

Abstract
The integration of a range of technologies including microfluidics, surface-enhanced Raman scattering and confocal microspectroscopy has been successfully used to characterize in situ single living CHO (Chinese hamster ovary) cells with a high degree of spatial (in three dimensions) and temporal (1 s per spectrum) resolution. Following the introduction of a continuous flow of ionomycin, the real time spectral response from the cell was monitored during the agonist-evoked Ca2+ flux process. The methodology described has the potential to be used for the study of the cellular dynamics of a range of signalling processes.
Figure.
Spectral mapping of a single CHO cell
Keywords: Microfluidics, Chinese hamster ovary (CHO) cells, Confocal microspectroscopy, Surface-enhanced Raman scattering (SERS), Dynamic monitoring
Introduction
The advance of miniaturized microfluidic systems for chemical and/or biochemical applications based on so-called Lab-on-a-Chip technology has demonstrated that such micro systems represent the ability to “shrink” conventional bench systems to the size of a few square centimetres with major advantages of speed, performance, integration, portability, reduced sample/solvent quantity, automation, hazard control and lower cost [1–6]. These merits are important for a variety of applications in analytical chemistry, biochemistry, clinical diagnosis, medical chemistry and industrial chemistry [1, 2]. Consequently, numerous micro total analysis systems (μ-TAS) and micro reactor systems have been developed, and many more are currently under investigation [2].
For the study of cellular and subcellular systems, a wide range of analytical methods have been used with fluorescence techniques being the most common. In addition, fluorescence-based imaging is a highly attractive methodology for the study of organelle dynamics, identifying subcellular compartments and monitoring biological kinetics [7]. Over the last two decades, Raman spectroscopy has become an increasingly important technology with ability to study the biophysics and biochemical processes involving cells [4, 8–12]. Since Raman spectroscopy is based on vibrational transitions where frequency shifts are associated with specific molecular vibrations within the sample of interest, it enables the identification of polarizable bio/chemical species, the elucidation of molecular structure and the investigation of interface reactions, all in a non-destructive manner. In addition, unlike fluorescence-based techniques, Raman spectroscopy does not require labelling dyes and since water is almost Raman “transparent”, the technique is ideally suited for analysing cell-based biological systems. Coupling a Raman spectrometer with a confocal microscope enables the acquisition of full spectral information with a high spatial (<1 μm) resolution in three dimensions. Raman spectroscopy, however, suffers from an inherent poor level of sensitivity compared for example to fluorescence, which can be 12–14 orders of magnitude more sensitive. As a consequence, despite advances in detector technologies, the technique can be less than ideal for the direct detection of intracellular components present at low concentrations, which may take from a few tens of seconds to a few minutes for spectral acquisition [12]. With the unexpected discovery of surface-enhanced Raman scattering (SERS) by Fleischman et al. [13], Raman intensity can now be dramatically increased (a factor of up to 105–1010) with the inclusion of metallic nano structures either on a substrate surface or in a colloidal solution [8]. For cellular and subcellular analysis with SERS, colloid nanoparticles (e.g. silver or gold) are normally loaded into cells by different means such as general incubation (fluid-phase uptake) or ultrasonication-assisted uptake [14, 15]. Due to its chemical inactivity, gold nanoparticles are generally regarded to be more suitable for incorporation within living cells [14].
To analyse living cells using Raman spectroscopy, the cells are usually fixed at a specific location. The most commonly used method for cell fixation is to seed or grow cells on a substrate, e.g. a microscope cover slide. Other approaches such as optical tweezers have also been reported [9]. However, the conventional batch operation in a static system limits the in situ Raman analysis, especially when a series of reagent treatments are required. With those limitations in mind, the development of microfluidic and associated Lab-on-a-Chip technologies provides unique opportunities for delivering and immobilizing cells on a microchannel surface, prior to introducing different reagents with a continuous flow in a given sequence [3, 4] whilst under spectroscopic investigation.
In this study, we report the successful integration of a range of techniques including microfluidics, surface-enhanced Raman scattering (SERS) and confocal microspectroscopy which enables in situ characterization of single living CHO cells with high spatial and temporal resolution. By using the microfluidic methodology, cells and reagents were introduced into the chip in a continuous flow as a series of plugs in a given sequence where agonist (ionomycin) evoked intracellular Ca2+ fluxes and the cell’s real time spectral response was recorded.
Materials and methods
Confocal Raman microspectroscopy
All Raman spectra were acquired with a LabRam inverted microscope spectrometer, manufactured by Jobin Yvon Ltd. Figure 1 shows the schematic of the experimental setup. The spectrometer was equipped with dual laser sources at wavelengths of 780 nm (diode laser, 70 mW) and 633 nm (He–Ne laser, 20 mW), confocal optics, a holographic transmission grating, and a charge coupled device (CCD) detector with 1,024 × 256 pixels. The instrument included a precision motorized X–Y sample stage for automated mapping at spatial resolution down to less than 1 μm and extensive software support (LabSpec 4.18) for data processing. In this study, an objective lens of ×50 magnification, 17-mm working distance and numerical aperture (NA) of 0.45 was used (L Plan SLWD 50, Nikon, Japan). This objective lens was mounted on a PI-721.10 piezo actuator (Physik Instrumente, Germany) for automatic focussing of the microscope objective at different depths in the Z direction enabling 3D mapping. A grating with 1,800 grooves mm−1, a confocal aperture of 300 μm and an entrance slit of 150 μm were selected for the experiments. The Raman spectrometer wavelength range was calibrated using the centre frequency of the silicon band from a silicon sample (520.2 cm−1). Using these conditions, a typical acquisition time of 1 s was used to collect SERS spectra from cells within the microchannel.
Fig. 1.

Schematic of experimental setup
Microfluidic device fabrication
The microfluidic device was constructed using a manifold clamping method according to published procedures with some adaptations [16–18]. The assembly of the device is illustrated in Fig. 2a. Briefly, the microchip consisted of a PARAFILM® sheet (thickness 130 μm, American National Can Company, US) with a channel network and two glass plates which sandwiched the polymer film. The Y-shaped channel network (Fig. 2b) cut through the film was 500-μm wide. The top glass plate (B-270, 25 × 25 × 3 mm) had three holes (diameter 1.5 mm) drilled through at appropriated positions in order to link the ends of the channels with inlet/outlet tubing. The bottom glass plate was a thin quartz coverslip (22 × 22 mm, Agar Scientific Ltd, UK) which had a thickness of 250 μm in order to minimise the glass background during Raman measurements. This sandwich chip was then clamped using two aluminium frames with screws. The windows on the frames were designed for tubing connections (through top frame) and for optical passage (through bottom frame).
Fig. 2.

(a) Assembly of microfluidic device and (b) Y-shaped channel network (channel depth 100 μm, width 500 μm) with cells loaded (c) for examination
Two KDS 200 syringe pumps (KD Scientific Inc., USA) were used to deliver cells in suspension and test solutions into the microchip channel (Fig. 2c). Ethylene tetrafluoroethylene (ETFE) polymer tubing with an inner diameter of 250 μm, on–off valves, and appropriate fittings and connectors, all obtained from Upchurch (Upchurch Scientific Inc., USA), were used for plumbing to link the chip and the syringes.
Cell culture and assay reagents
CHO-K1 (Chinese hamster ovary, Cricetulus griseus) cells were supplied by ATCC/LGC Promochem (ATCC® No. CCL-61™, LGC Promochem, UK). The cells were cultured routinely in DMEM/F-12 medium without L-glutamine (Invitrogen Ltd, UK) which was supplemented with fetal bovine serum (Invitrogen Ltd) to a final concentration of 10%, and L-glutamine (Invitrogen Ltd) to a final concentration of 4 mM. An incubator was used at 37 °C supplying 5% CO2. The concentration of cells used in loading the chips during this experiment was in the range of 7.5 × 106 cells mL−1.
A wash solution consisting of a modified Tyrodes buffer was used to wash the cells and to prepare the test solution. The Tyrodes buffer was composed of 145 mM NaCl, 2.5 mM KCl, 10 mM HEPES, 10 mM D-glucose and 1.2 mM MgCl2. CaCl2 (99.5%, BDH AnalaR, 150 mM dissolved in Tyrodes buffer) and probenecid (98%, Sigma, 0.834 M dissolved in 1 M NaOH aqueous solution) were then added to the Tyrodes buffer giving final concentrations of 1.5 mM and 2.5 mM for CaCl2 and probenecid, respectively. Ionomycin test solutions were made by adding ionomycin stock (1 mM in DMSO) into wash solutions for a concentration of 50 μM. Ionomycin was obtained from Calbiochem (Calbiochem of EMD Biosciences, Inc., USA), and DMSO from Sigma–Aldrich (99%, D2650). The flow rate for the introduction of ionomycin test solution into the microchip was 2 μL min−1.
Gold colloid with a particle size of 50 nm (EM.GC50), suspended in water, was supplied by BBinternational Ltd, UK. The gold nanoparticles were introduced into cells by a passive uptake method where the cells were incubated with gold colloid solution at desired concentration (20% colloid solution, v/v) and room temperature for 50 mins. Prior to loading into the microfluidic chip, the cells were washed with wash solution to remove the culture media and gold nanoparticles outside cells. The cells were re-suspended in the wash solution for measurements.
Results and discussion
SERS effects
After loading cells into the microfluidic channel, Raman spectra were taken from a selected single cell (Fig. 2c). By using the near-infrared laser excitation (780 nm) the optical and thermal effects of laser illumination on the living cells were minimised [19, 20]. In addition, using a near-infrared laser can significantly reduce the fluorescence interference background and light scattering from the quartz base plate. It has been reported that silver or gold nanoparticles with an individual size in the range of 20–60 nm can yield significant enhancement of Raman scattering for cellular analysis when nanoparticles are introduced into cells [14]. This has been confirmed in this study by using gold nanoparticles with a size of 50 nm.
Figure 3 shows a typical spectrum (upper) taken from a cell incubated with gold colloid solution. The spectrum was recorded in the range from 300 to 2,800 cm−1 with an acquisition time of 1 s. We observed that with the introduction of gold nanoparticles the Raman spectra were clearly detectable under those conditions. For comparison, a spectrum obtained from a cell incubated in medium without gold colloid solution is also shown in Fig. 3 (lower). It can be seen that even though the spectra acquisition time was increased to 10 s it was still unable to obtain a good quality spectrum without gold nanoparticles present. This observation confirmed the significant enhancement of Raman scattering with the presence of gold nanoparticles which also enabled a fast spectra acquisition although the exact mechanism of the SERS enhancement of Raman signals is not fully understood [21]. The introduction of gold nanoparticles therefore significantly shortened signal acquisition times mainly owing to the increase in sensitivity which in turn allowed an array of spectra to be obtained in a relatively short time period.
Fig. 3.

Raman spectra from cells following incubation in medium with gold colloid (upper spectrum) and without gold colloid (lower spectrum)
Characterization of single living cells by 3D mapping
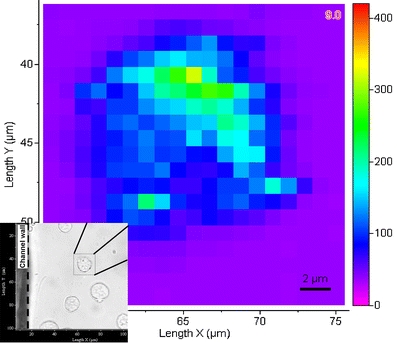
Using the SERS technique described above it was possible to obtain spectra from a single living cell which represented a “fingerprint” from which various chemical constituents in the cell can be assigned. Combined with an automated microscope stage which offers spatial resolution (i.e. 1 μm), mapping of an entire cell area at a specific plane was carried out. It has been suggested from previous studies that most of the spectral bands associated with living cells occur in the range 800–1,700 cm−1 [11, 22, 23], hence this range was used in this study for mapping. Figure 4a shows an “image” of Raman spectra obtained from the middle layer across the single cell by mapping an area of 21 × 21 μm2 with a spatial step of 1 μm in both X and Y directions. The mapping provided information on the distribution of selected bands, as seen in Fig. 4b in the range from 1,290 to 1,370 cm−1, which represent most of the significant bands associated with DNA and proteins within a cell’s nucleus and cytoplasm [11, 23].
Fig. 4.

a Spectral mapping of a single CHO cell on an X–Y plane and b corresponding spectra from three positions in the area of nucleus, cytoplasm and membrane, respectively
In general, the Raman spectra of single CHO cells showed contributions from all its cellular components including nucleic acids, proteins, lipids and carbohydrates. Table 1 summarises the band assignment for the Raman spectra taken from CHO cells based on the published data [8, 9, 12, 23–27]. Comparison of the spectra taken from different positions across the cell on an X–Y plane (Fig. 4) indicated that strong peaks from the nucleus spectrum corresponding to DNA sugar–phosphate backbone (895 and 1,142 cm−1), and bases G (1,320 and 1,487 cm−1), A (1,420 and 1,578 cm−1), T (1,176 and 1,376 cm−1) and C (1,420 cm−1) were noticeably reduced in the cytoplasm and membrane spectra (Table 1). This change was expected as the nucleus contains high densities of DNA, whilst the cytoplasm also had significant quantities of RNA contributing to the corresponding peaks. As expected, the spectrum taken from membrane area showed significant peaks corresponding to lipids (1,068 and 1,453 cm−1).
Table 1.
Band assignment for Raman spectra of CHO cells
| Bands (cm−1) | Assignments | ||
|---|---|---|---|
| DNA/RNA | Proteins | Lipids | |
| 830 | Phosphodiester BkB [12, 23, 24] | Tyr [12, 23, 24] | |
| 895 | Phosphodiester BkB, deoxyribose [12, 23, 24] | ||
| 940 | ν (C–C), α-helix [9, 12, 23] | ||
| 1,004 | Phenylalanine [9, 12, 23] | ||
| 1,065 | ν (C–O) [12, 23] | ν (C–C) chian [25] | |
| 1,126 | ν (C–N) BkB [9, 12, 23] | ν (C–C) chain [25] | |
| 1,144 | Ribose–phosphate [12, 23] | ||
| 1,157 | Ribose–phosphate [20, 26] | ||
| 1,176 | T, C, G [9, 12, 23, 24] | Phenylalanine [12, 23] | |
| 1,230 | C [12, 23, 24] | ||
| 1,266 | Amide III [25] | δ (C=CH2) [25] | |
| 1,295 | δ (CH2) [25] | ||
| 1,320 | G [24] | ||
| 1,342 | A [12, 23, 24] | ||
| 1,376 | T, A, G [9, 12, 23, 24] | ||
| 1,448 | δ (CH) [9, 12, 23] | δ (CH) [9, 12, 23] | |
| 1,482 | A, G [12, 23, 24] | ||
| 1,566 | Hemoglobin [25] | ||
| 1,578 | A, G, purine [12, 23, 24] | ||
| 1,603 | A, C [24] | ||
| 1,643 | Amide I [27] | ||
Abbreviations: BkB DNA sugar–phosphate backbone, Tyr tyrosine, A adenine, T thymine, G guanine, C cytosine, ν stretching vibrations, δ deformation vibrations
Using the confocal optics of the microscope system, the chemical concentration distribution in the Z axis at three levels in the nucleus was examined (Fig. 5) and indicated that the main peaks positions were generally identical but the peak heights were markedly different, indicating a concentration difference. From Fig. 5a significant a peak was observed at 1,320 cm−1 on the spectrum taken from Z = 3 μm (near to the bottom of the cell) which was assigned to DNA bases G [23], whilst the peak at 1,450 cm−1 from Z = 6 μm indicated a strong deformation from a C–H stretch in proteins [8, 12, 23]. Thus, by collecting spectra from different spatial positions, it provided an approach to build a 3D mapping of the distribution of chemicals within a single cell.
Fig. 5.

Raman spectra taken at three levels along the Z direction in the nucleus area
It should be noted that the spectra taken from different positions are distinct in terms of both Raman band amplitudes and band positions. This spectral variation can be attributed to the native chemical inhomogeneity within a cell. However, the possibility of non-uniform distribution of gold nanoparticles within the cell cannot be excluded [15]. In addition, this non-uniform distribution can also take place during the time-resolved monitoring because dynamic processes inside the cell can lead to local fluctuations of the particle densities and hence changes of the spectra. To further develop this spectroscopic technique for both qualitative and quantitative analysis, technologies need to be explored in order to deliver and position nanoparticles within cells in a controllable format for a uniform distribution. Nevertheless, it is still possible using this promising methodology to examine a specific point within a cell in a dynamic way with time, especially when applying stimulation, e.g. an agonist, under microfluidic control conditions.
In situ monitoring of cellular chemical dynamics
Finally, we monitored the chemical dynamics of the cell when exposed to the agonist ionomycin [3, 28]. After loading cells into the chip channel, a 40-min period of settlement was allocated before introducing the ionomycin solution at a constant flow rate of 2 μL min−1. Whilst focussed on a region close to the cell nucleus, a series of spectra were taken at an interval of 1 s in the range from 800 to 1,700 cm−1 (Fig. 6a). It can be seen from the spectra (Fig. 6b) that most of the peaks corresponding to nucleic acids, proteins and lipids remain visible with time but the peak heights vary noticeably, indicating the concentration change of these compounds. One of the most significant changes is the appearance of a peak at 1,643 cm−1 which can be assigned to amide I [27]. The time profile of this peak (Fig. 6c) reveals the concentration change of amide I within the cell, showing a similar trend to that of the Ca2+ flux evoked by the agonist ionomycin [3] which is commonly used in biomedical research to stimulate the intracellular production of proteins such as interferon [29]. Since amide I contributes spectrally to the Raman spectrum of interferon, it follows that the in situ monitoring of such molecules could in future be used to produce characterization of protein expression dynamics at subcellular levels. Clearly, more studies are required in order to understand the mechanism and cause of the concentration change of amide I within the cell when exposed to agonist ionomycin.
Fig. 6.

In situ monitoring of cellular dynamics by recording a series of Raman spectra (a) from a specific area at an interval of 1 s. The first spectrum, at time 0, was taken when no agonist was introduced (b); (c) shows the time profile of the peak at 1,643 cm−1 corresponding to the change of amide I
Conclusions
The SERS technique has been used for characterisation of single CHO (Chinese hamster ovary) cells with a microfluidic device where gold nanoparticles were introduced into cells for Raman enhancement. The use of an inverted microscope optical systems in combination with a charge coupled device (CCD) detector allows the measurement of Raman spectra for simultaneous analysis of bio/chemical species within cells. In addition to a precision automated X–Y sample stage, the confocal optics provides discrimination between points of different depths within the cell, enabling chemical mapping in three dimensions. By using microfluidic methodology, the cell manipulation and reagent delivery were performed in a controllable manner. By introducing cells and test reagent into the chip in a continuous flow as a series of plugs in a given sequence, it enabled the in situ chemical characterization of single CHO cells with a high degree of spatial and temporal resolution. This allows the real time monitoring of the dynamics of the agonist-evoked Ca2+ flux response. The approach described has the potential to be used for the study of the spatial dynamics of a range of intercellular processes.
Acknowledgements
We thank Ms Huguette Savoie of the Department of Chemistry at the University of Hull for help with the cell culture. The DTI (MNT Project 366) is acknowledged for funding this research.
References
- 1.Janasek D, Franzke J, Manz A (2006) Nature 442:374–380 [DOI] [PubMed]
- 2.Kitamori T, Fujita H, Hasebe S (eds) Proceedings of the 10th international conference on miniaturized systems for chemistry and life sciences (μ-TAS 2006), Tokyo, Japan, 5–9 November 2006
- 3.Zhang X, Yin H, Cooper JM, Haswell SJ (2006) Electrophoresis 27:5093–5100 [DOI] [PubMed]
- 4.Ramser K, Enger J, Goksor M, Hanstorp D, Logg K, Kall M (2005) Lab Chip 5:431–436 [DOI] [PubMed]
- 5.Huh D, Kamotani Y, Grotberg JB, Takayama S (2005) Physiol Meas 26:R73–R98 [DOI] [PubMed]
- 6.Monaghan PB, McCarney KM, Ricketts A, Littleford RE, Docherty F, Smith WE, Graham D, Cooper JM (2007) Anal Chem 79:2844–2849 [DOI] [PubMed]
- 7.Olson KJ, Ahmadzadeh H, Arriaga EA (2005) Anal Bioanal Chem 382:906–917 [DOI] [PubMed]
- 8.Lee S, Kim S, Choo J, Shin SY, Lee YH, Choi HY, Ha S, Kang K, Oh CH (2007) Anal Chem 79:916–922 [DOI] [PubMed]
- 9.Jess PRT, Garcés-Chávez V, Smith D, Mazilu M, Paterson L, Riches A, Herrington CS, Sibbett W, Dholakia K (2006) Opt Express 14:5779–5791 [DOI] [PubMed]
- 10.Krafft C, Knetschke T, Funk RHW, Salzer R (2006) Anal Chem 78:4424–4429 [DOI] [PubMed]
- 11.van Manen H-J, Kraan YM, Roos D, Otto C (2005) PNAS 102:10159–10164 [DOI] [PMC free article] [PubMed]
- 12.Puppels GJ, de Mul FF, Otto C, Greve J, Robert-Nicoud M, Arndt-Jovin DJ, Jovin TM (1990) Nature 347:301–303 [DOI] [PubMed]
- 13.Fleischman M, Hendra PJ, McQuillan AJ (1974) Chem Phys Lett 26:163–166 [DOI]
- 14.Kneipp J, Kneipp H, McLaughlin M, Brown D, Kneipp K (2006) Nano Lett 6:2225–2231 [DOI] [PubMed]
- 15.Kneipp K, Haka AS, Kneipp H, Badizadegan K, Yoshizawa N, Boone C, Shafer KE, Motz JT, Dasari RR, Feld MS (2002) Appl Spectrosc 56:150–154 [DOI]
- 16.He P, Watts P, Marken F, Haswell SJ (2005) Electrochem Commun 7:918–924 [DOI]
- 17.Kantak AS, Gale BK, Lvov Y, Jone SA (2003) Biomed Microdev 5:207–215 [DOI]
- 18.Ma N, Koelling KW, Chalmers JJ (2002) Biotech Bioeng 80:428–437 [DOI] [PubMed]
- 19.Notingher I, Jones JR, Verrier S, Bisson I, Embanga PP, Edwards P, Polak JM, Hench LL (2003) Spectroscopy 17:275–278
- 20.Notingher I, Verrier S, Romanska H, Bishop AE, Polak JM, Hench LL (2002) Spectroscopy 16:43–51
- 21.Chowdhury MH, Gant VA, Trache A, Baldwin AM, Meininger GA, Coté GL (2006) J Biomed Opt 11:24004 [DOI] [PubMed]
- 22.Notingher I, Verrier S, Haque S, Polak JM, Hench LL (2003) Biopolymers 72:230–240 [DOI] [PubMed]
- 23.Puppels GJ, Garritsen HSP, Segers-Nolten GMJ, Mul FFM, de Greve J (1991) Biophys J 60:1046–1056 [DOI] [PMC free article] [PubMed]
- 24.Ruiz-Chica AJ, Medina MA, Sánchez-Jiménez F, Ramírez FJ (2004) Nucleic Acids Res 32:579–589 [DOI] [PMC free article] [PubMed]
- 25.Krafft C, Sobottka SB, Schackert G, Salzer R (2005) Analyst 130:1070–1077 [DOI] [PubMed]
- 26.Overman SA, Aubrey KL, Reilly KE, Osman O, Hayes SJ, Serwer P, Thomas GJ Jr (1998) Biospectroscopy 4:S47–S56 [DOI] [PubMed]
- 27.Williams RW (1985) J Biol Chem 260:3937–3940 [PubMed]
- 28.Gergely L, Cook L, Agnello V (1997) Clin Diagn Lab Immunol 4:70–74 [DOI] [PMC free article] [PubMed]
- 29.Osna N, Elliott K, Khan MM (2001) Int Immunopharmacol 1:135–145 [DOI] [PubMed]