

Abstract
Elevated cardiac troponin-I (cTnI) levels have been demonstrated in serum of patients without acute coronary syndromes, potentially via a stretch-related process. We hypothesize that this cTnI release from viable cardiomyocytes is mediated by stimulation of stretch-responsive integrins. Cultured cardiomyocytes were treated with (1) Gly–Arg–Gly–Asp–Ser (GRGDS, n = 22) to stimulate integrins, (2) Ser–Asp–Gly–Arg–Gly (SDGRG, n = 8) that does not stimulate integrins, or (3) phosphate-buffered saline (control, n = 38). Cells and media were analyzed for intact cTnI, cTnI degradation products, and matrix metalloproteinase (MMP)-2. Cell viability was examined by assay of lactate dehydrogenase (LDH) activity and by nuclear staining with propidium iodide. GRGDS-induced integrin stimulation caused increased release of intact cTnI (9.6 ± 3.0%) as compared to SDGRG-treated cardiomyocytes (4.5 ± 0.8%, p < 0.001) and control (3.0 ± 3.4%, p < 0.001). LDH release from GRGDS-treated cardiomyocytes (15.9 ± 3.8%) equalled that from controls (15.2 ± 2.3%, p = n.s.), indicating that the GRGDS-induced release of cTnI is not due to cell necrosis. This result was confirmed by nuclear staining with propidium iodide. Integrin stimulation increased the intracellular and extracellular MMP2 activity as compared to controls (both p < 0.05). However, despite the ability of active MMP2 to degrade cTnI in vitro, integrin stimulation in cardiomyocytes was not associated with cTnI degradation. The present study demonstrates that intact cTnI can be released from viable cardiomyocytes by stimulation of stretch-responsive integrins.
Keywords: Mechanoreceptor, Stretch, Cardiomyocyte, Cell death, Cell culture
Introduction
Troponins are myofibrillar proteins involved in the regulation of actin–myosin interaction, thereby controlling contraction and relaxation. Cardiac troponins (cTnT, cTnT, and cTnC) are predominantly bound, via tropomyosin, to actin filaments of sarcomeres, with only a small proportion of cTnT (6–8%) and cTnI (3–8%) found in the soluble cytoplasmic pool [1]. Due to high cardiac specificity, serum concentrations of cTnI and cTnT are well-established diagnostic and prognostic markers of irreversible myocardial damage in acute coronary syndromes [9]. However, elevated serum levels of cardiac troponins have also been observed in patients without acute coronary syndromes in whom irreversible myocardial cell injury was not a prominent aspect [8, 11, 20]. Several studies reported elevated serum levels of troponins in patients with cardiomyopathy [29], heart failure [18], unstable angina pectoris [14], pulmonary embolism [17], renal insufficiency [5], and in ultra-endurance athletes [19]. The exact mechanism underlying the release of troponins in the absence of necrosis, without lethal disruption of the sarcolemma, remains to be elucidated and most likely differs from the release of troponins due to necrotic cell death.
A potential explanation for the cTnI release without lethal sarcolemmal disruption is the cellular release of proteolytic cTnI degradation products. Indeed, in isolated rat hearts, Feng et al. [6] showed degradation of cTnI upon increasing preload, which was independent of ischemia. They proposed that the cTnI degradation might be caused by increased myocardial stretch per se.
Mechanical stretch of cardiomyocytes, as occurs during pressure or volume overload, initiates a cascade of intracellular signals, including increased intracellular calcium concentration, increased intracellular NO formation, and the activation of intracellular proteases such as MMP2 and MMP14 [28, 36]. MMP2 is able to degrade cTnI intracellularly [39] and may be involved in the stretch-induced release of cTnI and its degradation products. In contrast, other studies suggest that cTnI leaks from reversibly damaged cardiomyocytes as an intact non-degraded molecule [8, 20].
Mechanical stretch of cardiomyocytes is sensed by stimulation of integrins, which are transmembrane glycoprotein receptors that link the extracellular matrix (ECM) to the intracellular cytoskeleton [26]. Integrins act as mechanotransducers and bidirectional signaling molecules and participate in overload-induced hypertrophic and post-infarct remodeling [26]. The Arg–Gly–Asp (RGD) sequence that is present in fibronectin and other ECM proteins is an agonist for integrins [27], which can be used to simulate myocardial mechanical stretch in vitro.
We hypothesize that the stretch-related process through which cTnI can be released from viable cardiomyocytes is mediated by integrin stimulation. The purpose of the present study is therefore to investigate whether (1) RGD-induced integrin stimulation causes release of intact and/or fragmented cTnI from cardiomyocytes in the absence of necrotic cell death, (2) integrin stimulation is associated with MMP2 activation and MMP2-related cTnI degradation, and (3) cTnI release in the absence of necrosis differs from the release of cTnI in the presence of necrosis.
Materials and methods
Primary cultures of neonatal cardiomyocytes
Primary cultures of cardiomyocytes were prepared from the ventricles of 2-day-old Wistar rats as described previously [24]. Briefly, ventricles were minced, and cells were isolated enzymatically using collagenase type I (CLS, Lakewood, NJ, USA) at 37°C. The cell suspension was centrifuged, and the cell pellet was resuspended in growth medium containing Ham’s F10 (Flow Laboratories, Irvine, UK), 10% fetal bovine serum, 10% horse serum (HS), 100 U/ml penicillin, and 100 μg/ml streptomycin (all from Invitrogen, Breda, The Netherlands). The cells were seeded in 6-cm diameter Primaria®-coated plastic culture dishes (Falcon, Becton Dickinson, Etten-Leur, The Netherlands), and after 45 min, non-adherent cells representing the cardiomyocytes were collected and plated in six-wells plates (∅ 35 mm per well, Costar, Corning, NY, USA) at a density of ≈1.5 × 106 cells per well. Cardiomyocytes were incubated in a humidified incubator at 37°C and 5% CO2 and culture medium [1:1 (v/v) mixture of DMEM (Invitrogen) and Ham’s F10 supplemented with 5% HS, penicillin (100 U/ml) and streptomycin (100 μg/ml)] was refreshed after 20 h and 48 h. After 3 days of culturing, a monolayer of spontaneously beating cardiomyocytes had formed. The investigations had the approval of the Animal Experiments Committee of the LUMC according to Dutch law.
Experimental protocol for integrin stimulation
Three days after cell isolation, cardiomyocyte cultures were washed twice in HEPES-buffered salt solution (HBSS), containing (in millimole per liter) NaCl (125), KCl (5), MgSO4 (1), KH2PO4 (1), CaCl2 (2.5), NaHCO3 (10), d-glucose (5), HEPES (20), pH 7.4 at 37°C. Cardiomyocytes were incubated for 24 h in 1 ml of HBSS supplemented with (1) the pentapeptide Gly–Arg–Gly–Asp–Ser containing the GRGDS sequence (Sigma, St. Louis, MO, USA) at various concentrations (100, 200, or 300 μg/ml) to stimulate integrins, with (2) Ser–Asp–Gly–Arg–Gly (SDGRG, the reverse sequence of GRGDS, Sigma), which does not affect integrin function, at a concentration of 300 μg/ml, or with (3) phosphate-buffered saline (PBS) as a control. After 24 h, medium was separated from the cells, cells were taken up in 1 ml of ice-cold lysis buffer (100 mmol/l Tris–HCl, 0.1% Tween, pH 7.5), both cell and medium samples were kept on ice awaiting assay of lactate dehydrogenase (LDH) activity on the same day, and subsequently stored at −20°C for later cTnI and MMP2 assays.
Troponin-I ELISA
Purified human heart cTnI (Calbiochem, LaJolla, CA, USA) was dissolved in urea/Tris buffer according to the manufacturer’s instructions. Monoclonal mouse anti-cTnI clone 19C7 (directed against amino acid sequence 40–50) and monoclonal mouse anti-cTnI clone 6F9 (directed against amino acid sequence 189-195) were purchased from HyTest (Turku, Finland). The enzyme-linked immunosorbent assay (ELISA) was based on the sandwich principle as described previously [13]. The percentage cTnI released from each culture equalled 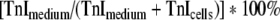 . Detection limit of the assay was 6 ng/ml. Inter-assay variability was 6 and 10% at a TnI concentration of 200 and 25 ng/ml, respectively. The intra-assay variability was 6% at TnI concentrations of 100 ng/ml.
. Detection limit of the assay was 6 ng/ml. Inter-assay variability was 6 and 10% at a TnI concentration of 200 and 25 ng/ml, respectively. The intra-assay variability was 6% at TnI concentrations of 100 ng/ml.
Western blot analysis
Protein extracts were size-fractionated on NuPage Novex 12% Bis–Tris gels (Invitrogen) and transferred to Hybond PVDF membranes (Amersham Biosciences, Roosendaal, The Netherlands). Non-specific binding sites were blocked using blocking solution consisting of 20 g/l ECL advance blocking agent (Amersham Biosciences) in TBS-Tween (10 mmol/l Tris–HCl, 150 mmol/l NaCl, 0.05% Tween pH 8.0). Membranes were incubated for 1 h with the anti-TnI antibody (K83085G, Biodesign, Saco, ME, USA), which detects various cTnI degradation products. After four washes in TBS-Tween, membranes were incubated with horseradish peroxidase-labeled secondary antibody (rabbit anti-goat IgG, Santa Cruz Biotechnology, Heidelberg, Germany). Chemiluminescence was induced by ECL advance detection reagent (Amersham Biosciences) and detected by exposure to Hyperfilm ECL (Amersham Biosciences).
LDH activity assay
Necrotic cell death was quantified by assay of LDH activity released from cells into medium according to Wroblewski and Ladue [41]. LDH activities of medium and cell samples were determined using a spectrophotometer (Ultrospec 3000, Pharmacia Biotech, Roosendaal, The Netherlands) at 25°C. The total LDH activity in each culture, i.e., cell+medium, was determined, and the LDH activity in cell and medium samples was expressed as a percentage of total LDH activity. The detection limit of the LDH assay was 5 U/l, the intra-assay variability was 3% (110 U/l) and 6% (48 U/l), and inter-assay variability was 12% (110 U/l).
Nuclear staining with propidium iodide
An additional assessment of cell death was performed by quantifying the nuclei of necrotic cells using propidium iodide and fluorescence microscopy. For this purpose, cardiomyocyte cultures were treated with PBS, GRGDS, and SDGRG for 24 h, then incubated with propidium iodide (5 mg/l, Molecular Probes, Eugene, OR, USA) for 15 min in the dark, washed in PBS, and viewed under a fluorescence microscope (Zeiss, Hamburg, Germany) at 535/617-nm excitation/emission. Subsequently, cultures were incubated with digitonine (10 μmol/l) for 5 min to induce necrotic cell death in all cardiomyocytes. The extent of necrosis equalled the number of necrotic nuclei before digitonine incubation as a fraction of the number of necrotic nuclei after digitonine incubation (100%).
In vitro degradation of purified cTnI by active MMP2
To study whether active MMP2 is able to degrade cTnI in vitro, purified human cardiac TnI (500 pg/μl, Calbiochem) was incubated with human recombinant active MMP2 (1.5 ng/μl, Oncogene, San Diego, CA, USA) in enzyme buffer at 37°C for 0, 60, and 120 min. In a separate series of experiments, MMP2 was preincubated with an inhibitor of MMP2, o-phenanthroline (100 μmol/l) for 15 min. As a control, cTnI was incubated without active MMP2 at 37°C. Intact cTnI and cTnI degradation products were visualized by Western blotting.
Zymography
Gelatinolytic activities of MMP2 in cell and medium samples of control, GRGDS- and SDGRG-treated cardiomyocytes were analyzed by zymography as described previously [12, 33]. Briefly, cell samples (tenfold concentrated by freeze-drying for 18 h) and medium samples (unconcentrated) were prepared in sample buffer and were loaded on gels. After electrophoresis, the gels were washed twice in 2.5% Triton X-100, were washed in enzyme buffer, and were incubated in enzyme buffer at 37°C overnight. Gels were stained with amino black and subsequently destained. Gelatinase activity was quantified by measuring the extent of gelatin digestion using a scanning densitometer (2202 Ultrascan, LKB, Paramus, NJ, USA).
Experimental protocol for necrotic cell death
Three days after cell isolation, cardiomyocyte cultures were washed twice in HBSS, pH 7.4, at 37°C, and subsequently incubated in 1 ml of HBSS, supplemented with sodium azide (1 mmol/l, n = 7) at 37°C. After 24 h of incubation, medium was separated from the cells, cells were taken up in 1 ml of ice-cold lysis buffer, both cell and medium samples were kept on ice awaiting assay of lactate dehydrogenase (LDH) activity on the same day, and subsequently stored at −20°C for later cTnI assay.
Statistics
Results are expressed as mean ± SD. Statistical analysis was performed by Student’s t test and by one-way analysis of variance followed by Bonferroni’s post hoc test. Differences were regarded as statistically significant if p < 0.05. SPSS14 for Windows (SPSS, Chicago, IL, USA) was used for statistical analysis.
Results
Effect of integrin stimulation on release of cTnI
Cell and medium samples were assayed for intact cTnI by ELISA (Fig. 1a) and for cTnI degradation products using Western blotting (Fig. 2a–d). Cellular cTnI content in ten cultures was 0.27 ± 0.10 μg per culture (100%). Control cardiomyocytes had released ≈3% of total cTnI, probably as a result of cell death of a few cardiomyocytes during 24 h of incubation. GRGDS treatment at a concentration of 100 or 200 μg/ml did not show considerable release of cTnI (≈2%). However, a concentration of 300 μg/ml GRGDS resulted in a significant extra release of intact cTnI (to ≈10%), compared with control cardiomyocytes (p < 0.001) and cardiomyocytes treated with 100 or 200 μg/ml GRGDS (both p < 0.001; Fig. 1a). Incubation of cardiomyocytes with 300 μg/ml SDGRG, the reverse sequence of GRGDS that has no effect on integrin function, did not induce a significant extra release of intact cTnI (Fig. 1a). In addition, Fig. 2b and d shows that cTnI was released as an intact protein of 29 kDa and that integrin stimulation is not associated with the release of cTnI degradation products.
Fig. 1.

a Release of cardiac troponin-I (cTnI) from cardiomyocytes incubated with PBS (control, n = 38), GRGDS (100 μg/ml, n = 5; 200 μg/ml, n = 4; 300 μg/ml, n = 22), or SDGRG (300 μg/ml, n = 8), detected by ELISA. b Release of LDH from cardiomyocytes incubated with PBS (control, n = 38), GRGDS (300 μg/ml, n = 22), or SDGRG (300 μg/ml, n = 8). (*p < 0.001 vs control, #p < 0.001 vs GRGDS 100, $p < 0.001 vs GRGDS 200, ‡p < 0.001 vs GRGDS 300, and §p < 0.001 vs SDGRG 300)
Fig. 2.

Western blots of intact cTnI (29 kDa) and cTnI degradation products present in cell samples (left, 2.5× diluted) and medium samples (right, 10× concentrated) of cardiomyocytes incubated with PBS (control), GRGDS (300 μg/ml, a and b), SDGRG (300 μg/ml, c and d), or sodium azide (1 mmol/l, e and f) for 24 h
Effect of integrin stimulation on cell viability
Necrotic cell death was quantified by the release of LDH activity from cardiomyocytes into the medium (Fig. 1b). Cellular LDH activity of ten cultures was 0.47 ± 0.24 U per culture (100%). In control cardiomyocytes, ≈15% of total LDH was released in 24 h, which confirms previous findings of cellular necrosis in cardiomyocytes after 24 h in serum-free medium [25]. LDH release from integrin-stimulated cardiomyocytes (GRGDS, 300 μg/ml) equalled LDH release from control cardiomyocytes (Fig. 1b). Surprisingly, cardiomyocytes treated with SDGRG (300 μg/ml) for 24 h showed significantly less LDH release (9.5%) than control and GRGDS-treated cardiomyocytes (both p < 0.001).
Assessment of cell death using nuclear staining with propidium iodide showed a similar pattern. The percentage of necrotic cells in controls (7.2 ± 2.1%, n = 8) equalled that of GRGDS-treated cardiomyocytes (8.5 ± 2.2%, n = 8, p = N.S.). Again, the percentage of necrotic cells in SDGRG-treated cardiomyocytes was significantly lower (6.2 ± 1.9%, n = 8) compared with GRGDS-treated cardiomyocytes (p < 0.05), confirming the results of the LDH assay.
These results demonstrate that integrin stimulation by GRGDS participates in the release of cTnI from cardiomyocytes in the absence of necrosis.
Effect of integrin stimulation on MMP2 activity
In control experiments, we verified that recombinant active MMP2 is able to degrade purified cTnI in vitro in a number of fragments of 26, 14 and 8 kDa (Fig. 3). However, as we observed no cTnI degradation upon integrin stimulation (Fig. 2), we studied whether this was due to the lack of MMP2 activation during integrin stimulation, measuring the zymographic gelatinolytic activities of MMP2. In control cardiomyocytes, gelatinolytic MMP2 activities were detected in both cell and medium samples, indicating a baseline level of MMP2 in cultured cardiomyocytes (Fig. 4). Total MMP2 levels were higher in medium than in cells (≈90 vs ≈10% of total MMP2, respectively).
Fig. 3.

Western blot of intact cTnI (29 kDa) and cTnI degradation products after in vitro incubation of purified intact cTnI without and with active MMP2 and with active MMP2 in combination with a MMP2 inhibitor (o-phenanthroline) for 0, 60, and 120 min
Fig. 4.

Gelatinolytic activities of MMP2 isoforms in cell samples (left, 10× concentrated) and medium samples (right, unconcentrated) of cardiomyocytes incubated with PBS (control, n = 12), GRGDS (300 μg/ml, n ≥ 9), or SDGRG (300 μg/ml, n ≥ 3) for 24 h
In cells samples, several gelatinolytic activities of MMP2 were detected corresponding to a rodent-specific glycosylated MMP2 (75 kDa), proMMP2 (72 kDa, the major gelatinase activity), an active intermediate MMP2 (64 kDa), and active MMP2 (62 kDa; Fig. 4, left). GRGDS-treated cardiomyocytes demonstrated significantly higher levels of cellular proMMP2 (72 kDa) and active MMP2 (64 + 62 kDa) than control and SDGRG-treated cardiomyocytes (Fig. 4, left), whereas levels of glycosylated MMP2 (75 kDa) were unaffected by GRGDS. In medium samples (Fig. 4, right), several gelatinolytic activities of MMP2 were detected, corresponding to a rodent-specific glycosylated MMP2 (75 kDa), proMMP2 (72 kDa), and active MMP2 (62 kDa), respectively. GRGDS-induced integrin stimulation resulted in significantly higher medium levels of glycosylated MMP2 (75 kDa) and active MMP2 (62 kDa) compared to medium samples of control and SDGRG-treated cardiomyocytes (both p < 0.05; Fig. 4, right). Medium levels of proMMP2 (72 kDa) were unaffected by integrin stimulation.
Release of cTnI from necrotic cardiomyocytes
To compare the release of cTnI from viable cardiomyocytes during GRGDS-induced integrin stimulation with the release of cTnI upon necrotic cell death, cardiomyocyte cultures were treated with sodium azide (1 mmol/l) to induce necrotic cell death. After 24 h of azide-treatment, LDH release from cardiomyocytes was 73.4 ± 10.5% (n = 7), indicating extensive necrotic cell death. Release of intact cTnI from these necrotic cardiomyocytes was 32.7 ± 13.8% (n = 7). Notably, in contrast to the release of intact cTnI from integrin-stimulated cardiomyocytes (Fig. 2b), cardiomyocyte necrosis leads to the release of at least four cTnI degradation products with molecular weights of 26, 20, 17, and 12 kDa (Fig. 2f).
Discussion
The major findings of this study are that integrin stimulation in cardiomyocytes by GRGDS leads to (1) release of intact cTnI from viable cardiomyocytes in the absence of necrosis and (2) increased levels of intracellular and extracellular MMP2 activity that, however, does not result in degradation of cTnI into its fragments. This study therefore indicates that the release mechanism of cTnI from viable cardiomyocytes upon integrin stimulation differs from cTnI release from necrotic cardiomyocytes, which is associated with extensive cTnI degradation.
Elevated plasma cTnI levels are frequently found in pathological conditions in which irreversible myocardial cell injury is not a prominent aspect. In patients with heart failure, elevated plasma cTnI levels have been reported with normal plasma levels of CK-MB [2, 18]. Some studies have suggested that elevated serum levels of cTnI may be the result of myocardial strain [15, 20], as occurs during pressure or volume overload. Logeart et al. [15] studied 71 patients with heart failure and found in 19 patients elevated cTnI concentrations associated with LV remodeling and increased plasma brain natriuretic peptide levels. They postulated that increased LV wall strain may have led to the increased cTnI release, but necrotic cell death was not excluded in that study. In acute pulmonary embolism, several reports showed elevated cTnI levels [17, 21] possibly related to the presence of myocardial strain. In addition, Feng et al. [6] demonstrated that increased preload in isolated rat hearts was sufficient to cause release of cTnI as the result of myocardial stretch independent of ischemia.
Overload-induced stretch at the cardiomyocyte level is sensed by integrins, mechanotransducers molecules that link the extracellular matrix to the intracellular cytoskeleton [32]. In the present study, we showed that GRGDS-induced integrin stimulation of cardiomyocytes resulted in a threefold increased release of cTnI that occurred in the absence of necrosis, as cell viability remained unchanged by GRGDS treatment compared with control cardiomyocytes. Control cardiomyocytes demonstrated baseline levels of necrosis after 24 h, consistent with previous findings of cell death in cardiomyocytes after 24 h in serum-free medium [25]. Surprisingly, we found significantly less necrosis in SDGRG-treated cardiomyocytes than in control cardiomyocytes with both LDH assay and nuclear staining with propidium iodide. The mechanism responsible for this protective effect is unlikely to be related to integrin function but may be related to protection against necrosis exerted by certain proteins such as serum components.
Several studies have suggested that release of cTnI from viable cardiomyocytes, without lethal disruptions of the cardiomyocyte sarcolemma, does not involve intact cTnI (29 kDa) but rather proteolytic degradation products of cTnI formed by intracellular proteases as MMP2 and calpain-I [6, 7]. In the present study, we demonstrated that purified intact cTnI can indeed be degraded by active MMP2 in vitro. MMPs are a family of zinc-containing enzymes involved in degradation of the extracellular matrix during tissue remodeling [34]. However, MMP2 also acts intracellularly and may, therefore, be responsible for degradation of myofilament proteins including cTnI and myosin light chain-1 (MLC-1) [30, 39]. MMPs are synthesized by a variety of cells, including cardiomyocytes [4], in a latent form (proMMP) that is activated by either proteolytic cleavage or by conformational changes induced by cytokines, reactive oxygen species, and peroxynitrite [22, 35, 38]. Previously, Wang et al. [37] have demonstrated that mechanical stretch of neonatal cardiomyocytes induced expression and activation of MMP2. In addition, integrin stimulation by GRGDS has been reported to upregulate MMP2 expression in fibroblasts and certain tumor cell lines [31, 40], suggesting an important role for integrins in regulating MMP2 activity. In the present study, we showed that GRGDS-induced integrin stimulation of cultured cardiomyocytes was indeed associated with increased activity of intracellular proMMP2 and active MMP2 and increased release of active MMP2 into the medium. In cardiomyocytes incubated with SDGRG, neither cellular levels nor medium levels of proMMP2 or active MMP2 were altered compared to control cardiomyocytes, indicating that increased MMP2 activity is specific for GRGDS-induced integrin stimulation.
However, despite the increase of MMP2 activity during integrin stimulation and the ability of active MMP2 to degrade purified cTnI in vitro, integrin stimulation was not associated with cTnI degradation. If cTnI degradation had occurred, we would have been able to detect that in this system (Figs. 3 and 2f). The reason for the lack of cTnI degradation in the presence of GRGDS-induced MMP2 activation is unclear. The discrepancy between the in vitro degradation of purified cTnI by active MMP2 and the lack of cTnI degradation in GRGDS-treated cardiomyocytes may be explained by a difference in specific MMP2 activity. The MMP2 activity per nanogram cTnI in the study with purified cTnI was fourfold higher than that in cardiomyocytes treated with RGD. In addition, purified cTnI may be more susceptible to MMP2 cleavage than cTnI that is complexed to cTnT, cTnC, and tropomyosin. In cardiomyocytes, 93–97% of cTnI is complexed to cTnT, cTnC, and tropomyosin which may protect the MMP2 cleavage sites on the cTnI molecule.
The mechanism responsible for the release of intact cTnI from viable integrin stimulated cardiomyocytes may be leakage of free intact cTnI from the cytosolic pool. Several studies have demonstrated that mechanically induced transient disruptions (wounding) of the sarcolemma are a constitutive event in vivo [3, 10, 16, 23]. This mechanism is responsible for the release of proteins, such as myocyte-derived growth factors (fibroblast growth factors 1 and 2), which are released despite the lack of a classic signal peptide sequence normally associated with exocytotic secretion. These mechanically induced alterations in cardiomyocyte’s sarcolemmal permeability may similarly be involved in the release of cTnI from the cytosolic pool of cardiomyocytes in the absence of necrotic cell death. Further study is needed to address this issue.
Conclusions
The present study demonstrates that viable cardiomyocytes release cTnI as an intact protein by a stretch-related mechanism mediated by integrins. This finding may possibly explain why, in several pathological conditions, plasma cTnI levels are elevated in the absence of myocardial necrosis.
Acknowledgments
This study was supported by the Dutch Heart foundation grant 2001.B124. We thank Wim van Duijn (Dept. of Gastro-enterology, LUMC) for assistance with zymography.
References
- 1.Bleier J, Vorderwinkler KP, Falkensammer J, Mair P, Dapunt O, Puschendorf B, Mair J (1998) Different intracellular compartmentations of cardiac troponins and myosin heavy chains: a causal connection to their different early release after myocardial damage. Clin Chem 44:1912–1918 [PubMed]
- 2.Chen YN, Wei JR, Zeng LJ, Wu MY (1999) Monitoring of cardiac troponin I in patients with acute heart failure. Ann Clin Biochem 36:433–437 [DOI] [PubMed]
- 3.Clarke MS, Caldwell RW, Chiao H, Miyake K, McNeil PL (1995) Contraction-induced cell wounding and release of fibroblast growth factor in heart. Circ Res 76:927–934 [DOI] [PubMed]
- 4.Coker ML, Doscher MA, Thomas CV, Galis ZS, Spinale FG (1999) Matrix metalloproteinase synthesis and expression in isolated LV myocyte preparations. Am J Physiol 277:H777–H787 [DOI] [PubMed]
- 5.Diris JH, Hackeng CM, Kooman JP, Pinto YM, Hermens WT, Dieijen-Visser MP (2004) Impaired renal clearance explains elevated troponin T fragments in hemodialysis patients. Circulation 109:23–25 [DOI] [PubMed]
- 6.Feng J, Schaus BJ, Fallavollita JA, Lee TC, Canty JM Jr (2001) Preload induces troponin I degradation independently of myocardial ischemia. Circulation 103:2035–2037 [DOI] [PubMed]
- 7.Gao WD, Atar D, Liu Y, Perez NG, Murphy AM, Marban E (1997) Role of troponin I proteolysis in the pathogenesis of stunned myocardium. Circ Res 80:393–399 [PubMed]
- 8.Hamm CW, Giannitsis E, Katus HA (2002) Cardiac troponin elevations in patients without acute coronary syndrome. Circulation 106:2871–2872 [DOI] [PubMed]
- 9.Katus HA, Remppis A, Neumann FJ, Scheffold T, Diederich KW, Vinar G, Noe A, Matern G, Kübler W (1991) Diagnostic efficiency of troponin T measurements in acute myocardial infarction. Circulation 83:902–912 [DOI] [PubMed]
- 10.Kaye D, Pimental D, Prasad S, Maki T, Berger HJ, McNeil PL, Smith TW, Kelly RA (1996) Role of transiently altered sarcolemmal membrane permeability and basic fibroblast growth factor release in the hypertrophic response of adult rat ventricular myocytes to increased mechanical activity in vitro. J Clin Invest 97:281–291 [DOI] [PMC free article] [PubMed]
- 11.Khan IA, Tun A, Wattanasauwan N, Win MT, Hla TA, Hussain A, Vasavada BC, Sacchi TJ (1999) Elevation of serum cardiac troponin I in noncardiac and cardiac diseases other than acute coronary syndromes. Am J Emerg Med 17:225–229 [DOI] [PubMed]
- 12.Kleiner DE, Stetler-Stevenson WG (1994) Quantitative zymography: detection of picogram quantities of gelatinases. Anal Biochem 218:325–329 [DOI] [PubMed]
- 13.Li L, Hessel M, van der Valk L, Bax M, van der Linden I, van der Laarse A (2004) Partial and delayed release of troponin-I compared with the release of lactate dehydrogenase from necrotic cardiomyocytes. Pflügers Arch 448:146–152 [DOI] [PubMed]
- 14.Lindahl B, Venge P, Wallentin L (1996) Relation between troponin T and the risk of subsequent cardiac events in unstable coronary artery disease. The FRISC Study Group. Circulation 93:1651–1657 [DOI] [PubMed]
- 15.Logeart D, Beyne P, Cusson C, Tokmakova M, Leban M, Guiti C, Bourgoin P, Solal AC (2001) Evidence of cardiac myolysis in severe nonischemic heart failure and the potential role of increased wall strain. Am Heart J 141:247–253 [DOI] [PubMed]
- 16.McNeil PL, Khakee R (1992) Disruptions of muscle fiber plasma membranes. Role in exercise-induced damage. Am J Pathol 140:1097–1109 [PMC free article] [PubMed]
- 17.Meyer T, Binder L, Hruska N, Luthe H, Buchwald AB (2000) Cardiac troponin I elevation in acute pulmonary embolism is associated with right ventricular dysfunction. J Am Coll Cardiol 36:1632–1636 [DOI] [PubMed]
- 18.Missov E, Calzolari C, Pau B (1997) Circulating cardiac troponin I in severe congestive heart failure. Circulation 96:2953–2958 [DOI] [PubMed]
- 19.Neumayr G, Gaenzer H, Pfister R, Sturm W, Schwarzacher SP, Eibl G, Mitterbauer G, Hoertnagl H (2001) Plasma levels of cardiac troponin I after prolonged strenuous endurance exercise. Am J Cardiol 87:369–371 [DOI] [PubMed]
- 20.Nunes JP (2001) Cardiac troponin I in systemic diseases. A possible role for myocardial strain. Rev Port Cardiol 20:785–788 [PubMed]
- 21.Nunes JP, Macedo F (2000) An analytical triad for the diagnosis of pulmonary embolism. Cardiology 94:264 [DOI] [PubMed]
- 22.Okamoto T, Akaike T, Nagano T, Miyajima S, Suga M, Ando M, Ichimori K, Maeda H (1997) Activation of human neutrophil procollagenase by nitrogen dioxide and peroxynitrite: a novel mechanism for procollagenase activation involving nitric oxide. Arch Biochem Biophys 342:261–274 [DOI] [PubMed]
- 23.Page E, Upshaw-Earley J, Goings G (1992) Permeability of rat atrial endocardium, epicardium, and myocardium to large molecules. Stretch-dependent effects. Circ Res 71:159–173 [DOI] [PubMed]
- 24.Persoon-Rothert M, Egas-Kenniphaas JM, van der Valk-Kokshoorn EJ, Mauve I, van der Laarse A (1990) Prevention of cumene hydroperoxide induced oxidative stress in cultured neonatal rat myocytes by scavengers and enzyme inhibitors. J Mol Cell Cardiol 22:1147–1155 [DOI] [PubMed]
- 25.Persoon-Rothert M, van der Wees KG, van der Laarse A (2002) Mechanical overload-induced apoptosis: a study in cultured neonatal ventricular myocytes and fibroblasts. Mol Cell Biochem 241:115–124 [DOI] [PubMed]
- 26.Ross RS, Borg TK (2001) Integrins and the myocardium. Circ Res 88:1112–1119 [DOI] [PubMed]
- 27.Ruoslahti E, Pierschbacher MD (1986) Arg–Gly–Asp: a versatile cell recognition signal. Cell 44:517–518 [DOI] [PubMed]
- 28.Ruwhof C, van der Laarse A (2000) Mechanical stress-induced cardiac hypertrophy: mechanisms and signal transduction pathways. Cardiovasc Res 47:23–37 [DOI] [PubMed]
- 29.Sato Y, Yamada T, Taniguchi R, Nagai K, Makiyama T, Okada H, Kataoka K, Ito H, Matsumori A, Sasayama S, Takatsu Y (2001) Persistently increased serum concentrations of cardiac troponin T in patients with idiopathic dilated cardiomyopathy are predictive of adverse outcomes. Circulation 103:369–374 [DOI] [PubMed]
- 30.Sawicki G, Leon H, Sawicka J, Sariahmetoglu M, Schulze CJ, Scott PG, Szczesna-Cordary D, Schulz R (2005) Degradation of myosin light chain in isolated rat hearts subjected to ischemia-reperfusion injury: a new intracellular target for matrix metalloproteinase-2. Circulation 112:544–552 [DOI] [PubMed]
- 31.Seftor RE, Seftor EA, Gehlsen KR, Stetler-Stevenson WG, Brown PD, Ruoslahti E, Hendrix MJ (1992) Role of the alpha v beta 3 integrin in human melanoma cell invasion. Proc Natl Acad Sci U S A 89:1557–1561 [DOI] [PMC free article] [PubMed]
- 32.Shyy JY, Chien S (1997) Role of integrins in cellular responses to mechanical stress and adhesion. Curr Opin Cell Biol 9:707–713 [DOI] [PubMed]
- 33.Sier CF, Kubben FJ, Ganesh S, Heerding MM, Griffioen G, Hanemaaijer R, van Krieken JH, Lamers CB, Verspaget HW (1996) Tissue levels of matrix metalloproteinases MMP-2 and MMP-9 are related to the overall survival of patients with gastric carcinoma. Br J Cancer 74:413–417 [DOI] [PMC free article] [PubMed]
- 34.Spinale FG (2002) Matrix metalloproteinases: regulation and dysregulation in the failing heart. Circ Res 90:520–530 [DOI] [PubMed]
- 35.Spinale FG, Gunasinghe H, Sprunger PD, Baskin JM, Bradham WC (2002) Extracellular degradative pathways in myocardial remodeling and progression to heart failure. J Card Fail 8:S332–S338 [DOI] [PubMed]
- 36.van der Wees CG, Bax WH, van der Valk EJ, van der Laarse A (2006) Integrin stimulation induces calcium signalling in rat cardiomyocytes by a NO-dependent mechanism. Pflügers Arch 451:588–595 [DOI] [PubMed]
- 37.Wang TL, Yang YH, Chang H, Hung CR (2004) Angiotensin II signals mechanical stretch-induced cardiac matrix metalloproteinase expression via JAK-STAT pathway. J Mol Cell Cardiol 37:785–794 [DOI] [PubMed]
- 38.Wang W, Sawicki G, Schulz R (2002) Peroxynitrite-induced myocardial injury is mediated through matrix metalloproteinase-2. Cardiovasc Res 53:165–174 [DOI] [PubMed]
- 39.Wang W, Schulze CJ, Suarez-Pinzon WL, Dyck JR, Sawicki G, Schulz R (2002) Intracellular action of matrix metalloproteinase-2 accounts for acute myocardial ischemia and reperfusion injury. Circulation 106:1543–1549 [DOI] [PubMed]
- 40.Werb Z, Tremble PM, Behrendtsen O, Crowley E, Damsky CH (1989) Signal transduction through the fibronectin receptor induces collagenase and stromelysin gene expression. J Cell Biol 109:877–889 [DOI] [PMC free article] [PubMed]
- 41.Wroblewski F, Ladue JS (1955) Lactic dehydrogenase activity in blood. Proc Soc Exp Biol Med 90:210–213 [DOI] [PubMed]