c (0.220 g, 1.3 mmol) in a minimal amount of CH2Cl2 were added, the mixture stirred for 20 h, and the solvent was concentrated. The purification of the residue by column chromatography (cyclohexane/EtOAc 75:25) and recrystallization gave 5 as a white fluffy solid. Yield: 48% (0.140 g). Mp: 108-109°C (EtOH). MS (EI): 293 (M+); 169 (100) 1H NMR (CDCl3): 0.93-1.97 (m, 11H); 2.25 (d, 2H); 7.30-7.62 (m, 9H); 7.79 (s, 1H) ppm. IR (Nujol): 3283, 1649.
Biphenyl-3-ylthiocarbamic acid S-cyclohexyl ester (6)
To a solution of 1d (0.372 g, 2.2 mmol) in dry CH3CN (2 ml) were added CDI (0.648 g, 4 mmol) and TEA (0.515 g, 0.71 ml, 5.1 mmol). After refluxing for 4 h, cyclohexanethiol (0.232 g, 0.25 ml, 2 mmol) was added. The mixture was refluxed for 3 h, cooled, and evaporated. Purification of the residue by column chromatography (cyclohexane/EtOAc 9:1) and recrystallization gave 6 as pale yellow crystals. Yield: 70% (0.435 g). Mp: 108-111°C (EtOH, sealed capillary tube). MS (EI): 311 (M+); 195 (100). 1H-NMR (d6-DMSO): 1.22-2.09 (m, 10H); 3.57 (m, 1H); 7.09 (br s, 1H); 7.25 -7.71 (m, 9H) ppm. IR (Nujol): 3294, 3137, 3082, 1643.
3-(3'-Substituted)phenylanilines 1d,e
To a stirred solution of 3-bromoaniline (1.032 g, 6 mmol) in toluene (42 ml) Pd(PPh3)4 (0.282 mg, 0.244 mmol), a solution of Na2CO3 (3.990 g, 37.6 mmol) in H2O (18 ml), and a solution of the suitable 3-substituted phenylboronic acid (6.72 mmol) in EtOH (18 ml) were added. The mixture was vigorously stirred under reflux for 1 h, cooled, added to water and extracted with EtOAc. The combined organic layers were dried and concentrated. Purification of the residue by column chromatography (cyclohexane/EtOAc 8:2) and, in the case of 1e, recrystallization gave 1d,e.
3'-Methylbiphenyl-3-ylamine (1d)
Opaque yellow oil that solidifies in freezer. Yield: 56% (0.614 g). Mp: 45.5-46.5°C (trituration with petroleum ether). MS (EI): 183 (M+, 100). 1H NMR (CDCl3): 2.43 (s, 3H); 3.72 (br s, 2H); 6.70 (m, 1H); 6.93 (t, 1H); 7.01 (m, 1H); 7.15-7.40 (m, 5H) ppm. IR (Neat): 3457, 3376, 3213, 3028, 2919, 2843, 1622, 1600.
3'-Trifluoromethylbiphenyl-3-ylamine (1e)
Colorless needles. Yield: 43% (0.610 g). Mp: 40-42°C (petroleum ether). MS (EI): 237 (M+, 100). 1H NMR (CDCl3): 3.83 (br s, 2H); 6.74 (m, 1H); 6.97 (m, 2H); 7.27 (m, 1H); 7.56 (m, 2H); 7.78 (m, 2H) ppm. IR (Nujol): 3446, 3332, 3196, 1622.
3-(Biphenyl-3-yloxy)phenylamine (1f)
To a cooled (0°C) solution of potassium 3-phenylphenolate [obtained by adding 3-phenylphenol (1.81 g, 10.65 mmol) to a solution of potassium (0.410 g, 10.65 mmol) in dry MeOH (40 ml), stirring the mixture for 0.5 h at room temperature, evaporating the solvent, and drying the residue] 3-bromoaniline (1.672 g, 1.16 ml, 10.65 mmol) and copper (0.630 g, 10 mmol) were added. The mixture was stirred at 160-170°C for 12 h, cooled, and concentrated. Purification of the residue by column chromatography (cyclohexane/EtOAc 8:2) gave 1f as a brown oil. Yield: 23% (0.642 g). MS (EI): 261 (M+); 170 (100). 1H NMR (CDCl3): 3.70 (br s, 2H); 6.44 (m, 2H); 6.99-7.61 (m, 11H) ppm. IR (Neat): 3468, 3381, 3218, 3066, 3033, 2925, 2849, 1736.
MGL Cloning
Rat MGL cDNA was cloned out of the pBluescript-SK vector using the following primers: 5'-CGCGGCAGCCATATGCTGAGGCAAGTTCACC (forward primer) and 5'-AGCAGCCGGATCCTCTCAGGGTAGACACCTAG (reverse primer). These primers introduced a BamHI and an NdeI endonuclease restriction site at the 5' and 3' end of the cDNA, respectively. Platinum Pfx DNA polymerase (Invitrogen, Carlsbad, CA) was used to run the polymerase chain reaction (PCR), followed by BamHI/NdeI double digest of the PCR product and the pET15b vector (Novagen, La Jolla, CA) containing an n-terminal histidine tag. The vector and the PCR product were ligated using T4 DNA ligase (Promega, Madison, WI) following the dephosphorylation of the pET15b vector DNA.
The ligation setup was transformed into DH5α E.coli cells and plated on a Luria Broth agar plate containing 50 μg/ml ampicillin. Ten colonies were selected and their plasmid DNAs were purified according to the QIAGEN plasmid DNA miniprep Kit protocol (QIAGEN, Valencia, CA). Restriction digest analysis resulted in four positive clones, which were subjected to DNA sequencing, confirming the correct sequence.
MGL Expression and Purification
Clone 2 was expressed in Rosetta 2(DE3)pLysS E. coli cells (Novagen) at 37°C using 1 mM isopropyl-beta-D-thiogalactopyranoside (IPTG). This clone was used for all subsequent protein expression experiments. 4 L of LB + 50 μg/ml ampicillin were inoculated with 40 ml of overnight culture of Rosetta 2(DE3)pLysS cells transformed with pET15b/MGL grown in LB + 50 μg/ml ampicillin. Cells were grown at 37°C until the optical density (OD) at 600 nm reached 0.8. At this point, addition of IPTG to a final concentration of 1 mM induced MGL overexpression. After 4 h, the cells were harvested by centrifugation at 6,000g for 15 min and the bacterial pellet was resuspended in 100 ml of lysis buffer (50 mM HEPES pH 7.4, 300 mM NaCl, 10 mM MgCl2, 3 mM β-mercaptoethanol, 0.5 mM benzamidine, 10 μM E-64 and 10 μg/ml aprotinin). The cells were lysed using a French press and the cell lysate centrifuged at 3,000g for 1 h at 4°C to separate the membrane fraction from the cell debris. The supernatant (membrane fraction) was then subjected to another centrifugation at 35,000g for 1 h at 4°C. The pellet was resuspended in 50 mM HEPES pH 7.4., 300 mM NaCl, 3 mM β-mercaptoethanol, 1% Triton X-100, stirred for about 30 min, and centrifuged again at 5,000g for 1 h at 4°C. The supernatant was loaded onto a TALON column (Clontech, Mountain View, CA) equilibrated with 50 mM HEPES pH 7.4, 300 mM NaCl, 3 mM β-mercaptoethanol, 0.1% Triton X-100 (buffer A). The column was washed with 5 volumes of buffer A, and the protein subsequently eluted from the column using a step gradient of imidazole ranging from 10 to 200 mM imidazole in buffer A (5 column volumes each). The protein eluted at about 75 mM imidazole.
Protein Analysis
Protein concentration of the purified MGL was determined by Coomassie-blue staining using fatty acid-free bovine serum albumin (BSA) (Sigma-Aldrich) as a standard. SDS-PAGE and Western blotting were performed as previously described [35], using 4-20% Tris-Glycine gels (Invitrogen). For Western blotting, proteins were transferred onto an Immun-Blot PVDF membrane (BioRad, Hercules, CA) using a Semiphor Transphor Unit (Amersham, Piscataway, NJ), and incubated with a rabbit MGL antibody [12]. Immunoreactive bands were visualized using the ECL-Plus Kit (Amersham).
Cerebellar Membrane Preparation
Male Wistar rats were anesthetized by halothane, decapitated, and cerebella (minus brainstem) were dissected and placed immediately in 10 volumes of ice-cold 20mM Tris, pH 7.5 with 0.32 M sucrose. Tissue was homogenized and potterized, then centrifuged at 1000g for 10 minutes at 4°C. The supernatant was removed and subjected to further centrifugation at 27,000g for 30 minutes at 4°C. Pellet was resuspended in 50mM Tris, pH 7.5. Protein concentration was measured by Bradford Assay.
Measurement of MGL Activity
The final reaction consisted of 0.445 ml of assay buffer (50 mM Tris-HCL, pH 8.0, 0.5 mg/ml BSA, fatty acid-free) containing either 10 ng of purified E.coli MGL, 200 ng of non-purified HeLa MGL [12], or 50 μg of cerebellar membranes, 50 μl of 2-OG (prepared in assay buffer, 10 μM final), and 5 μl of URB602, MAFP, or NAM (prepared in DMSO), for a final total reaction volume of 0.5 ml. Final concentration of vehicle (1% DMSO) had no effect on MGL activity. After 10 min incubation at 37°C, reactions were stopped by addition of 3 ml of chloroform/methanol (2:1, vol:vol), containing heptadecanoic acid (5 nmol) (Nu-chek Prep, Elysian, MN) as an internal standard. After centrifugation at 2,000g at 4°C for 10 min, the organic layers were collected and dried under N2. The residues were suspended in 50 μl of chloroform/methanol (1:3, vol:vol) and analyzed by LC/MS. We used a reversed-phase XDB Eclipse C18 column (50x4.6 mm i.d., 1.8 μm, Agilent Technologies, Wilmington, DE) eluted with a linear gradient from 90% to 100% of A in B for 2.5 min at a flow rate of 1.5 ml/min with column temperature at 40°C. Mobile phase A consisted of methanol containing 0.25% acetic acid and 5 mM ammonium acetate; mobile phase B consisted of water containing 0.25% acetic acid and 5 mM ammonium acetate. ESI was in the negative mode, capillary voltage was set at 4 kV and fragmentor voltage was 100V. N2 was used as drying gas at a flow rate of 13 liters/min and a temperature of 350°C. Nebulizer pressure was set at 60 psi. For quantification purposes, we monitored the [M-H]− ions of m/z = 281.3 for oleic acid and m/z = 269 for heptadecanoic acid.
Measurement of URB602
Samples containing either URB602 (300 μM), MGL (1.4 pM), or both URB602 and MGL were incubated at 37°C for 30 min in assay buffer. At various time points, the reaction was stopped with an equal volume of ice-cold methanol and directly analyzed in positive ionization mode by LC/MS. We used a SB-CN column (150x2.1 mm i.d., 5 μm, Agilent) eluted with a linear gradient of methanol in water containing 0.25% acetic acid and 5 mM ammonium acetate (from 60% to 100% of methanol in 8 min) at a flow rate of 0.5 ml/min with column temperature at 50°C. Capillary voltage was set at 4 kV and fragmentor voltage was 100V. Nebulizer pressure was set at 60 psi. N2 was used as drying gas at a flow rate of 13 liters/min and a temperature of 350°C. ESI was in the positive mode and a full scan spectrum was acquired from m/z 100 to 600. Extracted ion chromatograms were used to quantify URB602 ([M+H]+, m/z 296).
Hippocampal Slice Cultures
Hippocampal organotypic slice cultures were prepared from Wistar rats as previously described [36]. Briefly, postnatal day-9 rats were sacrificed by cryoanesthesia, hippocampi were dissected from 0.4 mm coronal slices prepared using a vibratome and 3 hippocampi per well were incubated in Neurobasal medium on a Millicell culture inserts (Millipore, Billerica, MA). Slices were maintained for 8 days at 37°C with 5% CO2 before treatment with either URB602 (100 μM) or vehicle (0.1% DMSO in DMEM) for 25 min at 37°C. Reactions were stopped by a wash with ice-cold PBS, followed by fixation with 4% paraformaldehyde for 30 minutes at room temperature. Slices were then extracted in 1 ml of ice-cold methanol/chloroform/water (1:1:2, vol:vol:vol) containing 500 pmol of 2-[2H8]-AG, and 10 pmol each of [2H4]-anandamide, and [2H4]-palmitylethanolamide, added as internal standards. Organic phases were recovered, evaporated under N2, reconstituted in 50 μl chloroform/methanol (1:3, vol:vol) and analyzed by LC/MS as previously described [18].
Acknowledgements
The authors would like to thank Dr. Faizy Ahmed (Agilent Technologies) for his advice on LC/MS-related experiments. This work was supported by the National Institute on Drug Abuse (R01 DA-012447), UC Discovery (02-10337), the NIH Training Program in Cellular and Molecular Neuroscience (T32NS007444-7), and the Italian Ministry of University and Research (2005032713_002).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem. Biophys. Res. Commun. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- 2.Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388:773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- 3.Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, Piomelli D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- 4.Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol. Rev. 2003;83:1017–1066. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- 5.Piomelli D. The endocannabinoid system: a drug discovery perspective. Curr. Opin. Investig. Drugs. 2005;6:672–679. [PubMed] [Google Scholar]
- 6.Beltramo M, Stella N, Calignano A, Lin SY, Makriyannis A, Piomelli D. Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science. 1997;277:1094–1097. doi: 10.1126/science.277.5329.1094. [DOI] [PubMed] [Google Scholar]
- 7.Hillard CJ, Edgemond WS, Jarrahian A, Campbell WB. Accumulation of N-arachidonoylethanolamine (anandamide) into cerebellar granule cells occurs via facilitated diffusion. J. Neurochem. 1997;69:631–638. doi: 10.1046/j.1471-4159.1997.69020631.x. [DOI] [PubMed] [Google Scholar]
- 8.Piomelli D. The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- 9.Goparaju SK, Ueda N, Taniguchi K, Yamamoto S. Enzymes of porcine brain hydrolyzing 2-arachidonolglycerol, an endogenous ligand of cannabinoid receptors. Biochem. Pharmacol. 1999;57:417–423. doi: 10.1016/s0006-2952(98)00314-1. [DOI] [PubMed] [Google Scholar]
- 10.Muccioli GG, Xu C, Odah E, Cudaback E, Cisneros JA, Lambert DM, López Rodríguez ML, Bajjalieh S, Stella N. Identification of a novel endocannabinoid-hydrolyzing enzyme expressed by microglial cells. J. Neurosci. 2007;27:2883–2889. doi: 10.1523/JNEUROSCI.4830-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karlsson M, Contreras JA, Hellman U, Tornqvist H, Holm C. cDNA cloning, tissue distribution, and identification of the catalytic triad of monoglyceride lipase. Evolutionary relationship to esterases, lysophospholipases, and haloperoxidases. J. Biol. Chem. 1997;272:27218–27223. doi: 10.1074/jbc.272.43.27218. [DOI] [PubMed] [Google Scholar]
- 12.Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, Kathuria S, Piomelli D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc. Natl. Acad. Sci. USA. 2002;99:10819–10824. doi: 10.1073/pnas.152334899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 14.Tsuboi K, Sun YX, Okamoto Y, Araki N, Tonai T, Ueda N. Molecular characterization of N-acylethanolamine-hydrolyzing acid amidase, a novel member of the choloylglycine hydrolase family with structural and functional similarity to acid ceramidase. J. Biol. Chem. 2005;280:11082–11092. doi: 10.1074/jbc.M413473200. [DOI] [PubMed] [Google Scholar]
- 15.Gulyas AI, Cravatt BF, Bracey MH, Dinh TP, Piomelli D, Boscia F, Freund TF. Segregation of two endocannabinoid-hydrolyzing enzymes into pre- and postsynaptic compartments in the rat hippocampus, cerebellum and amygdala. Eur. J. Neurosci. 2004;20:441–458. doi: 10.1111/j.1460-9568.2004.03428.x. [DOI] [PubMed] [Google Scholar]
- 16.Dinh TP, Kathuria S, Piomelli D. RNA interference suggests a primary role for monoacylglycerol lipase in the degradation of the endocannabinoid 2-arachidonoylglycerol. Mol. Pharmacol. 2004;66:1260–1264. doi: 10.1124/mol.104.002071. [DOI] [PubMed] [Google Scholar]
- 17.Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker M, Holmes PV, Crystal JD, et al. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–1112. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- 18.Makara JK, Mor M, Fegley D, Szabo SI, Kathuria S, Astarita G, Duranti A, Tontini A, Tarzia G, Rivara S, et al. Selective inhibition of 2-AG hydrolysis enhances endocannabinoid signaling in hippocampus. Nat. Neurosci. 2005;8:1139–1141. doi: 10.1038/nn1521. [DOI] [PubMed] [Google Scholar]
- 19.Guindon J, Desroches J, Beaulieu P. The antinociceptive effects of intraplantar injections of 2-arachidonoyl glycerol are mediated by cannabinoid CB2 receptors. Br. J. Pharmacol. 2007;150:693–701. doi: 10.1038/sj.bjp.0706990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Comelli F, Giagnoni G, Bettoni I, Colleoni M, Costa B. The inhibition of monoacylglycerol lipase by URB602 showed an anti-inflammatory and anti-nociceptive effect in a murine model of acute inflammation. Br. J. Pharmacol. 2007 August 13; doi: 10.1038/sj.bjp.0707425. advance online publication. 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saario SM, Palomäki V, Lehtonen M, Nevalainen T, Järvinen T, Laitinen JT. URB754 has no effect on the hydrolysis or signaling capacity of 2-AG in the rat brain. Chem. Biol. 2006;13:811–814. doi: 10.1016/j.chembiol.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 22.Dinh TP, Freund TF, Piomelli D. A role for monoglyceride lipase in 2-arachidonoylglycerol inactivation. Chem. Phys. Lipids. 2002;121:149–158. doi: 10.1016/s0009-3084(02)00150-0. [DOI] [PubMed] [Google Scholar]
- 23.Deutsch DG, Omeir R, Arreaza G, Salehani D, Prestwich GD, Huang Z, Howlett A. Methyl arachidonyl flurophosphonate: a potent irreversible inhibitor of anandamide amidase. Biochem. Pharmacol. 1997;53:255–260. doi: 10.1016/s0006-2952(96)00830-1. [DOI] [PubMed] [Google Scholar]
- 24.Lio YC, Reynolds LJ, Balsinde J, Dennis EA. Irreversible inhibition of Ca2+-independent phospholipase A2 by methyl arachidonyl fluorophosphonate. Biochim. Biophys. Acta, Lipids Lipid Metab. 1996;1302:55–60. doi: 10.1016/0005-2760(96)00002-1. [DOI] [PubMed] [Google Scholar]
- 25.Saario SM, Salo OMH, Nevalainen T, Poso A, Laitinen JT, Järvinen T, Niemi R. Characterization of the sulfhydryl-sensitive site in the enzyme responsible for hydrolysis of 2-arachidonoyl-glycerol in rat cerebellar membranes. Chem. Biol. 2005;12:649–656. doi: 10.1016/j.chembiol.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 26.Copeland RA. Reversible modes of inhibitor interactions with enzymes. In: Copeland RA, editor. Evaluation of Enzyme Inhibitors in Drug Discovery. John Wiley and Sons, Inc.; Hoboken, NJ: 2005. pp. 56–63. [Google Scholar]
- 27.Kathuria S, Gaetani S, Fegley D, Valiño F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat. Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- 28.Alexander JP, Cravatt BF. Mechanism of carbamate inactivation of FAAH: Implications for the design of covalent inhibitors and in vivo functional probes for enzymes. Chem. Biol. 2005;12:1179–1187. doi: 10.1016/j.chembiol.2005.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saario SM, Poso A, Juvonen RO, Järvinen T, Salo-Ahen OMH. Fatty acid amide hydrolase inhibitors from virtual screening of the endocannabinoid system. J. Med. Chem. 2006;49:4650–4656. doi: 10.1021/jm060394q. [DOI] [PubMed] [Google Scholar]
- 30.Fegley D, Gaetani S, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. Characterization of the fatty acid amide hydrolase inhibitor cyclohexyl carbamic acid 3'-carbamoyl-biphenyl-3-yl ester (URB597): effects on anandamide and oleoylethanolamide deactivation. J. Pharmacol. Exp. Ther. 2005;313:352–358. doi: 10.1124/jpet.104.078980. [DOI] [PubMed] [Google Scholar]
- 31.Forbes DC, Ene DG, Doyle MP. Stereoselective synthesis of substituted 5-hydroxy-1,3-dioxanes. Synthesis. 1998:879–882. [Google Scholar]
- 32.Leung W-Y, Mao F, Haugland RP, Kalubert DH. Lipophilic sulfophenylcarbocyanine dyes: synthesis of a new class of fluorescent cell membrane probes. Bioorg. Med. Chem. Lett. 1996;6:1479–1482. [Google Scholar]
- 33.Marcinek A, Platz MS, Chan SY, Floresca R, Rajagopalan K, Golinski M, Watt D, Unusually long lifetimes of the singlet nitrenes derived from 4-azido-2,3,5,6-tetrafluorobenzamides. J. Phys. Chem. 1994;98:412–419. [Google Scholar]
- 34.Leardini R, Zanardi G. A new and facile synthesis of alkyl N-arylcarbamates. Synthesis. 1982:225–227. [Google Scholar]
- 35.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Vol. 3. Cold Spring Harbor Lab. Press; Plainview, NY: 1989. pp. 18.47–18.75. [Google Scholar]
- 36.Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]