

Abstract
Congenital hyperinsulinism (CHI) is a disease characterized by persistent insulin secretion despite severe hypoglycemia. Mutations in the pancreatic ATP-sensitive K+ (KATP) channel proteins sulfonylurea receptor 1 (SUR1) and Kir6.2, encoded by ABCC8 and KCNJ11, respectively, is the most common cause of the disease. Many mutations in SUR1 render the channel unable to traffic to the cell surface, thereby reducing channel function. Previous studies have shown that for some SUR1 trafficking mutants, the defects could be corrected by treating cells with sulfonylureas or diazoxide. The purpose of this study is to identify additional mutations that cause channel biogenesis/trafficking defects and those that are amenable to rescue by pharmacological chaperones. Fifteen previously uncharacterized CHI-associated missense SUR1 mutations were examined for their biogenesis/trafficking defects and responses to pharmacological chaperones, using a combination of immunological and functional assays. Twelve of the 15 mutations analyzed cause reduction in cell surface expression of KATP channels by >50%. Sulfonylureas rescued a subset of the trafficking mutants. By contrast, diazoxide failed to rescue any of the mutants. Strikingly, the mutations rescued by sulfonylureas are all located in the first transmembrane domain of SUR1, designated as TMD0. All TMD0 mutants rescued to the cell surface by the sulfonylurea tolbutamide could be subsequently activated by metabolic inhibition on tolbutamide removal. Our study identifies a group of CHI-causing SUR1 mutations for which the resulting KATP channel trafficking and expression defects may be corrected pharmacologically to restore channel function.
Congenital hyperinsulinism (CHI) of infancy is characterized by inappropriate insulin secretion in the face of severe hypoglycemia in neonates and infants (1–4). Clinical treatments involving the use of diazoxide or somatostatin to inhibit insulin secretion are not always effective, necessitating partial or subtotal pancreatectomy in many cases (5,6). Mutations in ABCC8 and KCNJ11, which encode the ATP-sensitive K+ (KATP) channel subunits sulfonylurea receptor 1 (SUR1) and inward rectifier K+ channel Kir6.2, respectively, are the most common cause of the disease (4,7). KATP channels are critical for coupling metabolic signals to electrical signals to control insulin secretion (8,9). Potassium currents through the channels at resting metabolic state maintain the cell membrane at hyperpolarized potential, but the channels close on glucose stimulation to depolarize the cell membrane and trigger insulin secretion. In CHI patients carrying SUR1 or Kir6.2 mutations, KATP channel function is reduced or lost, leading to constant depolarization of the cell membrane and persistent insulin secretion, even when the blood glucose level is dangerously low (4,9–11).
Recent studies have shown that a mechanism underlying loss of channel function in CHI is loss of channel expression at the cell surface due to channel biogenesis/trafficking defects (12–16). The β-cell KATP channel is an octameric complex of four Kir6.2 and four SUR1 subunits (17–19). Subunit coassembly occurs in the endoplasmic reticulum (20). To ensure that only correctly assembled channels traffic to the cell surface, an Arg-Lys-Arg (RKR) tripeptide endoplasmic reticulum retention signal is present in each SUR1 and Kir6.2 subunit to prevent their exit out of the endoplasmic reticulum before complete channel assembly (20). On successful assembly of the channel complex, the endoplasmic reticulum retention signals are masked to allow forward trafficking of the channel to the Golgi and the plasma membrane. For SUR1 mutations, the loss of channel surface expression, which is caused by inability of the mutant protein to exit the endoplasmic reticulum, mutating the RKR signal to AAA has been shown to allow a small fraction of the mutants to escape the endoplasmic reticulum quality control mechanism and reach the plasma membrane (12,14). Reduced temperature has also been reported to increase surface expression of some endoplasmic reticulum–retained mutants (21). In addition, pharmacological agents, such as the oral hypoglycemic drugs sulfonylureas commonly used to treat type 2 diabetes, and the K+ channel opener diazoxide have been documented to correct channel trafficking defects (13,16). Of the above rescue strategies, the pharmacological manipulations have the highest potential in clinical application (13,16).
Although numerous CHI-associated KATP channel mutations have been identified, only a handful have been characterized in terms of their effects on channel trafficking and function (4,10,11,22). The purpose of this study is to identify additional mutations that cause channel biogenesis/trafficking defects and those that are amenable to pharmacological rescue. Of the 15 missense SUR1 mutations examined, 12 led to >50% reduction in channel expression at the cell surface (for simplicity, they will be collectively referred to as trafficking mutations). Interestingly, we found that only trafficking mutations located in the first transmembrane domain (referred to as TMD0) of SUR1 were responsive to sulfonylurea rescue. Furthermore, all TMD0 mutants that exhibited trafficking defects were rescued by sulfonylureas. By contrast, diazoxide failed to rescue any of the trafficking mutants. Channels rescued pharmacologically were activated on metabolic inhibition, suggesting that they would be able to open in response to hypoglycemia in vivo.
RESEARCH DESIGN AND METHODS
Genetic and clinical analyses
Peripheral blood was obtained from patients for isolation of genomic DNA. Individual exons and adjacent intronexon boundaries of the ABCC8 gene were amplified by the PCR and screened for mutations by direct nucleotide sequencing as previously described (23). SUR1 cDNA and protein sequences were numbered according to Nestorowicz et al. (24), with nucleotide numbering beginning with the first methionine and including the alternatively spliced exon 17 sequence (NCBI accession no. L78224). The study protocol was reviewed and approved by the Institutional Review Board of The Children’s Hospital of Philadelphia, and written informed consent was obtained from the parents of the patients.
Molecular biology
FLAG epitope (DYKDDDDK) waps inserted at the NH2-terminus of the hamster SUR1 cDNA in the pECE plasmid as previously described (12). SUR1 point mutations were made using the QuickChange site-directed mutagenesis kit (Stratagene) and confirmed by DNA sequencing. Rat Kir6.2 cDNA (gift from Dr. Carol Vandenberg) is in pCDNAI vector. Multiple PCR clones were analyzed to avoid false results caused by undesired PCR-introduced mutations.
Immunoblotting
COSm6 cells were plated in six-well tissue culture plates and transfected with 0.6 μg fSUR1 and 0.4 μg rat Kir6.2 per dish using FuGene6 (Roche Applied Science). Cells were lysed in 20 mmol/l HEPES, pH 7.0, 5 mmol/l EDTA, 150 mmol/l NaCl, and 1% Nonidet P-40 with Complete protease inhibitors (Roche Applied Science) 48–72 h after transfection. Proteins were separated by SDS-PAGE (8%), transferred to nitrocellulose, analyzed by M2 anti-FLAG antibody (Sigma) followed by horseradish peroxidase (HRP)–conjugated anti-mouse secondary antibodies (GE Healthcare), and visualized by chemiluminescence (Super Signal West Femto; Pierce). Experimental procedures for the immunoblot shown in Fig. 5D using INS-1 cells are described in detail in the online appendix (available at http://dx.doi.org/10.2337/db07-0150).
FIG. 5.
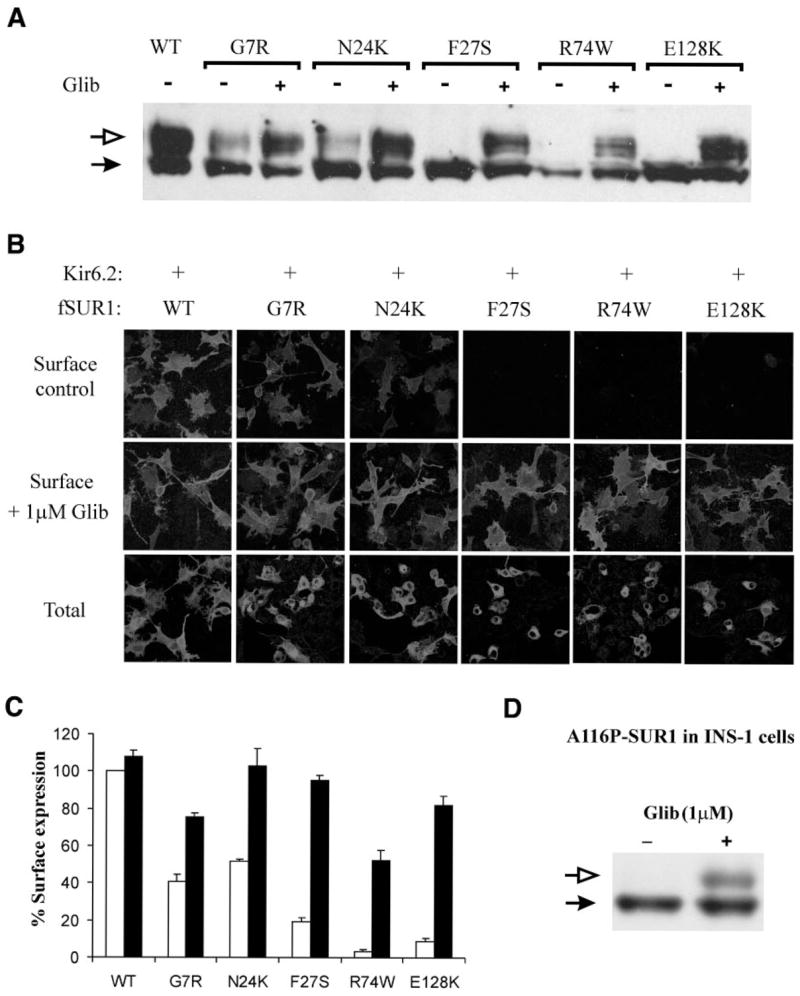
Sulfonylureas rescue the trafficking defects caused by TMD0 mutations. A: Western blot analysis of mutant fSUR1 in COS cells coexpressing Kir6.2. Glibenclamide (1 μmol/l, 24 h) increased the level of the upper fSUR1 band in all TMD0 mutants. B: Immunofluorescent staining demonstrates increased surface fSUR1 expression in cells treated with glibenclamide. C: Chemiluminescence assays show significantly higher surface channel expression in cells treated with glibenclamide for all TMD0 mutants (P < 0.001). □, control; ■, 1 μmol/l glibenclamide. D: Western blot showing that the A116P-SUR1 expressed in INS-1 cells lacks the complex-glycosylated band and that treatment of cells with glibenclamide led to appearance of the complex-glycosylated band, as observed in COS cells.
Immunofluorescence staining
COSm6 cells transfected with fSUR1 and Kir6.2 were plated onto coverslips for staining 48–72 h after transfection. For surface staining, living cells were incubated with anti-FLAG M2 mouse monoclonal antibody (10 μg/ml OptiMEM containing 0.1% BSA) for 1 h at 4°C, washed with ice-cold PBS, fixed with cold (−20°C) methanol for 10 min, and incubated with Cy-3–conjugated donkey anti-mouse secondary antibodies (Jackson) for 30 min at room temperature. After three 10-min washes in PBS/0.1% BSA and one 10-min wash in PBS, cells were viewed with an Olympus Fluoview confocal microscope. For total cellular staining, the same procedure was followed except the cells were fixed with cold (−20°C) methanol for 10 min before antibody incubation.
Chemiluminescence assay
Cells were fixed with 2% paraformaldehyde for 30 min at 4°C, preblocked in PBS/0.1% BSA for 30 min, incubated in M2 anti-FLAG antibody (10 μg/ml) for 1 h, washed four times for 30 min in PBS/0.1% BSA, incubated in HRP-conjugated anti-mouse (1:1,000 dilution; Jackson) for 20 min, and washed again four times for 30 min in PBS/0.1% BSA. Chemiluminescence was quantified using a TD-20/20 luminometer (Turner Designs) after 5-s incubation in Power Signal Elisa Femto luminol solution (Pierce). All steps after fixation were performed at room temperature. Results of each experiment are the average of two dishes, and each data point shown in figures is the average of three to six experiments.
86Rb+ efflux assay
Cells were incubated overnight in culture medium containing 86RbCl (1 μCi/ml) 48 h after transfection with fSUR1 and Kir6.2. Before measurement of 86Rb+ efflux, the cells were incubated for 30 min at 25°C in Krebs-Ringer solution with metabolic inhibitors (2.5 μg/ml oligomycin plus 1 mmol/l 2-deoxy-D-glucose). At selected time points, the solution was aspirated from the cells and replaced with fresh solution, and at the end of a 40-min period, cells were lysed. The 86Rb+ in the aspirated solution and the cell lysate was counted. The percentage of efflux at each time point was calculated as the cumulative counts in the aspirated solution divided by the total counts from the solutions and the cell lysates.
Patch-clamp recordings
Inside-out patch clamp recordings were carried out as previously described using an Axopatch 1D amplifier (Axon, Foster City, CA) (16). Micropipettes were pulled from nonheparinized Kimble glass (Fisher Scientific) on a horizontal puller (Sutter Instrument, Novato, CA) with ~1.5 MΩ electrode resistance. The bath and pipette solution (K-INT) had the following composition: 140 mmol/l KCl, 10 mmol/l K-HEPES, and 1 mmol/l K-EGTA, pH 7.3. ATP and ADP were added as the potassium salts. All currents were measured at membrane potential of −50 mV (pipette voltage = 50 mV) at room temperature. Data were analyzed using pCLAMP8 and Microsoft Excel.
Data analysis
Data were presented as means ± SE. Statistical analysis was performed using independent two-population two-tailed Student’s t test, with P < 0.05 considered statistically significant.
RESULTS
Mutations and clinical data
Previously, we reported several CHI-associated SUR1 mutations that cause endoplasmic reticulum retention and reduced surface expression of KATP channels (12,14,16). To extend these studies, we screened 15 additional SUR1 missense mutations that are distributed throughout the protein for their potential effects on surface expression of the channel. The mutations are divided into three groups based on locations according to the current SUR1 topology model (Fig. 1). The first group, including G7R, N24K, F27S, R74W, and E128K, is located in the first transmembrane domain TMD0; the second group, including R495Q, E501K, L503P, F686S, and G716V, is located in the second transmembrane domain TMD1 extending through the first nucleotide binding domain; the third group, including K1337N, L1350Q, S1387F, L1390P, and D1472H, is clustered in the second nucleotide binding domain and the COOH terminus of the protein. More than one-half of these mutations have been previously identified (for references, see Table 1) (22), although detailed analysis of how these mutations affect KATP channel biology is still lacking. The mutations that have not been previously reported in the literature include N24K, E128K, and L1390P. Clinical and genetic information available on patients carrying these mutations are summarized in Table 1.
FIG. 1.
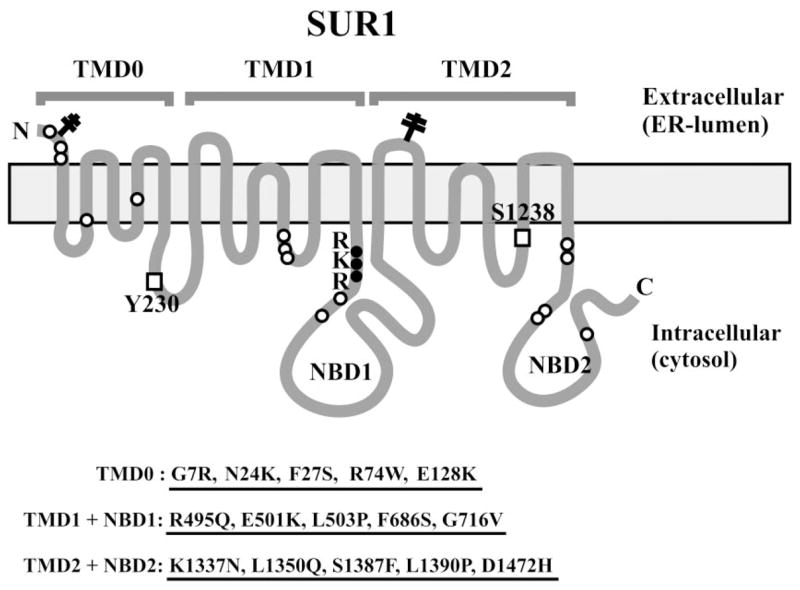
SUR1 topology model showing the three transmembrane domains, the -RKR–endoplasmic reticulum retention motif, sulfonylurea binding residues (□), and the locations of the CHI mutations.
TABLE 1.
Genetic and clinical information on patients carrying the CHI mutations
| Mutation | Disease | Haplotype | Diazoxide response | References |
|---|---|---|---|---|
| G7R | Focal | G7R | No | 44 |
| N24K | Diffuse | N24K/R1215W | No | Not reported |
| F27S | Focal | F27S | No | 39 |
| R74W | Focal | R74W/R1215Q | No | 39,45,46 |
| E128K | Diffuse | E128K | No | Not reported |
| R495Q | Diffuse | R495Q/R1215Q | No | 39 |
| E501K | Focal | E501K | No | 39 |
| L503P | Focal | L503P | No | 44 |
| F686S | Focal | F686S | No | 39 |
| G716V* | Diffuse | G716V/G716V | No | 47,48 |
| K1337N | Not done | g3992-9a/K1337N | Yes | 39 |
| L1350Q | Focal | L1350Q | No | 44 |
| S1387F | Diffuse | S1387F/NA | No | 9,24 |
| L1390P | NA | L1390P/NA | No | Not reported |
| D1472H | Diffuse | ΔF1388/D1472H | No | 39 |
Patient was from consanguineous mating and therefore was homozygous for the G716V mutation (48). NA, information not available.
Surface expression analysis of mutant KATP channels
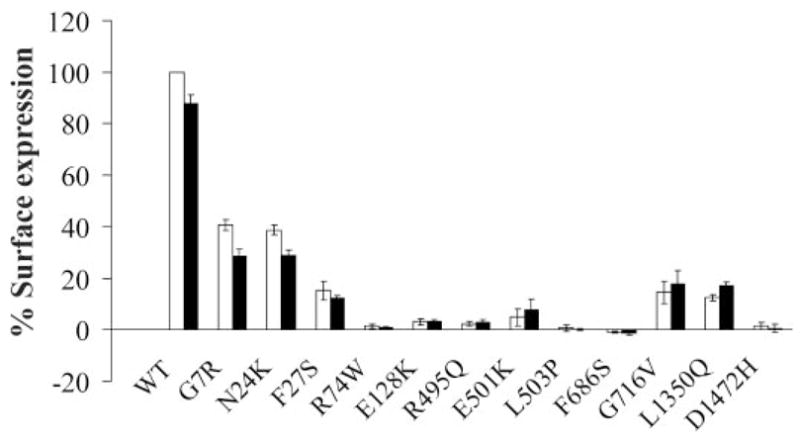
Cell surface expression of KATP channels was analyzed using immunoblotting, immunofluorescent staining, chemiluminescence assays, and electrophysiological recording of channel activity; these methods have been documented extensively in previous studies and provide both qualitative and quantitative measures of channel expression at the cell surface (12,14,16,25,26). All of the mutations were engineered into a SUR1 construct tagged at the extracellular NH2-terminus with a FLAG epitope (fSUR1) to facilitate detection at the cell surface; the FLAG tag has no discernable effect on either expression or function of wild-type channels (12). In immunoblots, wild-type fSUR1 coexpressed with Kir6.2 is detected as a core-glycosylated form of ~140 kDa and an complex-glycosylated form of ~180 kDa. Because complex glycosylation occurs in the medial Golgi, the upper fSUR1 band indicates fSUR1 that has passed the endoplasmic reticulum quality control checkpoint by forming channel complex with Kir6.2. The immunoblotting analysis revealed reduced level of the upper band in many SUR1 mutations (only selective mutants are shown in Fig. 2A), suggesting that these mutations might cause channel trafficking defects. To further investigate the subcellular localization of the mutants, we performed immunofluorescent staining of fSUR1 present at the cell surface and the total fSUR1 present in the cell (see RESEARCH DESIGN AND METHODS). The same mutations that exhibited no or reduced mature band in immunoblotting analysis also showed no or reduced surface fluorescence signals; total fluorescent staining revealed that the mutant proteins were present but were retained intracellularly (two mutants are shown in Fig. 2B; see also Supplemental Fig. 1). To better quantify channel expression level at the cell surface, we labeled surface fSUR1 with the anti-FLAG antibody, which was then detected using a chemiluminescence-based enzymatic reaction (14,16). Results from this assay showed that F27S, R74W, E128K, R495Q, E501K, L503P, F686S, G716V, L1350Q, and D1472H mutant channels had greatly reduced surface expression (<20% of wild-type level)—whereas G7R and N24K mutant channels displayed modestly decreased surface expression level (>30% but <50% of wild-type level) and K1337N, S1378F, and L1390P exhibited normal or mildly reduced expression (>60% of wild-type level; Fig. 3A). Channel density of the various mutants estimated using inside-out patch-clamp recording is in general agreement with chemiluminescence assay results (Fig. 3B; Supplemental Fig. 2), providing confirmation that the surface fSUR1 detected was co-assembled with Kir6.2 as KATP channel complex. Together, the above analyses identify new mutations of the group that likely cause hyperinsulinism by at least in part reducing surface expression of functional KATP channels.
FIG. 2.
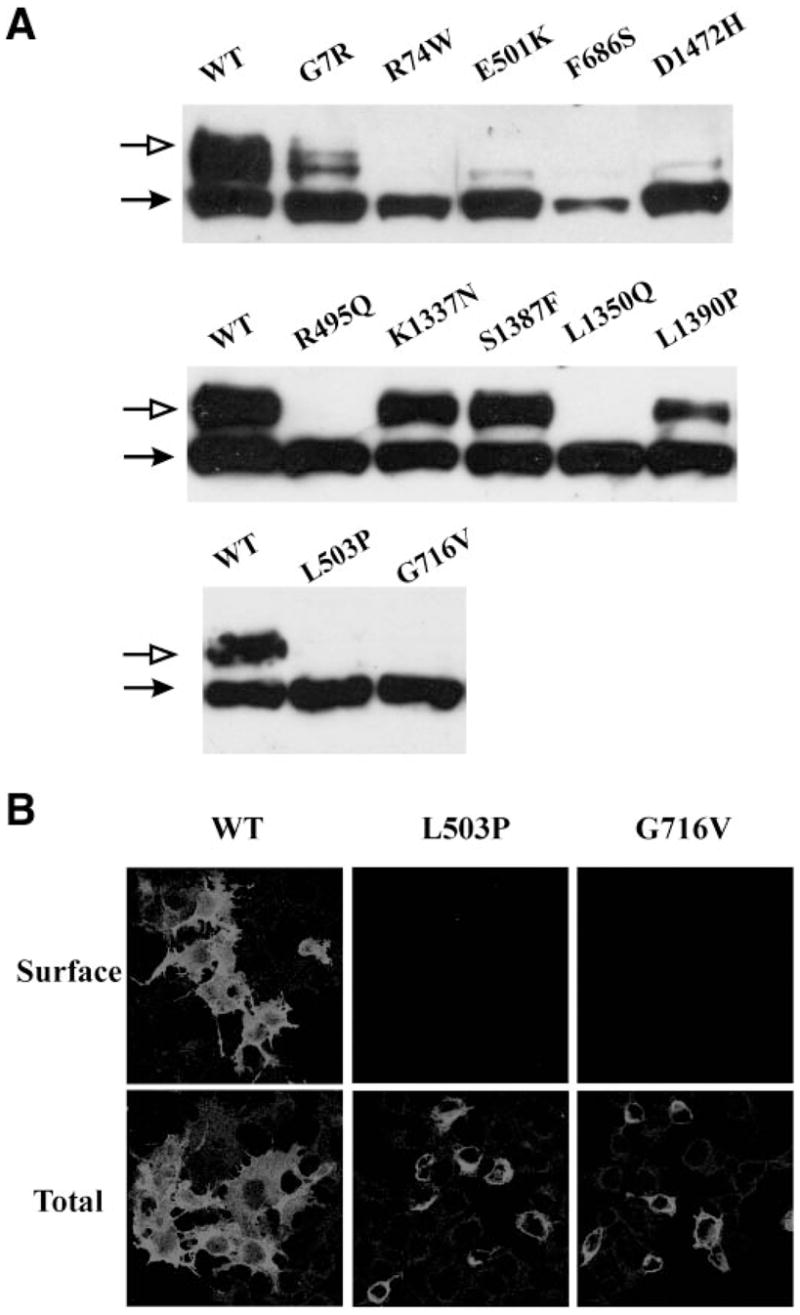
Analysis of mutant SUR1 by Western blotting and immunofluorescent staining in cells coexpressing Kir6.2. A: In Western blots, wild-type fSUR1 was detected as a lower core glycosylated and an upper complex glycosylated band. The various SUR1 mutants exhibit different levels of the upper band, suggesting varied processing efficiency. Only a subset of mutants is shown, and the mutations are not in a particular order. B: Surface staining of fSUR1 is shown in the top panel, and total cellular staining is shown in the bottom panel.
FIG. 3.
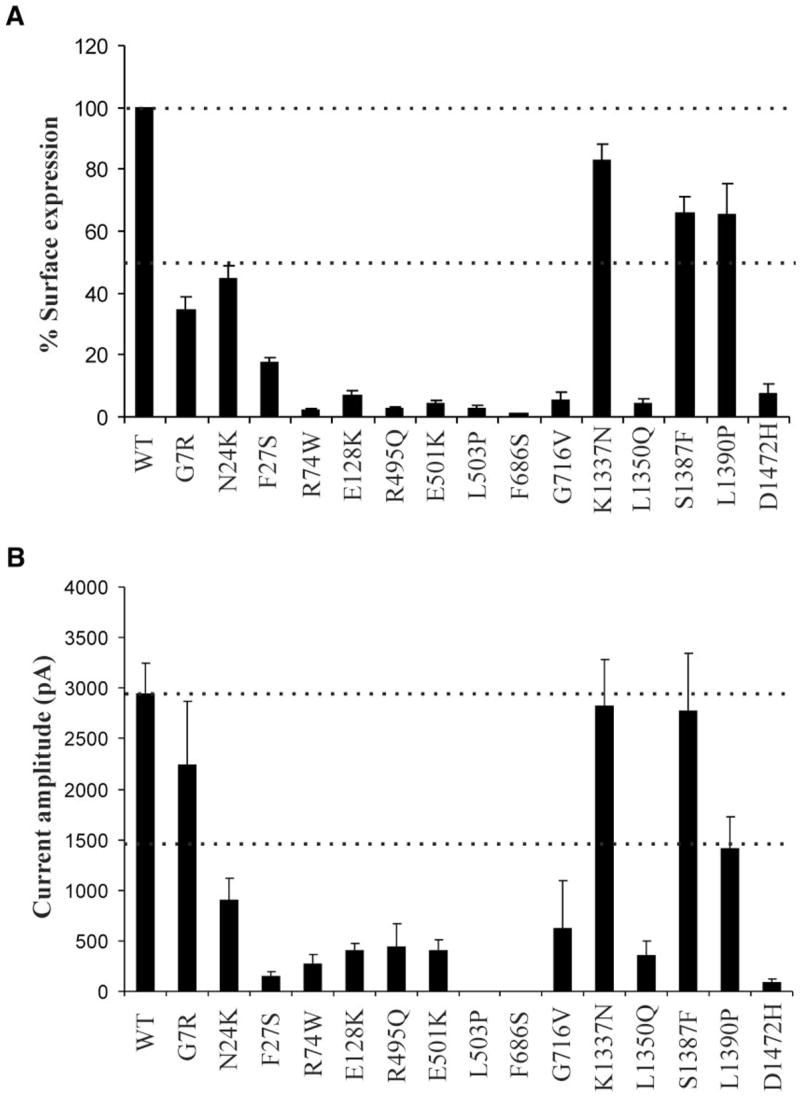
Analysis of mutant channel surface expression levels by chemiluminescence assays (A) and inside-out patch-clamp recordings (B). Surface expression in A is normalized to that of wild-type channels, with 50 and 100% expression levels marked by the dotted lines. Current amplitudes measured by patch-clamp recording are shown in pA, with the dotted lines marking 50 and 100% of wild-type currents. Note that because current density measurements by patching individual cells are subject to variation in expression efficiency from cell to cell, the errors tend to be larger.
Analysis of mutant KATP channel response to MgADP
In addition to loss of channel expression at the plasma membrane, inability of the channel to respond to MgADP stimulation is another prominent molecular defect that accounts for lack of KATP channel activity in hypoglycemic CHI patients (27,28). We therefore examined how those mutants that have good expression at the cell surface, including K1337N, S1387F, and L1390P, respond to MgADP using inside-out patch-clamp recording (27,28). In wild-type channels, exposure of the cytoplasmic face of the channel to 1 or 0.1 mmol/l ATP caused near total inhibition of channel activity; however, addition of 0.5 mmol/l MgADP to 0.1 mmol/l ATP antagonized the inhibition by 0.1 mmol/l ATP, demonstrating the stimulatory effect of MgADP on channel activity. This response to MgADP was abolished in the S1387F and L1390P mutations and slightly reduced (not statistically significant) in the K1337N mutation. Representative recordings from these mutants are shown in Fig. 4A, and the averaged data are summarized in Fig. 4B.
FIG. 4.
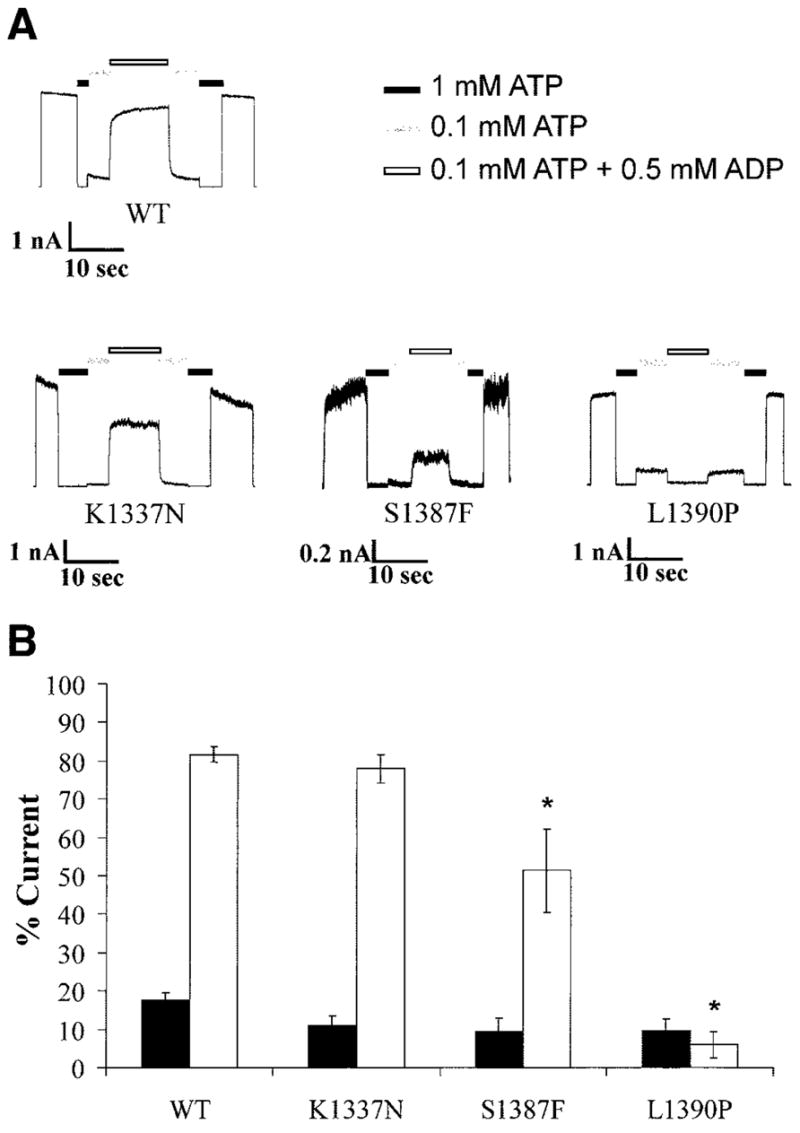
Reduced MgADP response in some mutants. A: Inside-out patch-clamp recordings of channels exposed to differing concentrations of ATP and ADP, as indicated by the bars above. Free [Mg2+] was at 1 mmol/l in all ATP-containing solutions. B: Quantification of currents in 0.1 mmol/l ATP (■) and 0.1 mmol/l ATP plus 0.5 mmol/l ADP (□) relative to that in the absence of ATP (expressed as percent current). Each bar is the average of four to seven patches ± SE. *P < 0.05.
Pharmacological correction of channel trafficking defects
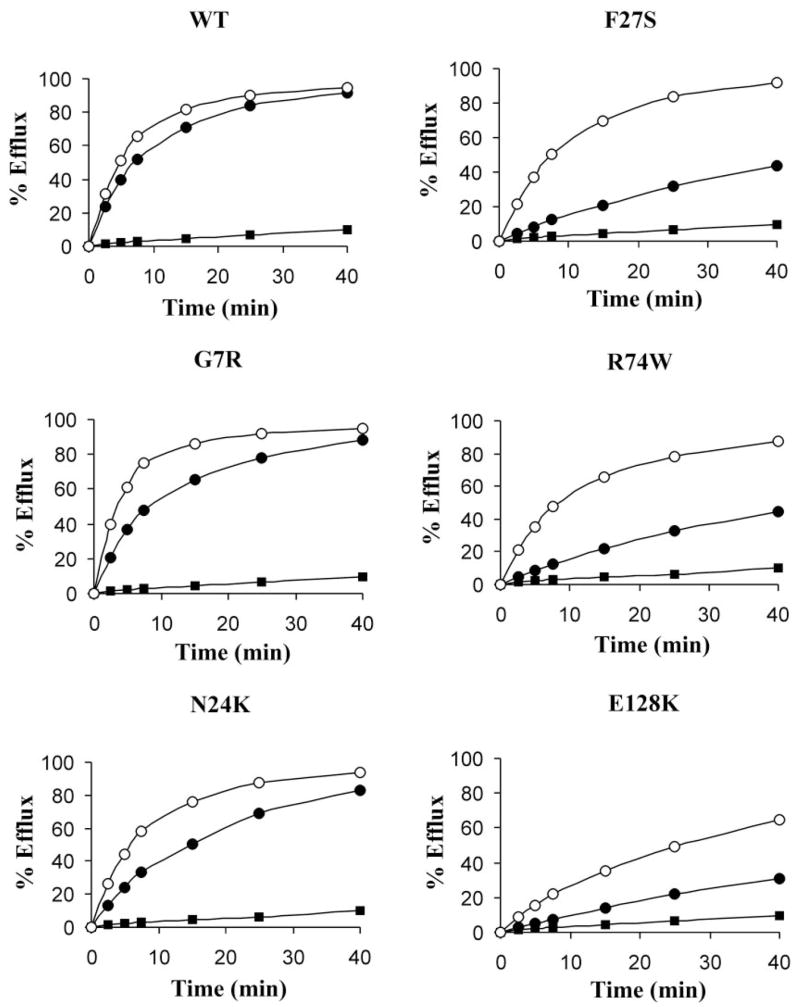
The observation that mutant SUR1 proteins lacking surface expression are present intracellularly prompted us to test whether the mutant proteins could be rescued to the cell surface using pharmacological agents that have been reported to correct channel trafficking defects (13,16). We first examined the effects of sulfonylureas, which were shown previously to improve surface expression of the A116P- and V187D-SUR1 mutants (16), on the trafficking mutants identified in this study (those that had surface expression <50% of wild type based on chemiluminescence assays shown in Fig. 3A). In Western blots, several mutants, including G7R, N24K, F27S, R74W, and E128K, all located in TMD0, exhibited increased complex-glycosylated SUR1 in cells coexpressing Kir6.2 on overnight treatment with 1 μmol/l glibenclamide (Fig. 5A). To determine whether the increased complex-glycosylated SUR1 corresponds to increased cell surface expression of mutant channels, immunofluorescent staining of surface fSUR1 was performed in cells treated with 1 μmol/l glibenclamide or DMSO control. As shown in Fig. 5B, glibenclamide treatment markedly increased surface expression of the aforementioned TMD0 mutants. Chemiluminescence assays yielded consistent results, with the surface expression level increased by 30–70% (Fig. 5C). Tolbutamide (at 300 μmol/l) similarly improved surface expression of the five TMD0 mutants in all three different assays (data not shown). To ensure that the trafficking defects of TMD0 mutations and their rescue by sulfonylureas are also seen in a cellular environment in which the channels normally reside, we examined whether A116P, a TMD0 trafficking mutant we documented previously, behaves the same in the insulin-secreting cell line INS-1 as in COS cells. Because INS-1 cells are difficult to transfect, we resorted to the adenoviral system for expression of the mutant protein (see online appendix for detailed procedures) (29). As shown in Fig. 5D, A116P-SUR1 expressed in INS-1 cells failed to mature into the complex-glycosy-lated form; however, overnight treatment of cells with 1 μmol/l glibenclamide overcame this processing defect and led to the appearance of the complex-glycosylated form. These observations recapitulate what we have reported previously for A116P-SUR1 expressed in COS cells (16), suggesting that our findings in COS cells are likely applicable to insulin-secreting cells.
We next examined whether diazoxide, a K+ channel opener that also binds SUR1, is able to rescue our trafficking mutants. In contrast to sulfonylureas, diazoxide (100 μmol/l) did not have significant rescue effects on surface expression of any of the trafficking mutants either in immunostaining experiments (not shown) or in chemiluminescence assays (Fig. 6). Because diazoxide has been reported by another group to correct channel trafficking defect caused by the R1394H-SUR1 mutation (13), we examined the expression pattern of the mutant and how it responds to diazoxide. In our hands, the R1394H mutant channels have expression levels very comparable with wild-type channels. The surface expression level is ~80% that of wild type, and diazoxide had no obvious effect on either expression level or cellular distribution of the mutant (data not shown).
FIG. 6.
Diazoxide failed to improve surface expression of any of the trafficking mutants. COSm6 cells coexpressing Kir6.2 and one of the fSUR1 constructs indicated below the x-axis were treated with (■) or without (□) 100 μmol/l diazoxide for 24 h before the chemiluminescence assay.
Functional restoration of TMD0 trafficking mutants rescued to the cell surface by pharmacological chaperones
The key defect in CHI caused by KATP channel mutations is inadequate channel activation by hypoglycemia. For channels expressed at the cell surface to perform their physiological function, they have to be able to open when cell metabolism is inhibited. We sought to determine whether the TMD0 mutant channels rescued to the plasma membrane are responsive to the metabolic signals associated with hypoglycemia. Channel activity in intact cells was assessed using the 86Rb+ efflux assay, which is well established for monitoring channel response to metabolic inhibition (23,28,30). In cells expressing wild-type KATP channel subunits, >90% efflux was observed in a 40-min time period after cells were preincubated for 30 min with the metabolic inhibitors 2-deoxy-D-glucose (1 mmol/l) and oligomycin (2.5 μg/ml). In contrast, untransfected cells showed <10% efflux in 40 min (Fig. 7). Consistent with the mutations causing hyperinsulinism, cells expressing one of the five TMD0 mutant channels exhibited reduced Rb+ efflux compared with cells expressing wild-type channels. To test whether sulfonylurea treatment improves the response of cells expressing the mutants to metabolic inhibition, we treated the cells with 300 μmol/l tolbut-amide overnight and removed the tolbutamide 4 h before the assay. Tolbutamide was chosen over glibenclamide for this experiment because we have previously shown that glibenclamide, which binds SUR1 with high affinity, remains bound to channels rescued to the cell surface, preventing the channel from opening on metabolic inhibition; tolbutamide can be easily washed off to recover channel function (16,31). Cells pretreated with tolbut-amide followed by subsequent removal showed greater efflux than untreated cells (Fig. 7; see also Supplemental Fig. 1). Our results demonstrate that reversible sulfonylurea drugs, such as tolbutamide, can be used to correct KATP channel trafficking defects and recover channel function in CHI patients carrying the TMD0-SUR1 mutations described in our earlier study (16) and the present study.
FIG. 7.
TMD0 mutant channels rescued to the cell surface are activated by metabolic inhibition. COSm6 cells coexpressing Kir6.2 and fSUR1 were treated with or without 300 μmol/l tolbutamide for 18 h and tolbutamide removed 4 h before the assay (similar results were obtained with 1- or 2-h tolbutamide washout). Representative Rb efflux profiles in response to metabolic inhibition in untransfected cells (■), control cells (●), and cells pretreated with tolbutamide (○) are shown.
DISCUSSION
KATP channel mutations that lead to partial or total loss of channel function are a major cause of CHI (4,6,11,32,33). Understanding how mutations affect molecular and cellular properties of the channel to blunt channel function may help develop new disease treatment strategies. Several reports have documented decreased KATP channel expression at the plasma membrane as mutation-induced defects that account for loss of channel function (14–16,25,33,34). Importantly, defective surface expression seen in some of these mutant channels may be corrected pharmacologically with the resulting channels being physiologically functional (13,16), raising the possibility of a novel treatment for patients carrying these mutations. In this study, by screening 15 disease-associated SUR1 missense mutations, we identified 12 additional SUR1 mutations that reduce KATP channel surface expression by >50%. Of these, five responded to sulfonylurea treatment with significantly improved surface channel expression. Moreover, mutant channels that were rescued by tolbutamide all showed improved activity in response to metabolic inhibition in 86Rb+ efflux assays, demonstrating restoration of channel function. Our findings reinforce the notion that mislocalization of KATP channels is a major mechanism contributing to loss of channel function in CHI and expand the number of mutations that may be targeted by pharmacological chaperones to restore channel expression and function.
TMD0 mutations
The first two trafficking mutations that we reported to be rescued by sulfonylurea drugs are A116P and V187D, both located in TMD0 of SUR1 (16). Strikingly, of the 12 new trafficking mutants that we identified in this study, only the TMD0 mutants responded to sulfonylurea rescue. TMD0 is a distinct structural feature of the SUR protein that separates it from other prototype ATP-binding cassette (ABC) transporter family members, such as CFTR (cystic fibrosis transmembrane conductance regulator) and P-glycoprotein, which contain only two transmembrane domains. TMD0 alone has been shown to associate with Kir6.2 and modulate its gating property, indicating that the domain is in and of itself a structural and functional entity (34,35). Recently, we have shown that sulfonylureas rescue TMD0 trafficking mutants via direct interactions with the channel complex (31). However, rather than binding to TMD0, sulfonylureas appear to bind to regions downstream of TMD0 to exert their chaperoning effects. First, a truncated SUR1 of TMD0 alone containing the A116P or V187D trafficking mutations failed to respond to sulfonylurea rescue. Second, two point mutations, Y230A and S1238Y, that are located downstream of TMD0 and are known to diminish or abolish glibenclamide and tolbutamide binding accordingly affected the ability of the drugs to rescue channel trafficking defects caused by TMD0 mutations (31,36,37). Thus, it appears that sulfonylurea binding to the ABC core region of SUR1 somehow overcomes the defect caused by TMD0 mutations. There is precedent that such substrate-induced transmembrane domain interactions occur: In another ABC transporter, the human P-glycoprotein, substrate-induced interactions between the two transmembrane domains promote superfolding of partially unfolded intermediates into the mature enzyme (38). An additional interesting and relevant finding from our recent study is that the Kir6.2 subunit coexpression is required for sulfonylureas to correct trafficking defects that resulted from SUR1-TMD0 mutations (31). It is possible that the TMD0 trafficking mutations interfere with normal association with Kir6.2 and that sulfonylurea binding promotes SUR1-Kir6.2 interactions to overcome channel biogenesis/trafficking defects.
Comparison between in vitro functional data and clinical data
The mutations that we examined here cause either significant reduction in channel expression level at the cell surface or channel response to MgADP when expressed as homozygous mutants, with the exception of the K1337N mutation, which only exhibited a slight reduction in surface expression and MgADP response that did not reach statistical significance. Therefore, in most cases, the in vitro channel phenotype is consistent with the loss of channel function phenotype in patients. Because the focus of this study is identifying new trafficking mutants and their rescue by pharmacological chaperones, we did not exhaustively analyze all of the mutants for their MgADP response or other channel properties that might affect channel function. However, it is likely that some of the mutations cause both expression and functional defects, as we have documented before (12,14). Also, some mutations are compound heterozygous with another SUR1 mutation in the other allele that may also reduce channel function. For example, both R74W and R495Q are compound heterozygous mutations with R1215Q. Based on in vitro functional phenotypes of mutant hamster SUR1/rat Kir6.2 channels expressed in COS cells, one would expect that in patients, in addition to the expression defects caused by the R74W or R495Q mutation, the R1215Q mutation would also reduce channel function by reducing channel response to MgADP (28). Another example is the combination of D1472H and ΔF1388, both of which result in nearly complete loss of channel expression at the cell surface (12). The functional data also show that in many mutants, channel activity is not completely lost but only partially reduced (Figs. 3, 4, and 7). This is consistent with the brisk acute insulin responses to tolbutamide observed in vivo in some of the diffuse cases of CHI (39). This was particularly the case in a patient with compound heterozygous R74W/R1215Q mutations who had a greater than normal insulin response to tolbutamide. Overall, the in vitro functional analysis provides valuable information by allowing prediction of phenotype based on genotype to facilitate disease diagnosis and management (40).
Therapeutic implications
Despite their defective biogenesis or trafficking, the TMD0 mutants tested so far all retain the ability to respond to metabolic inhibition in the Rb efflux assay, suggesting that they would improve β-cell function once rescued to the cell surface (16). However, it is important to recognize that the experiments described in this study on the identification of trafficking mutants and their response to sulfonylureas were all conducted in COS cells, which differ significantly from β-cells. In particular, recent studies have suggested that KATP channels are concentrated in insulin granules, which constitute the regulated secretory pathway absent in COS cells (41,42). Because there are many examples of cell-type–specific protein trafficking regulation (13,21,43), what we found in COS cells may not extrapolate directly to β-cells. In this regard, our results that a previously published TMD0 trafficking mutant, A116P, exhibits the same trafficking defect and response to sulfonylurea rescue in INS-1 cells as in COS cells provide some assurance that the TMD0 mutants are likely to behave similarly in their native environment. Nevertheless, it is imperative to confirm our findings in β-cells for all other trafficking mutants in the future. Another important point to consider is that because sulfonylureas also inhibit KATP channel activity, channel function can only be restored after drug removal. Thus, a future challenge in harnessing the sulfonylurea chaperoning effect to benefit CHI patients carrying TMD0 mutations would be to develop sulfonylurea administration strategies or analog compounds that will maximize the chaperoning effect of sulfonylureas but minimize their inhibitory effect on channel activity.
Supplementary Material
Acknowledgments
C.A.S. has received National Institutes of Health (NIH) Grants R01-DK-56268 and 5-MO1-RR-000240. S.-L.S. has received NIH Grants R01-DK-57699 and DK-66485.
We thank Jeremy Bushman, Jillene Casey, Malini Koch-har, and Emily Pratt from the Shyng laboratory for technical assistance and comments on the manuscript. We are grateful to the patients, the General Clinical Research Center staff, and the Children’s Hospital of Philadelphia nurses for participating in this research.
- ABC
ATP-binding cassette
- CHI
congenital hyperinsulinism
- HRP
horse- radish peroxidase
- KATP channel
ATP-sensitive K+ channel
- SUR1
sulfonylureka receptor 1
Footnotes
Published ahead of print at http://diabetes.diabetesjournals.org on 15 June 2007.
Additional information for this article can be found in an online appendix at http://dx.doi.org/10.2337/db07-0150.
References
- 1.Aynsley-Green A, Dunne MJ, James RF, Lindley KJ. Ions and genes in persistent hyperinsulinaemic hypoglycaemia in infancy: a commentary on the implications for tailoring treatment to disease pathogenesis. J Pediatr Endocrinol Metab. 1998;11(Suppl 1):121–129. doi: 10.1515/jpem.1998.11.s1.121. [DOI] [PubMed] [Google Scholar]
- 2.Glaser B. Hyperinsulinism of the newborn. Semin Perinatol. 2000;24:150–163. doi: 10.1053/sp.2000.6365. [DOI] [PubMed] [Google Scholar]
- 3.Stanley CA. Hyperinsulinism in infants and children. Pediatr Clin North Am. 1997;44:363–374. doi: 10.1016/s0031-3955(05)70481-8. [DOI] [PubMed] [Google Scholar]
- 4.Dunne MJ, Cosgrove KE, Shepherd RM, Aynsley-Green A, Lindley KJ. Hyperinsulinism in infancy: from basic science to clinical disease. Physiol Rev. 2004;84:239–275. doi: 10.1152/physrev.00022.2003. [DOI] [PubMed] [Google Scholar]
- 5.Stanley CA. Advances in diagnosis and treatment of hyperinsulinism in infants and children. J Clin Endocrinol Metab. 2002;87:4857–4859. doi: 10.1210/jc.2002-021403. [DOI] [PubMed] [Google Scholar]
- 6.Hussain K, Aynsley-Green A, Stanley CA. Medications used in the treatment of hypoglycemia due to congenital hyperinsulinism of infancy (HI) Pediatr Endocrinol Rev. 2004;2(Suppl 1):163–167. [PubMed] [Google Scholar]
- 7.Giurgea I, Bellanne-Chantelot C, Ribeiro M, Hubert L, Sempoux C, Robert JJ, Blankenstein O, Hussain K, Brunelle F, Nihoul-Fekete C, Rahier J, Jaubert F, de Lonlay P. Molecular mechanisms of neonatal hyperinsulinism. Horm Res. 2006;66:289–296. doi: 10.1159/000095938. [DOI] [PubMed] [Google Scholar]
- 8.Ashcroft FM. K(ATP) channels and insulin secretion: a key role in health and disease. Biochem Soc Trans. 2006;34:243–246. doi: 10.1042/BST20060243. [DOI] [PubMed] [Google Scholar]
- 9.Aguilar-Bryan L, Bryan J. Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr Rev. 1999;20:101–135. doi: 10.1210/edrv.20.2.0361. [DOI] [PubMed] [Google Scholar]
- 10.Ashcroft FM. ATP-sensitive potassium channelopathies: focus on insulin secretion. J Clin Invest. 2005;115:2047–2058. doi: 10.1172/JCI25495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huopio H, Shyng SL, Otonkoski T, Nichols CG. K(ATP) channels and insulin secretion disorders. Am J Physiol Endocrinol Metab. 2002;283:E207–E216. doi: 10.1152/ajpendo.00047.2002. [DOI] [PubMed] [Google Scholar]
- 12.Cartier EA, Conti LR, Vandenberg CA, Shyng SL. Defective trafficking and function of KATP channels caused by a sulfonylurea receptor 1 mutation associated with persistent hyperinsulinemic hypoglycemia of infancy. Proc Natl Acad Sci U S A. 2001;98:2882–2887. doi: 10.1073/pnas.051499698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Partridge CJ, Beech DJ, Sivaprasadarao A. Identification and pharmacological correction of a membrane trafficking defect associated with a mutation in the sulfonylurea receptor causing familial hyperinsulinism. J Biol Chem. 2001;276:35947–35952. doi: 10.1074/jbc.M104762200. [DOI] [PubMed] [Google Scholar]
- 14.Taschenberger G, Mougey A, Shen S, Lester LB, LaFranchi S, Shyng SL. Identification of a familial hyperinsulinism-causing mutation in the sulfonylurea receptor 1 that prevents normal trafficking and function of KATP channels. J Biol Chem. 2002;277:17139–17146. doi: 10.1074/jbc.M200363200. [DOI] [PubMed] [Google Scholar]
- 15.Crane A, Aguilar-Bryan L. Assembly, maturation, and turnover of K(ATP) channel subunits. J Biol Chem. 2004;279:9080–9090. doi: 10.1074/jbc.M311079200. [DOI] [PubMed] [Google Scholar]
- 16.Yan F, Lin CW, Weisiger E, Cartier EA, Taschenberger G, Shyng SL. Sulfonylureas correct trafficking defects of ATP-sensitive potassium channels caused by mutations in the sulfonylurea receptor. J Biol Chem. 2004;279:11096–11105. doi: 10.1074/jbc.M312810200. [DOI] [PubMed] [Google Scholar]
- 17.Clement JP, Kunjilwar K, Gonzalez G, Schwanstecher M, Panten U, Aguilar-Bryan L, Bryan J. Association and stoichiometry of K(ATP) channel subunits. Neuron. 1997;18:827–838. doi: 10.1016/s0896-6273(00)80321-9. [DOI] [PubMed] [Google Scholar]
- 18.Inagaki N, Gonoi T, Seino S. Subunit stoichiometry of the pancreatic beta-cell ATP-sensitive K+ channel. FEBS Lett. 1997;409:232–236. doi: 10.1016/s0014-5793(97)00488-2. [DOI] [PubMed] [Google Scholar]
- 19.Shyng S, Nichols CG. Octameric stoichiometry of the KATP channel complex. J Gen Physiol. 1997;110:655–664. doi: 10.1085/jgp.110.6.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zerangue N, Schwappach B, Jan YN, Jan LY. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron. 1999;22:537–548. doi: 10.1016/s0896-6273(00)80708-4. [DOI] [PubMed] [Google Scholar]
- 21.Yang K, Fang K, Fromondi L, Chan KW. Low temperature completely rescues the function of two misfolded K ATP channel disease-mutants. FEBS Lett. 2005;579:4113–4118. doi: 10.1016/j.febslet.2005.06.039. [DOI] [PubMed] [Google Scholar]
- 22.Gloyn AL, Siddiqui J, Ellard S. Mutations in the genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) in diabetes mellitus and hyperinsulinism. Hum Mutat. 2006;27:220–231. doi: 10.1002/humu.20292. [DOI] [PubMed] [Google Scholar]
- 23.Magge SN, Shyng SL, MacMullen C, Steinkrauss L, Ganguly A, Katz LE, Stanley CA. Familial leucine-sensitive hypoglycemia of infancy due to a dominant mutation of the beta-cell sulfonylurea receptor. J Clin Endocri-nol Metab. 2004;89:4450–4456. doi: 10.1210/jc.2004-0441. [DOI] [PubMed] [Google Scholar]
- 24.Nestorowicz A, Glaser B, Wilson BA, Shyng SL, Nichols CG, Stanley CA, Thornton PS, Permutt MA. Genetic heterogeneity in familial hyperinsulinism. Hum Mol Genet. 1998;7:1119–1128. doi: 10.1093/hmg/7.7.1119. [DOI] [PubMed] [Google Scholar]
- 25.Cartier EA, Shen S, Shyng SL. Modulation of the trafficking efficiency and functional properties of ATP-sensitive potassium channels through a single amino acid in the sulfonylurea receptor. J Biol Chem. 2003;278:7081–7090. doi: 10.1074/jbc.M211395200. [DOI] [PubMed] [Google Scholar]
- 26.Lin CW, Lin YW, Yan FF, Casey J, Kochhar M, Pratt EB, Shyng SL. Kir6.2 mutations associated with neonatal diabetes reduce expression of ATP-sensitive K+ channels: implications in disease mechanism and sulfonylurea therapy. Diabetes. 2006;55:1738–1746. doi: 10.2337/db05-1571. [DOI] [PubMed] [Google Scholar]
- 27.Nichols CG, Shyng SL, Nestorowicz A, Glaser B, Clement JP, Gonzalez G, Aguilar-Bryan L, Permutt MA, Bryan J. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science. 1996;272:1785–1787. doi: 10.1126/science.272.5269.1785. [DOI] [PubMed] [Google Scholar]
- 28.Shyng SL, Ferrigni T, Shepard JB, Nestorowicz A, Glaser B, Permutt MA, Nichols CG. Functional analysis of novel mutations in the sulfonylurea receptor 1 associated with persistent hyperinsulinemic hypoglycemia of infancy. Diabetes. 1998;47:1145–1151. doi: 10.2337/diabetes.47.7.1145. [DOI] [PubMed] [Google Scholar]
- 29.Lin CW, Yan F, Shimamura S, Barg S, Shyng SL. Membrane phosphoinositides control insulin secretion through their effects on ATP-sensitive K+ channel activity. Diabetes. 2005;54:2852–2858. doi: 10.2337/diabetes.54.10.2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aguilar-Bryan L, Nichols CG, Rajan AS, Parker C, Bryan J. Co-expression of sulfonylurea receptors and KATP channels in hamster insulinoma tumor (HIT) cells: evidence for direct association of the receptor with the channel. J Biol Chem. 1992;267:14934–14940. [PubMed] [Google Scholar]
- 31.Yan FF, Casey J, Shyng SL. Sulfonylureas correct trafficking defects of disease-causing ATP-sensitive potassium channels by binding to the channel complex. J Biol Chem. 2006;281:33403–33413. doi: 10.1074/jbc.M605195200. [DOI] [PubMed] [Google Scholar]
- 32.Sharma N, Crane A, Gonzalez G, Bryan J, Aguilar-Bryan L. Familial hyperinsulinism and pancreatic beta-cell ATP-sensitive potassium channels. Kidney Int. 2000;57:803–808. doi: 10.1046/j.1523-1755.2000.00918.x. [DOI] [PubMed] [Google Scholar]
- 33.Tornovsky S, Crane A, Cosgrove KE, Hussain K, Lavie J, Heyman M, Nesher Y, Kuchinski N, Ben-Shushan E, Shatz O, Nahari E, Potikha T, Zangen D, Tenenbaum-Rakover Y, de Vries L, Argente J, Gracia R, Landau H, Eliakim A, Lindley K, Dunne MJ, Aguilar-Bryan L, Glaser B. Hyperinsulinism of infancy: novel ABCC8 and KCNJ11 mutations and evidence for additional locus heterogeneity. J Clin Endocrinol Metab. 2004;89:6224–6234. doi: 10.1210/jc.2004-1233. [DOI] [PubMed] [Google Scholar]
- 34.Chan KW, Zhang H, Logothetis DE. N-terminal transmembrane domain of the SUR controls trafficking and gating of Kir6 channel subunits. EMBO J. 2003;22:3833–3843. doi: 10.1093/emboj/cdg376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Babenko AP, Bryan J. Sur domains that associate with and gate KATP pores define a novel gatekeeper. J Biol Chem. 2003;278:41577–41580. doi: 10.1074/jbc.C300363200. [DOI] [PubMed] [Google Scholar]
- 36.Ashfield R, Gribble FM, Ashcroft SJ, Ashcroft FM. Identification of the high-affinity tolbutamide site on the SUR1 subunit of the K(ATP) channel. Diabetes. 1999;48:1341–1347. doi: 10.2337/diabetes.48.6.1341. [DOI] [PubMed] [Google Scholar]
- 37.Bryan J, Vila-Carriles WH, Zhao G, Babenko AP, Aguilar-Bryan L. Toward linking structure with function in ATP-sensitive K+ channels. Diabetes. 2004;53(Suppl 3):S104–S112. doi: 10.2337/diabetes.53.suppl_3.s104. [DOI] [PubMed] [Google Scholar]
- 38.Loo TW, Clarke DM. Superfolding of the partially unfolded core-glycosylated intermediate of human P-glycoprotein into the mature enzyme is promoted by substrate-induced transmembrane domain interactions. J Biol Chem. 1998;273:14671–14674. doi: 10.1074/jbc.273.24.14671. [DOI] [PubMed] [Google Scholar]
- 39.Henwood MJ, Kelly A, Macmullen C, Bhatia P, Ganguly A, Thornton PS, Stanley CA. Genotype-phenotype correlations in children with congenital hyperinsulinism due to recessive mutations of the adenosine triphosphate-sensitive potassium channel genes. J Clin Endocrinol Metab. 2005;90:789–794. doi: 10.1210/jc.2004-1604. [DOI] [PubMed] [Google Scholar]
- 40.Peranteau WH, Ganguly A, Steinmuller L, Thornton P, Johnson MP, Howell LJ, Stanley CA, Adzick NS. Prenatal diagnosis and postnatal management of diffuse congenital hyperinsulinism: a case report. Fetal Diagn Ther. 2006;21:515–518. doi: 10.1159/000095664. [DOI] [PubMed] [Google Scholar]
- 41.Geng X, Li L, Watkins S, Robbins PD, Drain P. The insulin secretory granule is the major site of K(ATP) channels of the endocrine pancreas. Diabetes. 2003;52:767–776. doi: 10.2337/diabetes.52.3.767. [DOI] [PubMed] [Google Scholar]
- 42.Varadi A, Grant A, McCormack M, Nicolson T, Magistri M, Mitchell KJ, Halestrap AP, Yuan H, Schwappach B, Rutter GA. Intracellular ATP-sensitive K+ channels in mouse pancreatic beta cells: against a role in organelle cation homeostasis. Diabetologia. 2006;49:1567–1577. doi: 10.1007/s00125-006-0257-9. [DOI] [PubMed] [Google Scholar]
- 43.Cowley DJ, Moore YR, Darling DS, Joyce PB, Gorr SU. N- and C-terminal domains direct cell type-specific sorting of chromogranin A to secretory granules. J Biol Chem. 2000;275:7743–7748. doi: 10.1074/jbc.275.11.7743. [DOI] [PubMed] [Google Scholar]
- 44.Suchi M, MacMullen CM, Thornton PS, Adzick NS, Ganguly A, Ruchelli ED, Stanley CA. Molecular and immunohistochemical analyses of the focal form of congenital hyperinsulinism. Mod Pathol. 2006;19:122–129. doi: 10.1038/modpathol.3800497. [DOI] [PubMed] [Google Scholar]
- 45.Fernandez-Marmiesse A, Salas A, Vega A, Fernandez-Lorenzo JR, Barreiro J, Carracedo A. Mutation spectra of ABCC8 gene in Spanish patients with Hyperinsulinism of Infancy (HI) Hum Mutat. 2006;27:214. doi: 10.1002/humu.9401. [DOI] [PubMed] [Google Scholar]
- 46.Suchi M, MacMullen C, Thornton PS, Ganguly A, Stanley CA, Ruchelli ED. Histopathology of congenital hyperinsulinism: retrospective study with genotype correlations. Pediatr Dev Pathol. 2003;6:322–333. doi: 10.1007/s10024-002-0026-9. [DOI] [PubMed] [Google Scholar]
- 47.Meissner T, Beinbrech B, Mayatepek E. Congenital hyperinsulinism: molecular basis of a heterogeneous disease. Hum Mutat. 1999;13:351–361. doi: 10.1002/(SICI)1098-1004(1999)13:5<351::AID-HUMU3>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 48.Thomas PM, Wohllk N, Huang E, Kuhnle U, Rabl W, Gagel RF, Cote GJ. Inactivation of the first nucleotide-binding fold of the sulfonylurea receptor, and familial persistent hyperinsulinemic hypoglycemia of infancy. Am J Hum Genet. 1996;59:510–518. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.