

Abstract
Background
In sickle cell disease, ischemia-reperfusion injury and intravascular hemolysis produce endothelial dysfunction and vasculopathy characterized by reduced nitric oxide and arginine bioavailability. Recent functional studies of platelets in patients with sickle cell disease reveal a basally activated state, which suggests that pathological platelet activation may contribute to sickle cell disease vasculopathy.
Methods and Results
Studies were therefore undertaken to examine transcriptional signaling pathways in platelets that may be dysregulated in sickle cell disease. We demonstrate and validate in the present study the feasibility of comparative platelet transcriptome studies on clinical samples from single donors by the application of RNA amplification followed by microarray-based analysis of 54 000 probe sets. Data mining an existing microarray database, we identified 220 highly abundant genes in platelets and a subset of 72 relatively platelet-specific genes, defined by >10-fold increased expression compared with the median of other cell types in the database with amplified transcripts. The highly abundant platelet transcripts found in the present study included 82% or 70% of platelet-abundant genes identified in 2 previous gene expression studies on nonamplified mRNA from pooled or apheresis samples, respectively. On comparing the platelet gene expression profiles in 18 patients with sickle cell disease in steady state to those of 12 black control subjects, at a 3-fold cutoff and 5% false-discovery rate, we identified ≈100 differentially expressed genes, including multiple genes involved in arginine metabolism and redox homeostasis. Further characterization of these pathways with real-time polymerase chain reaction and biochemical assays revealed increased arginase II expression and activity and decreased platelet polyamine levels.
Conclusions
The present studies suggest a potential pathogenic role for platelet arginase and altered arginine and polyamine metabolism in sickle cell disease and provide a novel framework for the study of disease-specific platelet biology.
Keywords: platelets, genes, enzymes, metabolism, thrombolysis, polymerase chain reaction, signal transduction
In sickle cell disease, hemoglobin S–containing erythrocytes become entrapped in the microcirculation, which leads to repetitive cycles of ischemia-reperfusion tissue injury and infarction.1 Primary and secondary inflammation, endothelial activation, oxidant stress, and adhesion molecule expression contribute to this process. Hemoglobin S–containing erythrocytes also hemolyze prematurely, releasing erythrocyte hemoglobin and arginase into plasma. The hemoglobin reacts with and inactivates endothelium-derived nitric oxide (NO), whereas the arginase converts arginine, the substrate for NO synthesis, to ornithine.2,3 Both processes produce a state of endothelial dysfunction that is associated with the clinical development of pulmonary hypertension.2,4–6 Pulmonary hypertension, now identified as the greatest risk factor for death in patients with sickle cell disease, is associated with intravascular hemolysis, iron overload, renal insufficiency, and possibly intravascular thrombosis, especially in the setting of surgical splenectomy or autosplenectomy.5 Pathological studies reveal in situ thrombosis in the pulmonary vasculature of sickle cell patients associated with pulmonary hypertension.7–9 Indeed, overt thromboembolism and hypercoagulability are thought to contribute to sickle cell disease pathogenesis, with a number of studies documenting increased activation of platelets, increased thrombotic events, and increased expression of endothelial and whole blood tissue factor.10–14
Beyond the measurement of basal and stimulated platelet activation and aggregation, the signaling pathways and pretranslational events that drive the global activation state of platelets remain unclear. An improved understanding of the intrinsic signaling pathways that affect the transcriptome of platelets in general and in patients with sickle cell disease in particular could potentially identify novel therapeutic targets or candidate gene-environment interactions. The recent advent of genomic technologies presents opportunities for cell-type–specific molecular profiling on a large scale, employing microarray technology to rapidly and globally identify the molecular alterations in cell systems associated with disease.15 However, such studies on platelets have largely been hampered by technical difficulties in analyzing the platelet gene transcripts owing to the low abundance of RNA in platelets.16 For example, to obtain the 1 to 4 μg of RNA from platelets that is necessary for expression profiling, more than 500 mL of whole blood has to be processed, thus limiting gene expression studies to pooled platelet RNA or analysis of large-volume apheresis samples.17,18 The recent development of high-fidelity RNA amplification technologies now enables us to analyze such small samples.
Exploiting and validating such approaches, we report here the analysis of the platelet transcriptome from single donors as a result of our ability to validate and develop large, statistically significant gene lists of differentially regulated genes in patients with disease. These results provide a window into the molecular basis for dysfunctional platelets in sickle cell disease.
Methods
Subjects
The present study was approved by the National Heart, Lung, and Blood Institute's Institutional Review Board, and written informed consent was obtained from all study participants. Patients selected for the present study included 8 males and 10 females of mean age 41.6±10.1 years, and the controls (self-identified black subjects) included 4 males and 8 females of mean age 42.2±8.9 years. The patients' samples were collected in steady-state condition, and none of the controls or patients were taking antiplatelet medication.
Measurement of Platelet Activation by Flow Cytometry
Basal platelet activation in patients in the present study was assessed by flow cytometric measurement of P-selectin and glycoprotein IIb/IIIa expression in whole-blood samples as described by Tomer.19 In brief, 450 μL of citrated venous blood was mixed with either 50 μL of phosphate buffered saline, 50 μL of 1 to 10 μmol/L (final concentration) ADP, or 50 μL of 1 to 100 μmol/L (final concentration) thrombin receptor–activating peptide and allowed to incubate for 3 minutes at room temperature. Five microliters of blood was added to tubes containing saturating concentrations of either fluorescein isothiocyanate (FITC)-labeled PAC-1, phycoerythrin (PE)-labeled mouse IgG 1, and peridinin chlorophyll protein–labeled CD61 (Becton Dickinson, San Jose, Calif) or FITC-labeled PAC-1, PE-labeled CD62P, and peridinin chlorophyll protein–labeled CD61 (Becton Dickinson) with tetrapeptide adhesion ligand RGDS (arginine–glycine–aspartate–serine) as a competitive inhibitor for PAC-1 binding. Samples were analyzed on a FACScan flow cytometer. Platelets were distinguished by the characteristic light scatter and the platelet-specific antibody CD61 binding to glycoprotein IIb/IIIa or CD62P, which represented the surface expression of P-selectin and which was calculated from 20 000 events positive for CD61, with a fluorescence intensity greater than a threshold set at 1% from the respective negative control sample.
Platelet Preparation
Twenty milliliters of peripheral blood from patients and normal volunteers was collected in EDTA tubes and centrifuged at 150g for 10 minutes, and platelet-rich plasma was carefully aspirated and recentrifuged at 150g for 5 minutes to remove remaining red and white cells. Platelet-rich plasma was centrifuged again at 1500g for 10 minutes to separate the platelets into pellets. The cell pellet was lysed twice with erythrocyte lysis buffer to remove traces of contaminating red blood cells. The pellet was than washed with phosphate-buffered saline and checked for purity in a Cell-Dyn Coulter counter (Abbott Diagnostics, Abbott Park, Ill).
RNA Isolation
Total platelet RNA was extracted with an RNAqueous micro RNA isolation kit (Ambion, Austin, Tex) according to the manufacturer's directions. Platelets were lysed in lysis buffer containing guanidinium thiocyanate, and the cell lysate was mixed with ethanol and applied to a silica-based filter that selectively binds RNA. Genomic DNA was removed by DNase treatment. The concentration of the isolated RNA was determined with the Nanodrop ND-1000 spectrophotometer (Nanodrop Technologies, Wilmington, Del). Quality and integrity of the total RNA isolated were assessed on the Agilent 2100 bioanalyzer (Agilent Technologies, Palo Alto, Calif).
Amplification of RNA for Gene Expression Studies
T7-based RNA amplification was performed on 10 ng of the isolated platelet total RNA (corresponding to ≈4 to 5 mL of collected blood) with the Riboamp OA 2-round amplification kit as suggested by the manufacturer (Arcturus, Mountain View, Calif). Briefly, total RNA was incubated with oligo dT/T7 primers and reverse-transcribed into double-stranded cDNA. In vitro transcription of the purified cDNA was performed with T7 RNA polymerase at 42°C for 6 hours. The amplified RNA was purified and subjected to a second round of amplification and biotin labeling with Affymetrix's IVT labeling kit according to the manufacturer's directions (Affymetrix, Santa Clara, Calif). The yield and integrity of the biotin-labeled cRNA were determined with the Nanodrop ND-1000 spectrophotometer and the Agilent 2100 bioanalyzer. Twenty micrograms of biotin-labeled RNA was fragmented to ≈200-bp size by incubation in fragmentation buffer containing 200 mmol/L Tris-acetate pH 8.2, 500 mmol/L potassium acetate, and 500 mmol/L magnesium acetate for 35 minutes at 94°C before hybridization. Fragmented RNA was assessed for relative length on Agilent 2100 bioanalyzer and hybridized to Affymetrix Human Genome (HG) U133 Plus 2.0 chips for 16 hours, washed, stained on an Affymetrix fluidics station, and scanned with an Affymetrix GeneChip scanner.
Microarray Data Processing and Analysis
Affymetrix GeneChip operating software version 1.4 was used to calculate the signal intensity and the percent present calls on the hybridized Affymetrix chip. To select genes differentially expressed between patients and healthy control subjects, the signal-intensity values obtained for probe sets in the microarrays were transformed with an adaptive variance-stabilizing, quantile-normalizing transformation (P.J. Munson, GeneLogic Workshop of Low Level Analysis of Affymetrix GeneChip Data, 2001, software available at http://abs.cit.nih.gov/geneexpression.html). The transform, termed “S10,” is scaled to match the logarithm transform, base 10. Transformed data from all the chips were subjected to a principal component analysis to detect outliers. One-way ANOVA and post hoc t tests were performed to evaluate each probe set. Normality testing (Shapiro-Wilk test) of the residuals from ANOVA was performed on any genes ultimately selected, to ensure that the required assumptions for ANOVA were met. If data for a gene failed the normality test, a nonparametric test (Wilcoxon-Kruskal-Wallis test) was also performed and compared with the ANOVA result. The probability value for differences between 2 groups was calculated for each of the 54 675 probe sets. To address the multiple comparisons problem, fold cutoff filters and false-discovery rate analysis filters were applied.20,21 Two-way hierarchical clustering was used to bring together sets of samples and genes with similar expression patterns. The hierarchical cluster is run from the JMP statistical software package (www.jmp.com, SAS Institute, Cary, NC) using Ward's method.
To evaluate whether members of a given pathway or functional class of interest (gene set) are overrepresented in a list of differentially expressed genes or markers (in this case, enzymes in the arginine metabolic pathway), a statistical methodology called “gene-set enrichment analysis” (GSEA software, http://www.broad.mit.edu/gsea), as described by Subramanian et al,22 was performed.
Data Mining of Microarray Gene Expression Database for Identification of Platelet Genes
To identify platelet-abundant and relatively platelet-specific genes, we compared the gene expression profiles from the amplified platelet transcriptome to other amplified profiles from other cell types in our array database. The signal-intensity values of array data from 2 human chip types, HG-U133A and HG-U133 Plus, and from different cell types, including bronchoalveolar lavage cells (n=16), blood outgrowth endothelial cells (n=2), human microvascular endothelial cells (n=21), human apheresed circulating endothelial cells (n=3), human circulating endothelial cells (n=12), human endothelial progenitor cells and KDR (kinase-insert domain-containing receptor)–negative cells from peripheral blood (n=3), human normal bronchial epithelial cells (n=1), human peripheral blood mononuclear cells (n=4), human circulating T cells (n=11), human cultured T cells (n=4), human umbilical vein endothelial cells (n=7), and human platelets (n=40), were selected from our microarray gene expression database. Only probe sets available on both chip types (22 277 probe sets in total) were taken for comparison. The raw signal intensities reported by Affymetrix GeneChip operating software 1.4 were median-normalized and log10-transformed (“Lmed transform”). To characterize the expression in each cell type, the median Lmed of each probe set was then calculated for each cell type. The probe sets were ranked by platelet median expression (median Lmed) from high to low. The top 300 probe sets in this ranking were termed “platelet-abundant probe sets.” To further identify genes that are relatively specific for platelets, the relative platelet expression index (REI) in log10 scale was computed for each probe set as:
| REI | = median Lmed of platelet samples−median | |
| over cell types (median Lmed for that cell type) |
Platelet-abundant probe sets that also had REIs greater than 1.0, denoting 10-fold or higher expression in platelets compared with other cell types, were identified as platelet-abundant, relatively platelet-specific genes.
To validate the fidelity of the amplification procedure, platelet-abundant genes were then compared with previously published gene expression lists derived from pooled platelet mRNA and apheresis samples that had not been subjected to amplification.17,18 The lists of genes found in platelets from previous published studies were first mapped to the HG U133 Plus 2.0 chips used in the present study with the gene symbol identifier. Genes titled “unknown” or cDNA clones with no specific gene symbols in the published lists could not be mapped to the U133 Plus 2.0 chip. Such genes were removed and excluded from the comparative study. The genes that were identified as highly platelet abundant and relatively platelet specific were also subjected to gene ontology analysis with DAVID Bioinformatic Resources 2006 (National Institute of Allergy and Infectious Diseases/National Institutes of Health, Frederick, Md; http://david.abcc.ncifcrf.gov/tools.jsp).23
Validation of Gene Expression Data by Quantitative Polymerase Chain Reaction
First-strand cDNA was synthesized with 1 μg of amplified RNA and random primers in a 20-μL reverse-transcriptase reaction mixture using Invitrogen's Superscript cDNA synthesis kit (Invitrogen, Carlsbad, Calif) according to the manufacturer's directions. Quantitative real-time polymerase chain reaction assays were performed with the use of gene-specific double–fluorescently labeled probes in a 7900 Sequence Detector (PE Applied Biosystems, Norwalk, Conn). Probes and primers were obtained from Applied Biosystems. In brief, polymerase chain reaction amplification was performed in a 384-well plate with a 20-μL reaction mixture containing 300 nm of each primer, 200 nm of probe, 200 nm of dNTP in 1× real-time polymerase chain reaction buffer and passive reference (ROX) fluorochrome. The thermal cycling conditions were 2 minutes at 50°C and 10 minutes at 95°C, followed by 40 cycles of 15-second denaturation at 95°C and 1 minute of annealing and extension at 60°C. Samples were analyzed in duplicate, and the cycle threshold values obtained were normalized to the housekeeping gene β-actin. The comparative cycle threshold method,24 which compares the differences in cycle threshold values between groups, was used to achieve the relative fold change in gene expression between subjects with sickle cell disease and normal healthy subjects.
Measurement of Arginase Activity in Platelets
Platelet proteins were extracted by suspending the platelets in protein extraction buffer containing 20 mmol/L Tris-HCl, pH 7.4, and 1 mg/mL protease inhibitors with broad specificity for the inhibition of serine, cysteine, and aspartic acid proteases and aminopeptidases. The cell suspension was centrifuged, and the supernatant was used for measuring protein and arginase activity. Arginase activity was determined as the conversion of l-arginine that is 14C-labeled on the guanidino carbon to 14C-labeled urea, which was converted to 14C-labeled carbon dioxide by urease and trapped as 14C-labeled sodium carbonate for scintillation counting, as described previously.25 Briefly, aliquots of platelet protein extract were incubated for 10 minutes at 55°C in complete assay mixture lacking arginine. The reaction was initiated by addition of labeled arginine, and incubation was continued at 37°C for 2 hours. The reaction was terminated by heating at 100°C for 3 minutes. Samples were incubated with urease at 37°C for 45 minutes, and 14C-labeled sodium carbonate was trapped on sodium hydroxide–soaked filters after acidification of the samples with hydrochloric acid to volatilize the 14C-labeled carbon dioxide. Protein measurement was made by the BCA protein assay (Pierce, Rockford, Ill) according to the manufacturer's protocol. Arginase specific activity is expressed as nanomoles per minute per milligram of protein.
Quantitation of Polyamines in Platelets
Polyamines from platelets were extracted with 0.2N perchloric acid and quantitated according to the procedure of Adibhatla et al.26 Extracted polyamines were dansylated and separated by reverse-phase liquid chromatography with C-18 columns. The resin-bound dansylated polyamines were eluted with 1.5 mL of acetonitrile. Fifty microliters of the purified polyamine was injected into a Hewlett-Packard high-performance liquid chromatograph (Hewlett-Packard, Wilmington, Del) fitted with a Nova-Pak column (Waters Corp, Milford, Mass). The polyamines bound to the column were again eluted by gradient elution with acetonitrile and sodium acetate and quantitated against internal and external standards with a fluorescence detector. Statistical analysis of data from patients and control subjects was made with an unpaired t test, 1-way ANOVA, and Bonferroni multiple-comparison tests, as appropriate.
The authors had full access to and take full responsibility for the integrity of the data. All authors have read and agree to the manuscript as written.
Results
Activation of Platelets in Sickle Cell Disease
Consistent with previously published studies,12,19,27–29 analysis of unstimulated platelets by flow cytometry in patients with sickle cell disease showed evidence of platelet activation, with significantly higher percentages of platelets expressing activated glycoprotein IIb/IIIa in 20 patients with sickle cell disease (17.1±16.3 versus 6.98±7.34 in 20 controls; P<0.05). Similarly, the percentage of platelets expressing P-selectin was increased in patients in steady state (1.45±1.9 versus 0.51±0.43 in controls; P<0.05).
Isolation and Amplification of Platelet RNA
Platelet preparation from each donor was used for the extraction of total RNA. Typically, from 20 mL of blood, the RNA yield was found to be in the range of 50 to 70 ng. Because gene expression profiling studies on microarrays require microgram quantities of RNA, we proceeded to amplify the platelet RNA by T7-based 2-round amplification. To apply the 2-round amplification process, 10 ng of the total RNA from platelets from each subject was amplified, and at the end of second-round amplification, 35 to 45 μg of biotinylated cRNA was obtained from these samples (corresponding to more than 700 000-fold amplification of RNA). The size distribution and integrity of the amplified RNA were found to be of sufficient quality for microarray hybridization.
Characterization of Platelet Gene Expression by Principal Component and Hierarchical Cluster Analysis
To validate the fidelity of gene expression results after RNA amplification and to rule out any possible contamination of other cell types in the platelet preparation, we performed global analysis on the transcript profiles generated by the 2-round amplification process on platelets and other archived cell types in our microarray database. Of the 54 675 probe sets imprinted onto the chip, 24±4.2% of transcripts were computationally identified as “present” in platelets, and nearly 72±5.8% were identified as absent. The percentage of platelet-expressed genes was generally found to be lower than that obtained from other human cell types, in which 35% to 50% of genes are “present”; this reflects the relatively limited transcriptome repertoire of circulating platelets. We used principal component analysis to determine the cell types on the basis of their expression profile. In this analysis, we compared only the transcript profiles generated by the 2-round amplification process to eliminate method-induced alterations in the expression pattern. The first 100 principal components, which captured 83% of the total variability of the expression matrix, were then subjected to a hierarchical cluster analysis. The dendrogram shown in Figure 1 from the hierarchical cluster analysis distinctly segregates cell types of the same phenotype on the basis of their gene expression pattern. All the platelet samples clustered together, thus revealing a distinct gene expression pattern in platelets compared with peripheral blood mononuclear cells, endothelial cells, and epithelial cells. None of the platelet samples showed up as outliers in the analysis, thus implying consistency in the processes of platelet separation, linear amplification, and hybridization. Examination of the present/absent calls from the microarray data revealed absence of expression for CD45 and CD5, which are markers for lymphocytes and T cells, whereas the platelet glycoprotein Ib and glycoprotein IIb/IIIa transcripts were significantly expressed.
Figure 1.
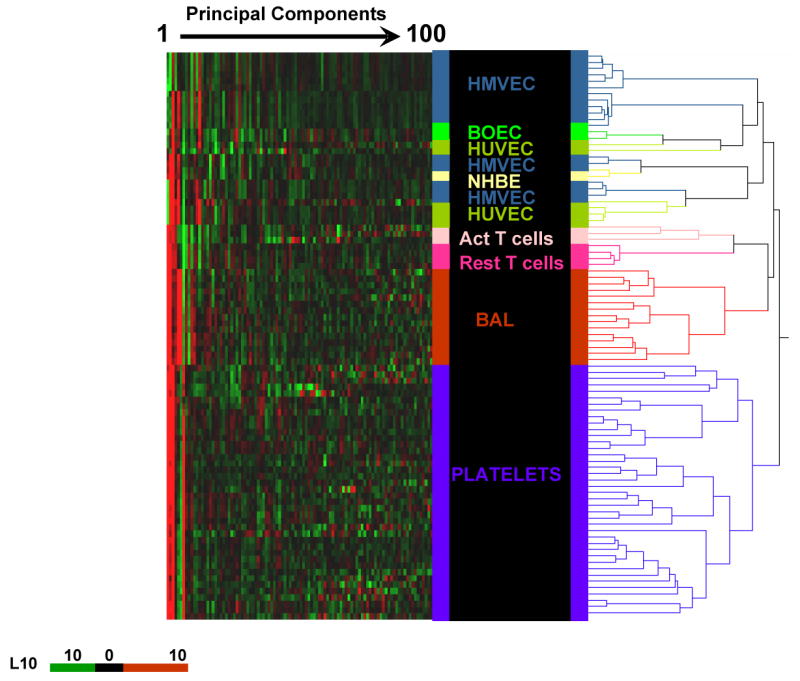
Principal component heat map analysis of the first 100 principal components representing 83% variability of 98 samples of different human cell types. Hierarchical cluster analysis was performed on transcriptome data derived from all samples in our database that were processed similarly by the 2-round amplification process and hybridized to HG-U133A or HG-U133 Plus 2.0 gene chips. Each principal component value for each sample is represented with a red, black, and green color scale. The dendrogram displays the clustering of samples according to their expression pattern and segregation of cell types of the same phenotype, thus revealing a distinct expression pattern for each of the cell types studied. HMVEC indicates human microvascular endothelial cells; BOEC, blood outgrowth endothelial cells; HUVEC, human umbilical vein endothelial cells; NHBE, normal human bronchial epithelial cells; PBMC, peripheral blood mononuclear cells; Act T, activated T lymphocytes; Rest T, resting T lymphocytes; and BAL, bronchoalveolar lavage cells.
Identification of Platelet-Specific/Abundant Genes
In an effort to identify transcripts that are highly abundant and relatively platelet-specific, a bioinformatics-based data mining experiment was performed with our microarray database archive of different gene expression profiles derived from several human cell types analyzed on HG-U133A and HG-U133 Plus 2.0 microarrays (Figure 1). We first sought to identify highly abundant messages in platelets to validate the amplification profile by comparing the abundant genes from the present study to 2 other published studies by Gnatenko and colleagues17 and McRedmond and colleagues18 using nonamplified apheresis samples and pooled platelet RNA, respectively. Platelet messages from the present study were ranked in order of abundance from high to low. The top 300 ranking probe sets, representing 220 genes, were identified as highly abundant in platelets. Comparison of these 220 platelet-abundant genes to previously identified platelet-abundant genes revealed that 41 of the abundant genes in the present study were found in either 1 or both of the previously published transcript lists, as shown in Figure 2.
Figure 2.
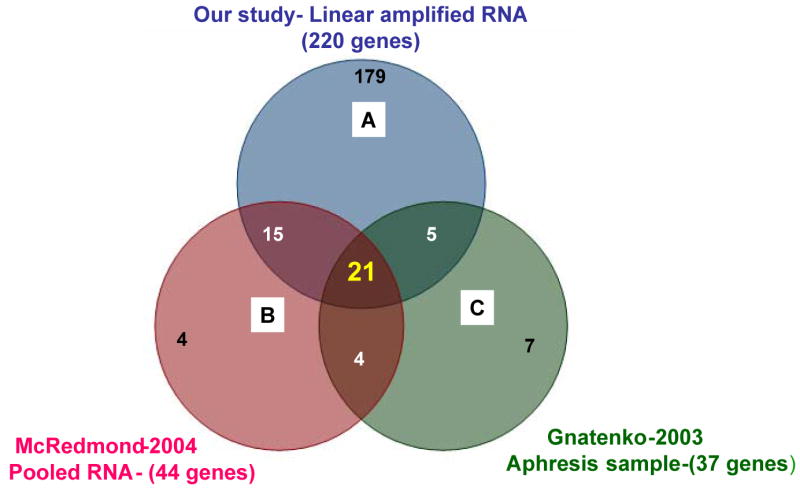
Venn diagram comparing 3 lists of genes. Comparison of (A) 220 platelet-abundant genes identified with amplified RNA from single donors, (B) 44 platelet-abundant genes identified with pooled RNA from several donors,18 and (C) 37 platelet-abundant genes identified with RNA from apheresis samples.17 In total, there were 21 genes that overlapped by all 3 methods. Nineteen percent of our gene list of 220 genes was reported in either or both of the previous studies cited.
The highly abundant platelet transcripts identified here included genes such as platelet factor-4, platelet basic protein and receptor glycoprotein Ib, glycoprotein IIb, and some universally expressed proteins, such as histones, actin, ferritin, myosin light chain, and microglobulin. A complete list of the 220 platelet-abundant genes is found in Table I in the Data Supplement.
These 220 genes were further filtered on the basis of their relative expression index (REI). Using a cutoff of 1.0 for REI, which means the median expression of the gene in question is 10-fold or higher in expression in platelets than the median of other cell types, we identified 72 relatively platelet-specific genes. We found that only 20 of these 72 genes overlap with 1 or the other published platelet gene list, which suggests that from the amplified single-donor transcriptome data, we could identify many novel abundant and relatively platelet-specific genes. Gene ontology analysis of these 72 abundant and relatively platelet-specific genes indicated them to fall into the category of carbohydrate binding (4.3%), cell adhesion/binding (6.2%), chemokine receptor (1.6%), chemokines (7.6%), cytokines (7.9%), enzyme activity (6.4%), protein binding (20.1%), receptor regulator activity (3.2%), receptor binding (7.8%), signal transduction (33.2%), and transcription regulators (1.7%). Table 1 lists representative genes from each of these categories. Table II in the Data Supplement shows the gene ontology analysis of the these 72 abundant and relatively platelet-specific genes.
TABLE 1. Platelet-Abundant Genes.
| Probe ID | Gene Symbol | Gene Title | REI |
|---|---|---|---|
| Binding | |||
| 215305_at | PDGFRA | Platelet-derived growth factor receptor | 2.77 |
| 202555_s_at | MYLK | Myosin, light polypeptide kinase | 2.65 |
| 207957_s_at | PRKCB1 | Protein kinase C, β1 | 2.47 |
| 213036_x_at | ATP2A3 | ATPase, Ca2+ transporting, ubiquitous | 2.27 |
| 219947_at | CLECSF6 | C-type lectin | 2.26 |
| 212531_at | LCN2 | Lipocalin 2 | 2.13 |
| 207766_at | CDKL1 | Cyclin-dependent kinase-like 1 | 2.10 |
| 203662_s_at | TMOD1 | Tropomodulin 1 | 2.01 |
| Coagulation/cell adhesion/chemokines | |||
| 207815_at | PF4V1 | Platelet factor-4 variant 1 | 3.42 |
| 214146_s_at | PPBP | Proplatelet basic protein | 3.39 |
| 206390_x_at | PF4 | Platelet factor-4 | 3.28 |
| 215240_at | ITGB3 | Integrin, β3 | 2.90 |
| 206655_s_at | GP1BB | Glycoprotein Ib (platelet) | 2.77 |
| 206493_at | ITGA2B | Integrin, α2b | 2.72 |
| 205898_at | CX3CR1 | Chemokine (C-X3-C motif) receptor 1 | 2.60 |
| 222043_at | CLU | Clusterin | 2.41 |
| 204081_at | NRGN | Neurogranin | 2.41 |
| 207926_at | GP5 | Glycoprotein V (platelet) | 2.40 |
| 205612_at | MMRN1 | Multimerin 1 | 2.35 |
| 205114_s_at | CCL3 | Chemokine (C-C motif) ligand 3 | 2.33 |
| 204563_at | SELL | Selectin L | 2.28 |
| 1405_i_at | CCL5 | Chemokine (C-C motif) ligand 5 | 2.18 |
| 206049_at | SELP | Selectin P | 2.14 |
| 214433_s_at | SELENBP1 | Selenium binding protein 1 | 2.06 |
| 201438_at | COL6A3 | Collagen, type VI, α3 | 2.04 |
| Catalytic activity/enzyme regulator | |||
| 211560_s_at | ALAS2 | Aminolevulinate, δ, synthase 2 | 3.21 |
| 203817_at | GUCY1B3 | Guanylate cyclase 1 | 2.69 |
| 203913_s_at | HPGD | Hydroxyprostaglandin dehydrogenase | 2.42 |
| 206963_s_at | NAT8 | N-acetyltransferase 8 (camello like) | 2.41 |
| 204041_at | MAOB | Monoamine oxidase B | 2.40 |
| 206177_s_at | ARG1 | Arginase, liver | 2.33 |
| 212588_at | PTPRC | Protein tyrosine phosphatase, receptor | 2.16 |
| 204446_s_at | ALOX5 | Arachidonate 5-lipoxygenase | 2.10 |
| 209676_at | TFPI | Tissue factor pathway inhibitor | 2.00 |
| Histones/structural molecule | |||
| 210387_at | HIST1H2BG | Histone 1, H2bg | 2.75 |
| 202708_s_at | HIST2H2BE | Histone 2, H2be | 2.71 |
| 215071_s_at | HIST1H2AC | Histone 1, H2ac | 2.71 |
| 202202_s_at | LAMA4 | Laminin, α4 | 2.40 |
| 217428_s_at | COL10A1 | Collagen, type X, α1 | 2.38 |
| 203471_s_at | PLEK | Pleckstrin | 2.27 |
| 208601_s_at | TUBB1 | Tubulin, β1 | 2.13 |
| 212873_at | HA-1 | Minor histocompatibility antigen HA-1 | 2.02 |
| 214469_at | HIST1H2AE | Histone 1, H2ae | 2.01 |
| Signal transduction | |||
| 219090_at | SLC24A3 | Solute carrier family 24 | 3.13 |
| 207651_at | H963 | Platelet activating receptor | 2.61 |
| 221491_x_at | HLA-DRB3 | Major histocompatibility complex | 2.05 |
| 336_at | TBXA2R | Thromboxane A2 receptor | 2.03 |
Differential Platelet Gene Expression in Sickle Cell Disease
We compared the platelet gene expression profiles in 18 patients with sickle cell disease and 12 healthy black volunteer subjects using stringent statistical filters of 5% false-discovery rate and fold-change greater than 3 (Figure 3). Examination of differentially regulated genes from this analysis revealed significant modulation of genes involved in arginine and nitrogen metabolism, redox homeostasis, cell growth, adhesion, and signaling pathways. Interestingly, we observed significant upregulation of mRNAs encoding arginase II and ornithine decarboxylase antizyme in patients with sickle cell disease compared with control subjects. Arginase II metabolizes arginine to ornithine and can divert arginine away from NO synthesis.30,31 Ornithine decarboxylase antizyme inhibits the enzyme ornithine decarboxylase, which is required to convert ornithine into the polyamines. Glutathione peroxidase 4, thioredoxin reductase, and superoxide dismutase, which are involved in redox homeostasis, were also significantly upregulated in sickle cell disease. Arginine metabolic enzymes and enzymes involved in redox homeostasis are highlighted in Figure 3.
Figure 3.
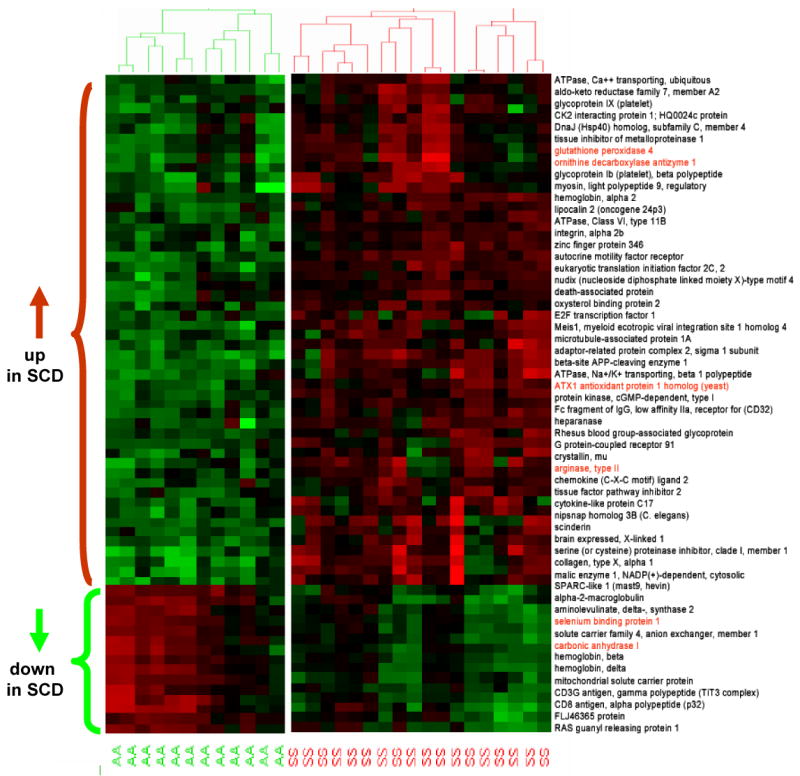
Heat map of differential gene expression in platelets in sickle cell disease (SCD) compared with control subjects. Cluster analysis was applied to gene expression data derived from all probes on HG-U133 Plus 2.0 at a false-discovery rate of 5% and fold change (FC) >3.0 from 18 SCD patients (SS) and 12 control subjects (AA). The level of expression of each gene in each sample relative to the mean level of expression of that gene across all samples is represented with a red, black, and green color scale (green indicates below mean; black, equal to mean; and red, above mean). The dendrogram displays the unsupervised clustering of patients and control subjects using the differentially expressed gene list. Gene names are displayed on the right side of the figure, and genes of interest are highlighted.
Altered Expression of Arginine Metabolic Enzymes in Sickle Cell Disease
Having observed significant changes in the expression of platelet enzymes involved in arginine metabolism, and considering the recent appreciation of a role for dysregulated arginine metabolism in the pathogenesis of sickle cell disease vasculopathy2 and the well-characterized role of NO in modulating platelet activation,32 we chose to further examine these pathways of interest (arginine/NO pathway) in more detail using a gene-set enrichment analysis with a false-discovery rate setting at 25%. Gene-set enrichment analysis with 384 predefined gene sets (pathways provided by GSEA at Broad Institute, Harvard University, Cambridge, Mass) were applied to the sickle cell platelet data generated from patients and healthy volunteers in the present study. Results from this analysis showed 95 gene sets to be enriched or overexpressed in sickle cell disease (P<0.001) and 45 gene sets to be significantly repressed (P<0.05). Examination of the significantly modulated genes by gene-set enrichment analysis after inclusion of a selected list of enzymes in pathways of arginine metabolism revealed upregulation of mRNAs encoding arginase II, cationic amino acid transporter-2, ornithine decarboxylase, ornithine decarboxylase antizyme, argininosuccinate synthetase, inducible NO synthase, spermine and spermidine synthases, and pyrroline-5-carboxylate reductase, which are all involved in the metabolism of arginine and the regulation of NO, polyamine, and proline synthesis.33
Further validation of the genes identified by microarrays to be differentially regulated in sickle cell disease was performed with quantitative real-time reverse-transcription polymerase chain reaction (TaqMan) studies on ABI 7900 for selected genes in the arginine/NO/polyamine metabolic pathway. Results are represented as fold change comparing sickle cell disease to control subjects. TaqMan fold changes were comparable and showed the same trends as the microarray data (Figure 4A; Table 2).
Figure 4.
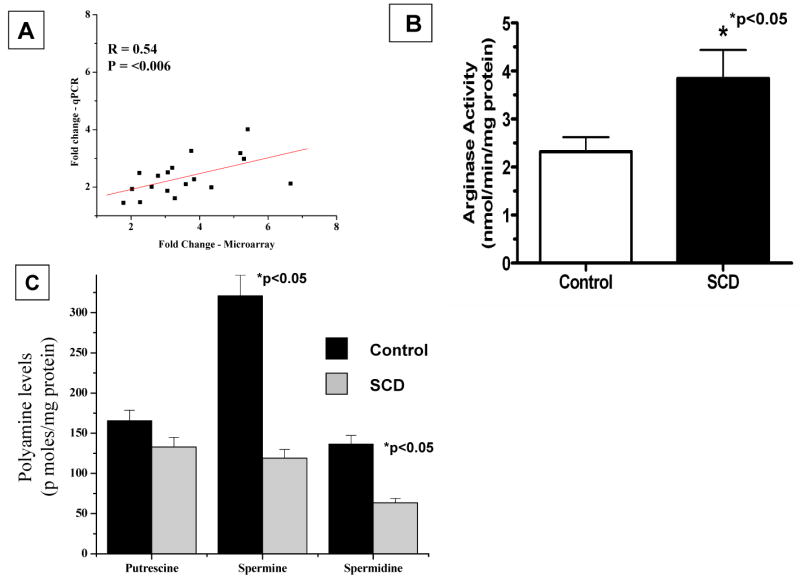
Modulation of arginase II mRNA abundance, arginase activity, and polyamine levels in platelets of patients with sickle cell disease (SCD). A, Correlation of fold change of arginase II mRNA abundance in SCD patients determined by microarray platform and TaqMan gene expression assay–based real-time polymerase chain reaction (PCR). X-axis represents log2-fold change determined by microarrays; y-axis represents log2-fold change determined by real-time PCR (qPCR). B, Platelet total arginase activity in SCD patients and control subjects. Arginase activity was determined as described in Methods. Values are expressed as mean±SD for 10 controls and 10 SCD patients. *P for comparison of SCD patients vs control subjects. C, Concentrations of polyamines in platelets from patients with SCD and control subjects. Polyamines were quantitated by high-performance liquid chromatography as described in Methods and are expressed as picomoles per milligram of protein (shown as mean±SD for 10 controls and 10 SCD patients). *P for comparison of SCD patients vs control subjects.
TABLE 2. Fold Change in Selected Genes, Sickle Cell Disease Versus Control Subjects.
| Fold Change
|
|||
|---|---|---|---|
| Gene Name | MA | QPCR | P |
| Arg2 | 3.29 | 2.83 | <0.006 |
| ODC | 1.33 | 1.31 | <0.005 |
| OAT | –1.08 | –1.21 | NS |
| CAT-2 | 1.22 | 1.44 | <0.01 |
| eNOS | –1.51 | –1.03 | NS |
| iNOS | 1.1 | 1.08 | NS |
| AL | –1.02 | –1.39 | NS |
| ASS | 1.33 | 1.78 | <0.05 |
| SS | 1.92 | 1.56 | <0.05 |
| SRM | 1.61 | 1.26 | <0.05 |
| P5CR | 2.09 | 1.65 | <0.005 |
MA indicates microarray; QPCR, quantitative polymerase chain reaction; Arg 2, arginase 2; ODC, ornithine decarboxylase; OAT, ornithine aminotransferase; CAT-2, cationic amino acid transporter-2; eNOS, endothelial NO synthase; iNOS, inducible NO synthase; AL, argininosuccinate lyase; ASS, argininosuccinate synthetase; SS, spermine synthase; SRM, spermidine synthase; and P5CR, pyrroline-5-carboxylate reductase.
Altered Platelet Arginase Activity and Polyamine Levels in Sickle Cell Disease
Determination of the specific activity of arginase in platelets corroborated the microarray data, with a significant increase in arginase activity in platelets from patients with sickle cell disease (Figure 4B). For comparison, arginase activities in alveolar macrophages and red blood cells from healthy human subjects averaged 1.8 and 23.5 nmol · min−1 · mg protein−1, respectively.2,34 We also found significantly reduced levels of total polyamines (putrescine+spermine+spermidine) in platelets of sickle cell patients compared with control subjects (622±38 versus 1107±52; P=0.056). Analysis of individual polyamines revealed significant (P<0.05) reductions specifically in spermine and spermidine content in platelets of sickle cell patients relative to controls (Figure 4C). Levels of putrescine also trended downward in patients, but the reduction did not reach statistical significance in this relatively small number of samples. The selective reductions in spermine and spermidine may reflect a decreased activity of S-adenosylmethionine decarboxylase35 that is not revealed by the transcriptome analysis.
Discussion
Overt thromboembolism and hypercoagulability are thought to contribute to sickle cell disease pathogenesis. Consistent with this notion, a number of studies have documented increased activation of platelets and increased expression of endothelial and whole-blood tissue factor, among other protean impairments in the thrombotic cascade.10–14
A direct analysis of the platelet transcriptome might be potentially valuable, allowing for a more sensitive and universal exploration of dysregulated or compensatory pathways that might contribute to the recognized thrombophilia. However, platelets are derivatives of megakaryocytes, are anucleate, and retain only small amounts of megakaryocyte-derived RNA,36 and thus, they have traditionally been considered to exhibit very limited variation of their mRNA repertoire. Therefore, the utility of transcriptome analysis in platelets not only has been unclear but also, from a technical standpoint, is very challenging. On the other hand, studies showing evidence of proteins synthesized in a regulated manner during platelet activation indicate that platelets are not phenotypically invariant but do have the ability to exhibit varying patterns of gene expression.37 Recent studies by Healy and colleagues38 on the platelet transcriptome of acute and stable coronary heart disease patients revealed the upregulated expression of CD69 and myeloid-related protein in the platelets of patients with acute cardiovascular events. This work provides evidence for the ability of circulating platelets to modulate their transcriptome in disease conditions in response to extracellular stress signaling.
We therefore sought to validate methodologies to characterize the global transcriptome of amplified platelet RNA from individuals with sickle cell disease. Using these tools, we observed a complex population of mRNAs in circulating platelets and were able to characterize the single-donor platelet transcriptome. Finally, in platelets from patients with sickle cell disease, we observed altered levels of mRNAs encoding various enzymes in arginine metabolism that potentially contribute to reductions in arginine bioavailability and polyamine levels that may play a role in increased basal activation of platelets. We speculate that although platelets are anuclear, the alterations in platelet gene transcripts in patients with sickle cell disease could be a reflection of (1) alterations in the stability of preformed mRNA, (2) a mixture of circulating platelets of different ages due to increased formation of platelets from megakaryocytes and the shortened life span of platelets, and (3) alterations in the megakaryocyte transcriptome.
The low mRNA content of platelets obligated the need for amplification of RNA, because high-throughput expression profiling generally requires microgram quantities of RNA. On careful evaluation of the available technologies for RNA amplification, we found T7-based 2-round amplification to be satisfactory, because it did not introduce major systemic biases during the amplification process in the model system tested. Our laboratory has evaluated and validated this amplification process on a well-established lipopolysaccharide-stimulated endothelial cell model system and observed high fidelity in gene expression relative to unamplified samples (Raghavachari et al, unpublished observations). Applying this technique to platelets from single donors, we generated microgram amounts of RNA from a few nanograms of total RNA for comparative gene expression studies.
Global transcriptome analysis of different cell types in our archived microarray database indicated that platelets have distinct gene expression patterns. Principal component analysis of the cell types processed by the 2-round amplification process on HG-U133A and HG-U133 Plus 2.0 gene chips clearly showed the relatedness of platelet samples based on their gene expression profile. In this analysis, as shown in the dendrogram in Figure 1, cell types from different subjects with the same phenotype clustered together. It is evident from this analysis that robust, reproducible signatures can be obtained from platelets from single donors. The related expression pattern of the platelets despite the amplification process, as shown by the dendrogram, further confirmed the purity of the platelet preparation and ruled out possible cross contamination of platelet samples with other blood cells, which contain orders-of-magnitude more mRNA than platelets. Such contamination would have the potential to greatly distort the gene expression pattern after amplification. The absence of markers specific for lymphocytes and T cells and the positive identification of platelet-specific genes in the platelet transcriptome further validated the purity of the platelet preparations used in the present study. These results indicate that homogenous preparations of platelets can easily be prepared by differential centrifugation without any additional cell-depletion techniques.
Data mining identified 220 transcripts to be of high abundance in platelets. When we compared these genes with platelet-abundant transcripts reported in either of 2 previous microarray-based analyses, 70% of the genes previously identified with apheresis samples17 and 82% of the genes identified with pooled platelet RNA samples18 were found among the 220 highly abundant platelet genes. Of note, among the highly expressed platelet transcripts were transcripts for universally expressed proteins such as actin, tubulin, ferritin and microglobulin, as well as proteins known to be in abundance in platelets, such as platelet factor-4, proplatelet basic protein, glycoprotein 1b, neurogranin, clusterin, collagen, coagulation factor XIII, integrin-αIIb, and several histones. Intriguingly, transcripts for hemoglobin-α, -β, and -γ were also found to be in abundance in platelets in the present data. Similar to our observation, Gnatenko and colleagues17 and McRedmond and colleagues18 also identified transcripts for globin genes. The late cell lineage differentiation into megakaryoblasts and erythroblasts may account for the abundant global transcripts, although further functional studies are indicated. Further examination of these highly abundant genes for specificity revealed 72 genes with a 10-fold increase (REI >1.0) in expression level compared with the median expression of other cell or tissue types. Overall classification of the identified platelet abundant and specific transcripts by gene ontology analysis showed them to be involved predominantly in binding and signal transducer activities.
Comparative analysis of the transcriptome in sickle cell disease revealed differential expression of ≈100 genes, based on stringent statistical filters that required less than a 5% false-discovery rate and >3.0-fold induction. Hierarchical cluster analysis of these significantly differentially expressed genes distinctly separated subjects into healthy controls and sickle cell patients, thereby indicating a unique platelet gene expression pattern in sickle cell disease. A more focused analysis on the modulated genes and associated pathways during these disease conditions implicated a global activation of genes that are involved in arginine uptake and catabolism. These results are interesting, considering the known critical roles for NO and polyamines in limiting platelet activation. Arginine is taken up by platelets via the cationic amino acid transporter (CAT-2) and converted by platelet “endothelial” and “inducible” NO synthase isoforms to NO and citrulline.2 The enzymes arginase I and II are upregulated in inflammatory diseases and can convert arginine to ornithine, thus limiting substrate availability for NO synthesis.2 A role for arginase activation in endothelial dysfunction, pulmonary hypertension, and asthma, among many other disease states, has recently been appreciated.2,31,39,40 The present study represents the first report of increased expression and activity of arginase in platelets in disease in general and sickle cell disease specifically. Circulating arginine is critically depleted in sickle cell disease for a number of reasons: Low arginine intake during vaso-occlusive crisis likely contributes to reductions during the course of acute illness, but more importantly, during intravascular hemolysis, arginase I is released from erythrocytes and catabolizes arginine to ornithine in plasma.2 The present study demonstrates that the intracellular environment is not shielded from dysregulated arginine metabolism.
It is striking that the expression of multiple genes involved in various aspects of arginine metabolism is altered in platelets of sickle cell patients, similar to changes seen in activated macrophages. These genes include enzymes involved in synthesis of NO, polyamines, and proline, as well as synthesis of arginine itself.33 These changes suggest a number of effects on platelet arginine metabolism and physiology that are particularly interesting, considering the known critical roles for NO and polyamines in limiting platelet activation (Figure 5). First, increased levels of mRNAs encoding CAT-2 and argininosuccinate synthetase could represent mechanisms to enhance arginine bioavailability within the cell by increasing arginine uptake and by increasing arginine synthesis from citrulline, which can be taken up from the circulation and also is generated by NO synthase enzymes within platelets. Opposing these mechanisms is increased arginase II expression, which can both limit arginine for NO synthesis and increase production of ornithine for synthesis of polyamines and/or proline. In fact, the increased expression of P5CR (pyrroline-5-carboxylate reductase) would be expected to facilitate synthesis of ornithine-derived proline. The consequences of concomitant increases in levels of mRNAs encoding antizyme-1 and ornithine decarboxylase are difficult to interpret, because these proteins are strongly regulated at translational and posttranslational levels, respectively.41 However, direct measurements demonstrating reduced polyamine levels in platelets of sickle cell patients suggest that ornithine decarboxylase activity is probably reduced in these cells, and selective reductions in spermine and spermidine further suggest reduced activity of S-adenosylmethionine decarboxylase. Increased levels of P5CR mRNA raise the possibility that ornithine may be diverted from polyamine synthesis to proline synthesis.2
Figure 5.
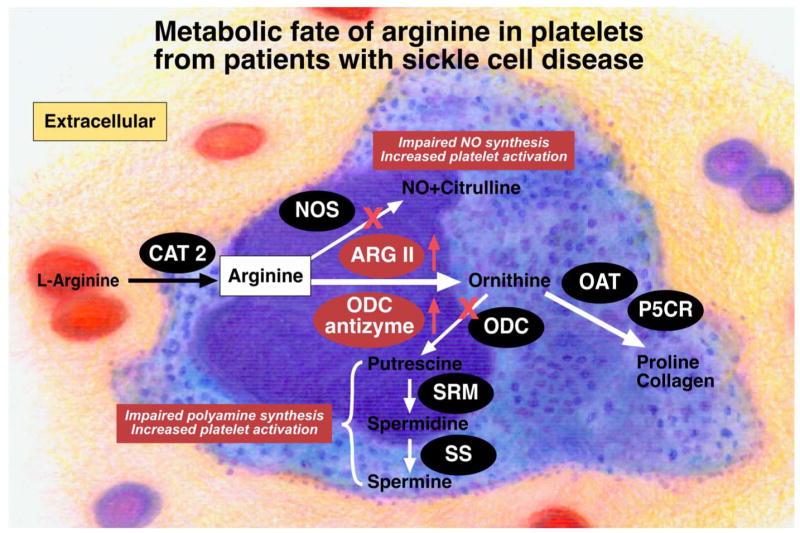
Schematic illustration of the metabolic fate of arginine in platelets in sickle cell disease. Under normal situations, arginine will be taken up by the platelets via cationic amino acid transporter-2 (CAT-2) for the synthesis of NO and polyamines, which protect cells from activation and aggregation. In sickle cell disease, increased arginase activity and consequent limitation in arginine bioavailability is expected to impair NO synthesis, whereas reduced polyamine levels will enhance platelet aggregation. The increased production of ornithine via arginase II (ARG-2), instead of going toward polyamine synthesis, would be diverted toward proline synthesis, consistent with increased levels of mRNA encoding pyrroline-5-carboxylate reductase (P5CR). Increased expression of enzymes is indicated by the red upward-pointing arrows and red highlighting of enzymes. ODC indicates ornithine decarboxylase; SRM, spermidine synthase; SS, spermine synthase; and antizyme, ornithine decarboxylase antizyme.
In summary, our findings of dysregulated arginine metabolism, combined with our analysis of platelet transcriptomes, indicates for the first time that platelet dysfunction in sickle cell disease can involve mechanisms at a pretranslational level. Concordant increases in arginase II mRNA and arginase activity indicate limitation of intracellular arginine bioavailability for platelet NO synthesis in sickle cell disease, further compounding the limitation in plasma arginine bioavailability in sickle cell patients.2 The discordance between reduced polyamine levels and increases in mRNAs encoding enzymes of polyamine synthesis, however, emphasizes the need to take into account additional mechanisms, such as translational control. Regarding this point, a recent serial analysis of gene expression (SAGE) of the platelet transcriptome identified potential elements in platelet mRNAs that may be involved in mRNA stabilization or translational control42
Polyamines (putrescine, spermidine, and spermine) have been shown to be potent inhibitors of platelet aggregation.43,44 Palanimurugan and colleagues45 have indicated that externally added polyamines (especially spermine) possess anti-aggregating effects on platelets that complement the effects of l-arginine. Consequently, decreased polyamine levels may increase the tendency of platelets to aggregate and thus increase the probability of thromboembolism. Although increased arginine uptake and/or synthesis from citrulline could potentially enhance intracellular arginine bioavailability, this could be counterbalanced by the increased arginase II expression, which can limit arginine availability for NO synthase. Consequently, platelet-mediated thrombotic events may be enhanced. Further studies with larger cohorts of patients will be required to evaluate levels of arginine metabolites in platelets, platelet activation, and risk of in situ embolic and thrombotic disease.
CLINICAL PERSPECTIVE.
In sickle cell disease, abnormal hemoglobin S–containing erythrocytes become entrapped in the microvasculature and hemolyze, which produces ischemia-reperfusion injury and infarction of tissue, endothelial dysfunction, and vasculopathy. From a clinical standpoint, thromboembolism and hypercoagulability contribute to disease pathogenesis. A number of studies have documented increased activation of platelets among other protean impairments in the thrombotic cascade. Recent functional studies of platelets in the sickle cell disease patients in the present study also revealed a basally activated state, which confirms that pathological platelet activation contributes to sickle cell disease vasculopathy. With the goal of deciphering transcriptional signaling pathways in platelets that may be dysregulated in sickle cell disease, we compared the platelet transcriptome derived from amplified RNA from single donors of sickle cell disease patients with that of black control subjects. Data analysis of their platelet transcriptome identified ≈100 differentially expressed genes in sickle cell disease, including multiple genes involved in arginine metabolism and redox homeostasis. Further characterization of these pathways with real-time polymerase chain reaction and biochemical assays revealed increased arginase II expression and activity and decreased platelet polyamine levels. Our findings suggest for the first time a potential pathogenic role for platelet arginase and altered arginine and polyamine metabolism in sickle cell disease. These studies also provide an experimental framework for the study of disease-specific platelet biology and identify novel therapeutic targets for managing complications in sickle cell disease–associated vasculopathy.
Supplementary Material
The online-only Data Supplement, consisting of tables I and II, is available with this article at http://circ.ahajournals.org/cgi/content/full/CIRCULATIONAHA.106.658641/DC1
Acknowledgments
The authors wish to thank Diane Kepka-Lenhart for excellent technical assistance and Dr Murali Adibhatla of the University of Wisconsin-Madison for his generous help in polyamine analysis.
Source of Funding: This work was supported in part by National Institutes of Health grant RO1 GM57384 to Dr Morris.
Footnotes
The online version of this article, along with updated information and services, is located on the World Wide Web at: http://circ.ahajournals.org
Subscriptions: Information about subscribing to Circulation is online at http://circ.ahajournals.org/subsriptions/
Permissions: Permissions & Rights Desk, Lippincott Williams & Wilkins, 351 West Camden Street, Baltimore, MD 21202-2436. Phone 410-5280-4050. Fax: 410-528-8550. Email: journalpermissions@lww.com
Reprints: Information about reprints can be found online at http://www.lww.com/static/html/reprints.html
Disclosures: Dr Morris served as a consultant to International Council on Amino Acid Science (ICAAS) and received honoraria for talks on arginase activity and function at Whitaker Cardiovascular Institute, Boston University School of Medicine, Children's Hospital Oakland Research Institute and Department of Nutrition, Case Western Reserve University. The remaining authors report no conflicts.
References
- 1.Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997;337:762–769. doi: 10.1056/NEJM199709113371107. [DOI] [PubMed] [Google Scholar]
- 2.Morris CR, Kato GJ, Poljakovic M, Wang X, Blackwelder WC, Sachdev V, Hazen SL, Vichinsky EP, Morris SM, Jr, Gladwin MT. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA. 2005;294:81–90. doi: 10.1001/jama.294.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reiter CD, Gladwin MT. An emerging role for nitric oxide in sickle cell disease vascular homeostasis and therapy. Curr Opin Hematol. 2003;10:99–107. doi: 10.1097/00062752-200303000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Castro O, Hoque M, Brown BD. Pulmonary hypertension in sickle cell disease: cardiac catheterization results and survival. Blood. 2003;101:1257–1261. doi: 10.1182/blood-2002-03-0948. [DOI] [PubMed] [Google Scholar]
- 5.Gladwin MT, Sachdev V, Jison ML, Shizukuda Y, Plehn JF, Minter K, Brown B, Coles WA, Nichols JS, Ernst I, Hunter LA, Blackwelder WC, Schechter AN, Rodgers GP, Castro O, Ognibene FP. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med. 2004;350:886–895. doi: 10.1056/NEJMoa035477. [DOI] [PubMed] [Google Scholar]
- 6.Rother RP, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA. 2005;293:1653–1662. doi: 10.1001/jama.293.13.1653. [DOI] [PubMed] [Google Scholar]
- 7.Vichinsky EP. Pulmonary hypertension in sickle cell disease. N Engl J Med. 2004;350:857–859. doi: 10.1056/NEJMp038250. [DOI] [PubMed] [Google Scholar]
- 8.Ataga KI, Moore CG, Jones S, Olajide O, Strayhorn D, Hinderliter A, Orringer EP. Pulmonary hypertension in patients with sickle cell disease: a longitudinal study. Br J Haematol. 2006;134:109–115. doi: 10.1111/j.1365-2141.2006.06110.x. [DOI] [PubMed] [Google Scholar]
- 9.Ataga KI, Sood N, De Gent G, Kelly E, Henderson AG, Jones S, Strayhorn D, Lail A, Lieff S, Orringer EP. Pulmonary hypertension in sickle cell disease. Am J Med. 2004;117:665–669. doi: 10.1016/j.amjmed.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 10.Blann AD, Marwah S, Serjeant G, Bareford D, Wright J. Platelet activation and endothelial cell dysfunction in sickle cell disease is unrelated to reduced antioxidant capacity. Blood Coagul Fibrinolysis. 2003;14:255–259. doi: 10.1097/01.mbc.0000061293.28953.8c. [DOI] [PubMed] [Google Scholar]
- 11.Francis RB., Jr Platelets, coagulation, and fibrinolysis in sickle cell disease: their possible role in vascular occlusion. Blood Coagul Fibrinolysis. 1991;2:341–353. doi: 10.1097/00001721-199104000-00018. [DOI] [PubMed] [Google Scholar]
- 12.Mohan JS, Lip GY, Bareford D, Blann AD. Platelet P-selectin and platelet mass, volume and component in sickle cell disease: relationship to genotype. Thromb Res. 2006;117:623–629. doi: 10.1016/j.thromres.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 13.Hebbel RP, Eaton JW, Steinberg MH, White JG. Erythrocyte/endothelial interactions in the pathogenesis of sickle-cell disease: a “real logical” assessment. Blood Cells. 1982;8:163–173. [PubMed] [Google Scholar]
- 14.Ozcan M, Morton CT, Solovey A, Dandelet L, Bach RR, Hebbel RP, Slungaard A, Key NS. Whole blood tissue factor procoagulant activity remains detectable during severe aplasia following bone marrow and peripheral blood stem cell transplantation. Thromb Haemost. 2001;85:250–255. [PubMed] [Google Scholar]
- 15.Kiechle FL. DNA technology, the clinical laboratory, and the future. Arch Pathol Lab Med. 2001;125:72–76. doi: 10.5858/2001-125-0072-DTTCLA. [DOI] [PubMed] [Google Scholar]
- 16.Bahou WF, Gnatenko DV. Platelet transcriptome: the application of microarray analysis to platelets. Semin Thromb Hemost. 2004;30:473–484. doi: 10.1055/s-2004-833482. [DOI] [PubMed] [Google Scholar]
- 17.Gnatenko DV, Dunn JJ, McCorkle SR, Weissmann D, Perrotta PL, Bahou WF. Transcript profiling of human platelets using microarray and serial analysis of gene expression. Blood. 2003;101:2285–2293. doi: 10.1182/blood-2002-09-2797. [DOI] [PubMed] [Google Scholar]
- 18.McRedmond JP, Park SD, Reilly DF, Coppinger JA, Maguire PB, Shields DC, Fitzgerald DJ. Integration of proteomics and genomics in platelets: a profile of platelet proteins and platelet-specific genes. Mol Cell Proteomics. 2004;3:133–144. doi: 10.1074/mcp.M300063-MCP200. [DOI] [PubMed] [Google Scholar]
- 19.Tomer A. Platelet activation as a marker for in vivo prothrombotic activity: detection by flow cytometry. J Biol Regul Homeost Agents. 2004;18:172–177. [PubMed] [Google Scholar]
- 20.Jison ML, Munson PJ, Barb JJ, Suffredini AF, Talwar S, Logun C, Raghavachari N, Beigel JH, Shelhamer JH, Danner RL, Gladwin MT. Blood mononuclear cell gene expression profiles characterize the oxidant, hemolytic, and inflammatory stress of sickle cell disease. Blood. 2004;104:270–280. doi: 10.1182/blood-2003-08-2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klipper-Aurbach Y, Wasserman M, Braunspiegel-Weintrob N, Borstein D, Peleg S, Assa S, Karp M, Benjamini Y, Hochberg Y, Laron Z. Mathematical formulae for the prediction of the residual beta cell function during the first two years of disease in children and adolescents with insulin-dependent diabetes mellitus. Med Hypotheses. 1995;45:486–490. doi: 10.1016/0306-9877(95)90228-7. [DOI] [PubMed] [Google Scholar]
- 22.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4(9):R60. I-II. [PubMed] [Google Scholar]
- 24.Bieche I, Parfait B, Tozlu S, Lidereau R, Vidaud M. Quantitation of androgen receptor gene expression in sporadic breast tumors by real-time RT-PCR: evidence that MYC is an AR-regulated gene. Carcinogenesis. 2001;22:1521–1526. doi: 10.1093/carcin/22.9.1521. [DOI] [PubMed] [Google Scholar]
- 25.Morris SM, Jr, Kepka-Lenhart D, Chen LC. Differential regulation of arginases and inducible nitric oxide synthase in murine macrophage cells. Am J Physiol. 1998;275(pt 1):E740–E747. doi: 10.1152/ajpendo.1998.275.5.E740. [DOI] [PubMed] [Google Scholar]
- 26.Adibhatla RM, Hatcher JF, Sailor K, Dempsey RJ. Polyamines and central nervous system injury: spermine and spermidine decrease following transient focal cerebral ischemia in spontaneously hypertensive rats. Brain Res. 2002;938:81–86. doi: 10.1016/s0006-8993(02)02447-2. [DOI] [PubMed] [Google Scholar]
- 27.Ibanga IA. Significance of platelet activation in sickle cell anaemia. Niger J Med. 2006;15:148–150. doi: 10.4314/njm.v15i2.37100. [DOI] [PubMed] [Google Scholar]
- 28.Inwald DP, Kirkham FJ, Peters MJ, Lane R, Wade A, Evans JP, Klein NJ. Platelet and leucocyte activation in childhood sickle cell disease: association with nocturnal hypoxaemia. Br J Haematol. 2000;111:474–481. doi: 10.1046/j.1365-2141.2000.02353.x. [DOI] [PubMed] [Google Scholar]
- 29.Wun T, Paglieroni T, Rangaswami A, Franklin PH, Welborn J, Cheung A, Tablin F. Platelet activation in patients with sickle cell disease. Br J Haematol. 1998;100:741–749. doi: 10.1046/j.1365-2141.1998.00627.x. [DOI] [PubMed] [Google Scholar]
- 30.Li H, Meininger CJ, Hawker JR, Jr, Haynes TE, Kepka-Lenhart D, Mistry SK, Morris SM, Jr, Wu G. Regulatory role of arginase I and II in nitric oxide, polyamine, and proline syntheses in endothelial cells. Am J Physiol Endocrinol Metab. 2001;280:E75–E82. doi: 10.1152/ajpendo.2001.280.1.E75. [DOI] [PubMed] [Google Scholar]
- 31.Xu W, Kaneko FT, Zheng S, Comhair SA, Janocha AJ, Goggans T, Thunnissen FB, Farver C, Hazen SL, Jennings C, Dweik RA, Arroliga AC, Erzurum SC. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J. 2004;18:1746–1748. doi: 10.1096/fj.04-2317fje. [DOI] [PubMed] [Google Scholar]
- 32.Loscalzo J. Nitric oxide insufficiency, platelet activation, and arterial thrombosis. Circ Res. 2001;88:756–762. doi: 10.1161/hh0801.089861. [DOI] [PubMed] [Google Scholar]
- 33.Wu G, Morris SM., Jr Arginine metabolism: nitric oxide and beyond. Biochem J. 1998;336(pt 1):1–17. doi: 10.1042/bj3360001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Erdely A, Kepka-Lenhart D, Clark M, Zeidler-Erdely P, Poljakovic M, Calhoun WJ, Morris SM., Jr Inhibition of phosphodiesterase 4 amplifies cytokine-dependent induction of arginase in macrophages. Am J Physiol Lung Cell Mol Physiol. 2006;290:L534–L539. doi: 10.1152/ajplung.00326.2005. [DOI] [PubMed] [Google Scholar]
- 35.Wallace HM, Fraser AV, Hughes A. A perspective of polyamine metabolism. Biochem J. 2003;376(pt 1):1–14. doi: 10.1042/BJ20031327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weyrich AS, Zimmerman GA. Platelets: signaling cells in the immune continuum. Trends Immunol. 2004;25:489–495. doi: 10.1016/j.it.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 37.Weyrich AS, Zimmerman GA. Evaluating the relevance of the platelet transcriptome. Blood. 2003;102:1550–1551. doi: 10.1182/blood-2003-05-1429. [DOI] [PubMed] [Google Scholar]
- 38.Healy AM, Pickard MD, Pradhan AD, Wang Y, Chen Z, Croce K, Sakuma M, Shi C, Zago AC, Garasic J, Damokosh AI, Dowie TL, Poisson L, Lillie J, Libby P, Ridker PM, Simon DI. Platelet expression profiling and clinical validation of myeloid-related protein-14 as a novel determinant of cardiovascular events. Circulation. 2006;113:2278–2284. doi: 10.1161/CIRCULATIONAHA.105.607333. [DOI] [PubMed] [Google Scholar]
- 39.Vercelli D. Arginase: marker, effector, or candidate gene for asthma? J Clin Invest. 2003;111:1815–1817. doi: 10.1172/JCI18908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zimmermann N, King NE, Laporte J, Yang M, Mishra A, Pope SM, Muntel EE, Witte DP, Pegg AA, Foster PS, Hamid Q, Rothenberg ME. Dissection of experimental asthma with DNA microarray analysis identifies arginase in asthma pathogenesis. J Clin Invest. 2003;111:1863–1874. doi: 10.1172/JCI17912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pegg AE. Regulation of ornithine decarboxylase. J Biol Chem. 2006;281:14529–14532. doi: 10.1074/jbc.R500031200. [DOI] [PubMed] [Google Scholar]
- 42.Dittrich M, Birschmann I, Stuhlfelder C, Sickmann A, Herterich S, Nieswandt B, Walter U, Dandekar T. Understanding platelets: lessons from proteomics, genomics and promises from network analysis. Thromb Haemost. 2005;94:916–925. doi: 10.1160/TH05-02-0121. [DOI] [PubMed] [Google Scholar]
- 43.Krishnamurthi S, Kakkar VV. Studies on the effect of platelet inhibitors on platelet adhesion to collagen and collagen-induced human platelet activation. Thromb Haemost. 1985;53:337–342. [PubMed] [Google Scholar]
- 44.Mendez JD, Zarzoza E. Inhibition of platelet aggregation by L-arginine and polyamines in alloxan treated rats. Biochem Mol Biol Int. 1997;43:311–318. doi: 10.1080/15216549700204091. [DOI] [PubMed] [Google Scholar]
- 45.Palanimurugan R, Scheel H, Hofmann K, Dohmen RJ. Polyamines regulate their synthesis by inducing expression and blocking degradation of ODC antizyme. EMBO J. 2004;23:4857–4867. doi: 10.1038/sj.emboj.7600473. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.