

Abstract
Very little is known about the role of histone H3 phosphorylation in malignant transformation and cancer development. Here, we examine the function of H3 phosphorylation in cell transformation in vivo. Introduction of small interference RNA (siRNA)-H3 into JB6 cells resulted in decreased EGF-induced cell transformation. In contrast, wildtype histone H3-overexpressing cells markedly stimulated EGF-induced cell transformation, whereas the H3 mutant S10A cells suppressed transformation. When wildtype histone H3 was overexpressed, EGF induction of c-fos and c-jun promoter activity was significantly increased compared to control cells, but not in the H3 mutant S10A or S28A cells. In addition, AP-1 activity in wildtype histone H3-overexpressing cells was markedly upregulated by EGF in contrast to the H3 mutant S10A or S28A cells. These results indicate that the phosphorylation of histone H3 at serine 10 is an essential regulatory mechanism for EGF-induced neoplastic cell transformation.
Introduction
Phosphorylation of histone H3 has been linked with mitotic chromatin condensation. Histone H3 is phosphorylated during mitosis on at least two serine residues, Ser10 (1, 2) and Ser28 (3). Phosphorylation at Ser10 begins in early G2 in the pericentromeric heterochromatin of each chromosome (4) and by metaphase has spread throughout all chromosomes, while phosphorylation on Ser28 starts to be evident only in early mitosis (3). Phosphorylation of histone H3 at Ser10 appears to be involved in the initiation of mammalian chromosome condensation, but not the maintenance (5). Increased phosphorylation of histone H3 at serine 10 was found in mitogen-stimulated and oncogene-transformed mouse fibrobasts (6). Moreover, the phosphorylation of histone H3 at N-terminal serine 10 is closely related to the induction of immediate-early (IE) response genes, including proto-oncogenes c-fos and c-jun (2, 7, 8). These and other IE genes are rapidly and transiently expressed in response to extracellular stimuli. The IE gene response has been implicated in differentiation, mitosis, and disease such an inflammation and cancer (9, 10). However, much less is known about the role of histone H3 in EGF-induced neoplastic cell transformation.
The epidermal growth factor (EGF) is a well-known tumor promotion agent used to study malignant cell transformation in cell and animal models of cancer (11). EGF induces activation of the transcription factor, activator protein-1 (AP-1; (12)). When treated with EGF, JB6 Cl41 skin epidermal cells showed an induction of AP-1 transcriptional activation in promotion-sensitive (P+) phenotypes but not in promotion-resistant (P−) phenotypes (13). Blocking AP-1 activation causes P+ cells to revert to the P− phenotype, indicating a unique requirement for AP-1 activation in EGF-induced cell transformation (14). In previous studies, phosphorylation of histone H3 at serine 10 was shown to be involved in different signal transduction pathways and to be dependent on the specific stimulation or stress. Epidermal growth factor (EGF) induces phosphorylation of H3 at serine 10, which is mediated by RSK2 (15). RSK2 mutation in humans is linked to Coffin-Lowery Syndrome and fibroblasts derived from a Coffin-Lowery Syndrome patient fail to exhibit EGF-stimulated phosphorylation of histone H3 at serine 10 (15). In addition, mitogen- and stress-activated protein kinase (MSK1) has been shown to mediate EGF or 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced phosphorylation of histone H3 at serine 10 (15). These results suggested that histone H3 might be regulated by mitogen-derived signal transduction induced by tumor promoters, such as EGF. Although EGF signaling cascades have been thoroughly studied, the involvement of histone H3 in the neoplastic cell transformation is unknown.
Recently, members of the Aurora kinase family, which are overexpressed in many human cancer cells, were shown to be responsible for mitotic phosphorylation of histone H3 at serine 10 in yeast and nematodes (16–18). Moreover, increased H3 histone phosphorylation as a result of AIM-1 (Aurora B) overexpression is a major precipitating factor of chromosome instability and, thus, may play a role in carcinogenesis (19). Because mitosis is the phase of the cell cycle most susceptible to various chemical and physical agents, the consistent observation of histone H3 phosphorylation at serine 10 and/or overexpression in human tumor cell lines (20) suggested that regulation of mitosis by phosphorylation of histone H3 at serine 10 may be a possible target for cancer treatment. Thus, although Aurora-B-mediated mitotic H3 phosphorylation seems to induce carcinogenesis, whether histone H3 directly is essential for neoplastic cell transformation and cancer development is still an intriguing question. In this study, we investigated whether phosphorylation of histone H3 is required for inducing neoplastic cell transformation using knockdown and dominant negative constructs of histone H3, which were stably transfected with psi-H3 or pV5-H3 mutant constructs (S10A, S28A, or S10/28A) respectively.
Materials and Methods
Reagents and Antibodies.
Eagle’s minimal essential medium (MEM), L-glutamine, and gentamicin were purchased from Invitrogen (Carlsbad, CA). EGF was purchased from EMD Biosciences, Inc (San Diego, CA) and fetal bovine serum (FBS) from Gemini Bio-Products (Calabasas, CA). Polyvinylidene difluoride (PVDF) membrane was from Millipore (Bedford, MA). The DNA ligation kit (version 2.0) was purchased from TAKARA Bio. Inc. (Otsu, Shiga, Japan). Antibodies against histone H2A or the V5-epitope were purchased from Upstate (Lake Placid, NY) or Invitrogen (Carlsbad, CA), respectively. Antibodies against c-Jun or c-Fos were purchased from Cell Signaling Tech., Inc. (Beverly, MA) or EMD Biosciences, Inc. (San Diego, CA), respectively. Control siRNA, c-Jun siRNA (mouse), or c-Fos siRNA (mouse) was purchased from Santa Cruz Biotech., Inc. (Santa Cruz, CA) and jetSITM-ENDO was from Qbiogene (Irvine, CA).
Cell Culture and Transfection.
JB6 Cl41 mouse epidermal cells were cultured in MEM supplemented with 5% FBS. psi-H3 or pV5-H3 stably transfected JB6 Cl41 cells were established in MEM supplemented with 5% FBS containing 600 μg/ml G418 and maintained in MEM supplemented with 5% FBS containing 200 μg/ml G418. Cells were transfected by a cationic polymer transfection method, jetPEI (Qbiogene, Carlsbad, CA). To transfect siRNA duplex with jetSITM-ENDO, we were performed following manufacture’s protocol (Qbiogene, Irvine, CA).
Construction of Mammalian Expression Vector and siRNA Vector.
A cDNA fragment encoding human histone H3 was inserted in-frame into the BamHI/EcoRI sites of the pcDNA3.1/V5-His vector (Invitrogen, San Diego, CA) to produce the V5-epitope tagged construct, pV5-H3. cDNAs encoding the S10A, S28A, and S10/28A mutants of histone H3 were generated using the QuickChange II site-directed mutagenesis kit (Stratagene, La Jolla, CA) and subcloned into the pcDNA3.1/V5-His vector to produce the V5-epitope tagged constructs, pV5-H3 S10A, pV5-H3 S28A, and pV5-H3 S10/28A, respectively. All of the positive clones containing cDNA inserts were identified by restriction enzyme mapping and sequenced at Genewiz, Inc. (North Brunswick, NJ). To construct the siRNA-histone H3 (psi-H3), the pU6pro vector (provided by David L. Turner, University of Michigan) was digested with XbaI and BbsI. The annealed synthetic primers were then introduced: sense siRNA: 5′-TTTGCAGACAGCTCGGAAATCCATTCA AGAGATGGATTTCCGAGCTGTCTGTTTTT; antisense siRNA: GTCTGTCGAGCCTTT AGGTAAGTTCTCTACCTAAAGGCT-CGACAGACAAAAAGATC-5′ following the recommended protocols (21). The psi-H3 was confirmed by agarose gel electrophoresis and DNA sequencing. pcDNA3.1-H-RasG12V were provided by UMR(University of Missouri-Rolla) cDNA resource center.
3-(4,5-Dimethylthiazol-2-yl)-5-(3-Carboxymethoxy-Phenyl)-2-(4-Sulfony)-2H-Tetrazolium Assay.
To estimate cell proliferation, JB6 Cl41 cells transfected with pU6pro, psi-H3, pV5, or pV5-H3 were seeded (1 × 103) in 96-well plates in 100 μl of 5% FBS-MEM in a 37°C, 5% CO2 incubator. After culturing for 12 hr, 20 μl of the CellTiter 96 Aqueous One Solution (Promega, Medison, WI) were added to each well, and cells were then incubated for 1hr at 37°C, 5% CO2. Absorbance was measured at 492 and 690 nm.
Anchorage-Independent Cell Transformation Assay (Soft Agar Assay).
EGF-induced cell transformation was investigated in mock, psi-H3 or pV5-H3 stably transfected cells. In brief, cells (8 × 103/ml) were exposed to EGF (0.1–10 ng/ml) in 1 ml of 0.3% basal medium Eagle (BME) agar containing 10% FBS. The cultures were maintained at 37 °C, in a 5% CO2 incubator for 10 days, and the cell colonies were scored using a microscope and the Image-Pro PLUS computer software program (Media Cybernetics, Silver Spring, MD) as described by Colburn et al (22).
Focus Forming Assay
Transformation of 3T3 Swiss cells was performed following standard protocols (23). 3T3 Swiss cells were plated in 60 mm dishes at a density 1 × 104 cells. 3T3 cells were transiently transfected with 100 ng H-RasG12V, 2.5 μg pV5, or 2.5 μg pV5-H3 plasmid. Cells were kept in EMEM with 2% FBS. The medium was changed every 3 days for a period of 3 weeks. Foci were enumerated by staining the monolayer with methanol for fixation and 0.4% crystal violet. Data show the obtained from three independent plates for each transfection.
Reporter Gene Assays.
The reporter gene assay for firefly luciferase activity was performed using lysates from transfected cells. In addition, the reporter gene vector pRL-SV40 (Promega) was co-transfected into each cell line and the renilla-luciferase activity generated by this vector was used to normalize the results for transfection efficiency. Cell lysates were prepared by first washing the transfected JB6 Cl41 cells (grown in 60-mm diameter dishes) once in PBS at 37 °C. After removing the PBS completely, 500 μl of passive lysis buffer (PLB, Promega Dual Luciferase Reporter Assay System) were added, and then cells were incubated for 1 hr with gentle shaking. The lysate was then transferred into a reaction tube and the cellular debris removed by centrifugation. The supernatant fraction was used for the measurement of firefly and renilla luciferase activities. Cell lysates (20 μl each) were mixed with 100 μl of luciferase assay II reagent and firefly luciferase light emission was measured by a Luminoskan Ascent (Thermo Electron Corp., Helsinki, Finland). Subsequently, 100 μl of colentiazine reagent containing the substrate for renilla luciferase light emission was mixed in order to normalize the firefly luciferase data. The results are expressed as relative c-Fos, c-Jun, or AP-1 activity (fold or %) and are presented as luciferase activity relative to the c-Fos-, c-Jun-, or AP-1-only transfected control cells. c-Fos promoter luciferase (pFos-WT GL3) and c-Jun promoter luciferase (pJC6 GL3) constructs were kindly provided by Dr. Ron Prywes (Columbia University, New York). The AP-1 luciferase reporter plasmid (−73/+63 collagenase-luciferase) was constructed as reported previously (24, 25).
Isolation of Histone Proteins.
For the isolation of histone proteins, cells (2 × 107 to 5 × 107) were homogenized in 1 ml of nuclear preparation buffer (10 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1.5 mM MgCl2, 0.65 % Nonidet P-40, and 1 mM PMSF) in the presence of protein phosphatase inhibitors (10 mM NaF, 1 mM sodium orthovanadate, 25 mM β-glycerophosphate). Nuclei were recovered by centrifugation at 1500xg for 10 min. All centrifugations were carried out at 4 °C. Nuclei were resuspended in 0.3 ml of resuspension buffer (10 mM Tris-HCl, pH 7.6, 3 mM MgCl2, 10 mM NaCl, 1 mM PMSF, and protein phosphatase inhibitors). Nuclei were extracted with 0.4 N H2SO4 to isolate total histones. The samples were precipitated with trichloroacetic acid (TCA) and then resuspended in double distilled H2O.
Immunoblotting.
The proteins were resolved by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto PVDF membranes. The membranes were blocked, hybridized with the appropriate primary antibody overnight at 4 °C. Histone H3, V5-epitope histone H3, or hstone H2A was detected with an antibody against anti-histone H3, anti-V5, or anti-histone 2A. Protein bands were visualized by the chemiluminescence detection kit (ECL of Amersham Biosciences Corp., Piscataway, NJ) after hybridization with the horseradish peroxidase (HRP)-conjugated secondary antibody from rabbit or mouse.
Results
EGF-Induced Neoplastic Cell Transformation is Suppressed by Knockdown of Histone H3.
To decrease endogenous histone H3 protein level by the introduction of U6 hairpin siRNA, we synthesized a duplex length of 19 nt with a 9 nt loop specific for histone H3 mRNA using the sites conserved on both the human and the mouse gene (Fig. 1, A and B). Our designed siRNA target sequence for histone H3 was based on an expression vector with a mouse U6 promoter, in which we could insert a sequence after the first nt of the U6 transcript (a G). The mU6 pro vector is expected to express 21-nt complementary single-stranded RNAs with 19 nt corresponding to the sense or antisense strands. In addition to JB6 Cl41 cells stably expressing histone H3 siRNA (psi-H3), we constructed JB6 Cl41 cells stably overexpressing the mock vector (pU6 pro) as a control. In our initial experiments, we observed up to a 50% silencing of histone H3 induced by the introduction of siRNA compared to cells expressing mock vector only (Fig. 1C). To test the knock-down effect of H3 in the cell proliferation, the 3-(4,5-Dimethylthiazol-2-yl)-5-(3-Carboxymethoxy-Phenyl)-2-(4-Sulfony)-2H-Tetrazolium assay was used to exam proliferation in cells stably transfected with pU6 pro or psi-H3. psi-H3 stably transfected cells showed a marked decrease in the rate of proliferation compared to pU6 pro stably transfected cells (**, p < 0.005; Fig. 1D). Using these cell lines, we studied differences in EGF-promoted cell transformation using our previously developed methods (26). Mock vector (pU6pro) cells and cells expressing siRNA against histone H3 (psi-H3) were treated separately with EGF (0.1, 1.0, or 10 ng/ml) in a soft agar matrix and incubated at 37 °C in a 5% CO2 incubator for 10 days. Our results showed that psi-H3 cells significantly inhibited the formation of EGF-promoted colonies compared with mock vector control cells (Fig. 1E). The inhibition was evident not only in colony number but also in colony size. Untreated pU6pro and psi-H3 cells showed no colony formation. These data indicated that histone H3 is involved in JB6 Cl41 cell proliferation as well as cell transformation as a positive regulator.
Figure 1.
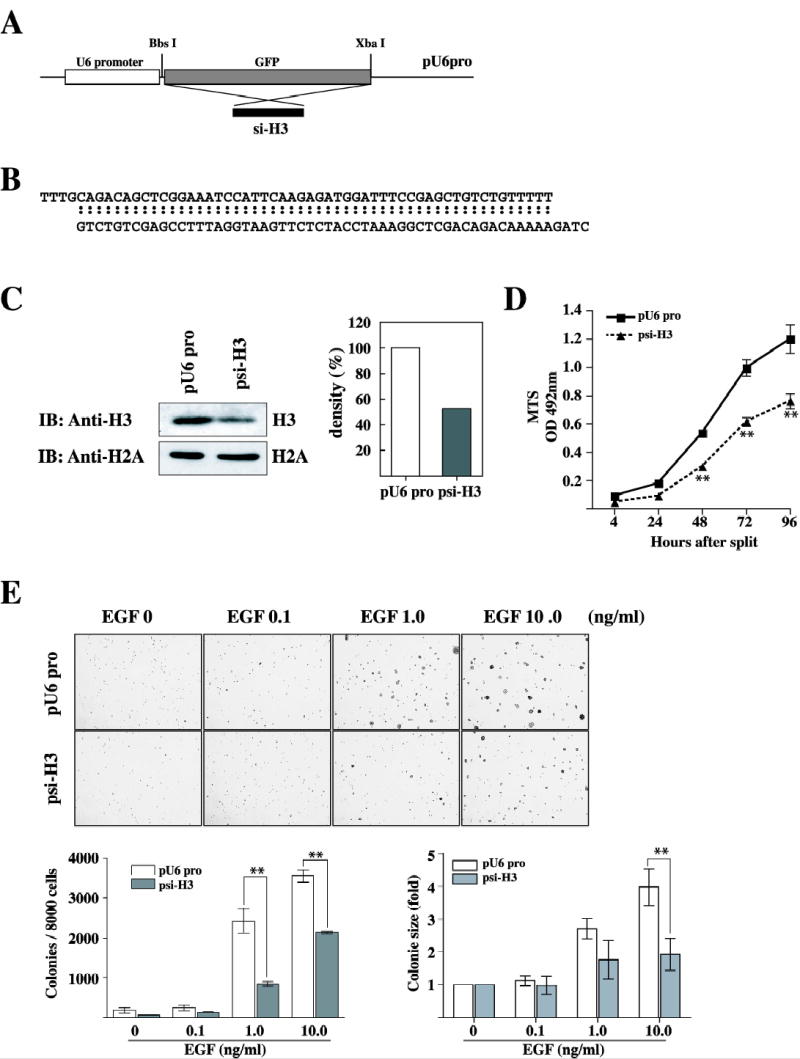
Introduction of histone H3 siRNA inhibits EGF-induced neoplastic cell transformation. (A) Schematic diagram for the construction of the si-RNA H3 stable expression vector. For the gene knockdown experiment, the siRNA-H3 expression vector was constructed using the pU6pro vector by replacing the Green Fluorescence Protein (GFP) region with siRNA-H3 synthetic oligo primers. (B) DNA sequence of the two synthetic oligo primers. (C) The JB6 Cl41 cells were co-transfected with pcDNA3.1neo and pU6pro control or psi-H3, selected with 600 μg/ml G418 for 10 days, and then pooled. The expression of histone H3 was analyzed in the stable transfectant JB6 Cl41 cells. Western blot analysis of histone H3 was performed as described in Materials and Methods. For visualizing equal loading of protein, total histone H2A was detected by western blotting. Knockdown of histone in psi-H3 cells was quantitated compared to pU6 pro cells. IB: immunoblot. (D) Cells stably transfected with the mock vector or psi-H3 were seeded (1 × 103/wells) in 96-well plates in 100 μl of 5% FBS-MEM, and cell proliferation was estimated using the CellTiter 96 Aqueous One Solution detection kit (Promega). Cell proliferation was estimated by absorbance (A492) at 24-h intervals up to 96 h. Each bar indicates the mean ± SD of values obtained from triplicate experiments. Significant differences were evaluated using the Student’s t test (**: p < 0.005). (E) pU6 pro cells and psi-H3 stably transfected cells were used for cell transformation in an anchorage-independent cell transformation assay (soft agar assay) in the presence of EGF. Cells (8 × 103/ml) transfected with mock vector or psi-H3 were exposed to EGF (0, 0.1, 1, 10 ng/ml) in 1 ml of 0.3 basal medium Eagle agar containing 10% FBS. The cultures were maintained at 37 °C in a 5% CO2 atmosphere for 10 days and then colonies were counted automatically using a microscope and the Image-Pro Plus computer software program. The average colony number was calculated and photographed from three separate experiments (bottom, left panel). Colony sizes were measured using Image J software program and compared between pU6 pro cells and psi-H3 cells stimulated by EGF (bottom, right panel). pU6pro or psi-H3 denotes cells transfected with the pU6pro vector or psi-H3, respectively. Significant differences were evaluated using the Student’s t test (**: p < 0.005), significant decrease in EGF-induced cell transformation in siRNA-H3 cells compared to pU6pro cells.
The Overexpression of Histone H3 Enhanced EGF-Induced Neoplastic Cell Transformation.
EGF stimulates the proliferation of a number of epithelial cells both in vivo and in vitro and is implicated in epithelial tumor formation (27). MAP kinases and histone H3 were rapidly and transiently phosphorylated upon EGF stimulation. An accumulation of recent evidence showed that phosphorylation of histone H3 is mediated by EGF (28). To better determine the role of histone H3 in neoplastic cell transformation, we cloned the human histone H3 cDNA, including the open reading frame, and recombined it into the mammalian expression vector, pcDNA3.1/V5 (pV5-H3). This plasmid was then introduced into JB6 Cl41 mouse skin epidermal cells, and neoplastic cell transformation was assessed. Overexpression of histone H3 was detected in histone H3 stably transfected cells (pV5-H3) using an anti-V5 antibody (Fig. 2A). The effect of histone H3 on cell proliferation was tested in the pV5-H3 cells compared with mock cells because knockdown of histone H3 by siRNA decreased cell proliferation as well as neoplastic cell transformation. The 3-(4,5-Dimethylthiazol-2-yl)-5-(3-Carboxymethoxy-Phenyl)-2-(4-Sulfony)-2H-Tetrazolium assay result indicated that histone H3 overexpressing cells proliferated at a significantly higher rate compared with mock vector stably transfected control cells (**: p < 0.005, Fig. 2B). This result confirmed that histone H3 is involved in cell proliferation. Next, cells (mock and pV5-H3) were employed to determine whether they were susceptible to cell transformation induced by EGF. Cells were treated with EGF (0.1, 1.0, or 10 ng/ml) in a soft agar matrix and incubated at 37 °C in a 5% CO2 incubator for 10 days and colony numbers determined as described. Results indicated increased colony numbers as well as colony sizes in cells overexpressing wildtype histone H3 promoted by EGF compared with control cells stimulated by EGF under the same conditions (Fig. 2C). The overexpression of histone H3 caused a dramatic increase in cell transformation without addition of EGF (Fig. 2C). This result strongly suggests that histone H3 also might induce cell transformation independent with EGF-induced signal transduction. To clarify this, we next decided to assess the focus forming activity elicited by histone H3 in a reduced serum condition (2% FBS) without EGF. Swiss 3T3 cells were transiently transfected with each expression vector (mock, pV5-H3, or H-RasG12V). Approximately 3 weeks later, the formation of small cell clusters became clearly visible. RasG12V transfected cells showed a large number of transformed foci and very importantly, the transfection of histone H3 (pV5-H3) into Swiss 3T3 cells readily induced the appearance of foci of transformation compared to mock cells (Fig. 2D). Taken together, these data strongly support the idea that histone H3 contributes to neoplastic cell transformation in epidermal mouse skin fibroblast cells stimulated or not stimulated with EGF.
Figure 2.
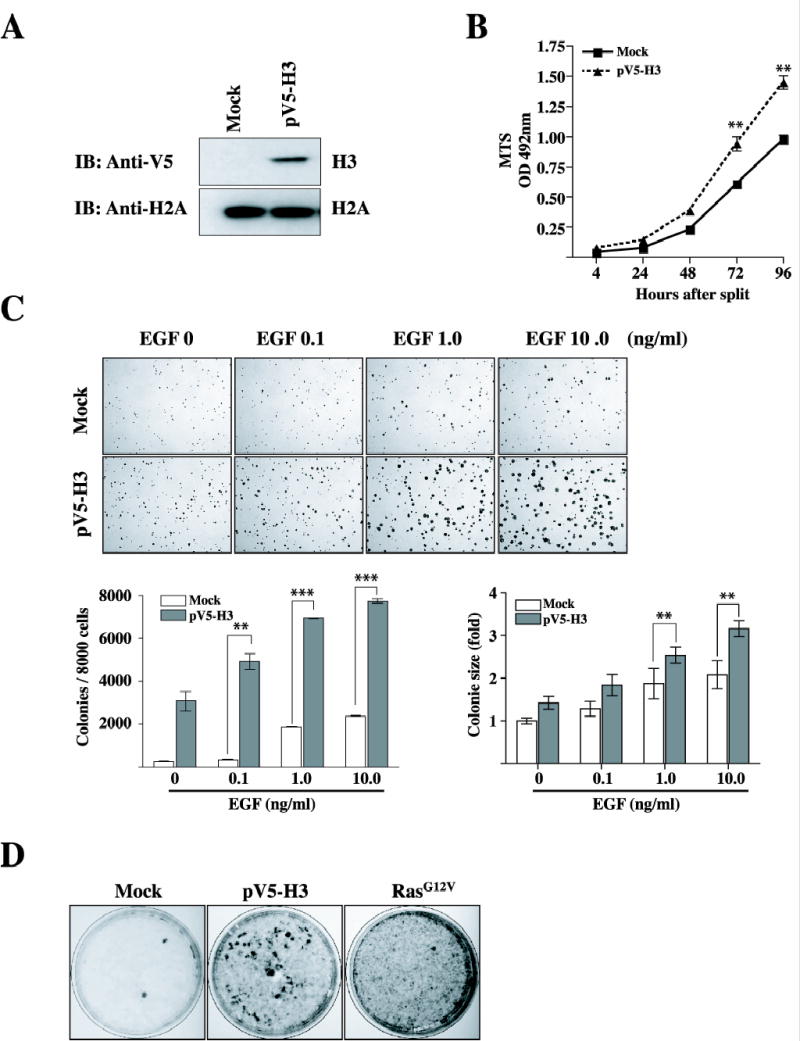
EGF-induced neoplastic cell transformation is increased in cells overexpressing wildtype histone H3. (A) The JB6 Cl41 cells were transfected with mock or pV5-H3, selected with 600 μg/ml G418 for 10 days, and then pooled. pV5-H3 expression was detected with an antibody against the V5 epitope. For visualizing equal loading of protein, total histone H2A was detected by western blotting. Mock or pV5-H3 denotes the pcDNA3.1/V5 transfected control cell line or pcDNA3.1/V5-H3, respectively. IB: immunoblotting. (B) Cells stably transfected with the mock vector or pV5-H3 were seeded (1 × 103/wells) in 96-well plates in 100 μl of 5% FBS-MEM, and cell proliferation was estimated using the CellTiter 96 Aqueous One Solution detection kit (Promega). Cell proliferation was estimated by absorbance (A492) at 24-h intervals up to 96 h. Each bar indicates the mean ± SD of values obtained from triplicate experiments. Significant differences were evaluated using the Student’s t test (**: p < 0.005). (C) Mock or pV5-H3 transfected cells were subjected to a soft agar assay in the presence of EGF. Cells (8 × 103/ml) were exposed to EGF (0, 0.1, 1, or 10 ng/ml) in 1ml of 0.3 basal medium Eagle agar containing 10% FBS. The culture was maintained at 37 °C in a 5% CO2 atmosphere for 10 days. The average colony number was calculated and photographed from three separate experiments (bottom, left panel). Colony sizes were using Image J software program and compared between mock cells and pV5-H3 cells stimulated by EGF (bottom, right panel). Significant differences were evaluated using the Student’s t test (**: P < 0.005; ***: p < 0.001); significant increase in EGF-induced cell transformation in pV5-H3 cells compared to mock cells. (D) Swiss 3T3 cells were transiently transfected with pV5-H3 (2.5 μg) or H-RasG12V (0.1 μg) as described in “Materials and Methods.” Cells transfected with the vector (pcDNA3.1) were served as negative controls.
Motif of Histone H3 Phosphorylation Responsible for Neoplastic Cell Transformation Promoted by EGF.
Serines 10 and 28 on the histone H3 tail are each proceeded by the same three amino acids (alanine-argininelysine) and both of these phosphorylatable motifs are very highly conserved, being identical in yeast and human (29). Two distinct modes of histone H3 phosphorylation have been characterized, mitotic and stimulus-inducible phosphorylation, both of which occur at these two serine residues. Furthermore, MAP kinase-mediated phosphorylation of serines 10 and 28 has also been demonstrated upon stimulation with EGF (28), UVB (30, 31), TPA or anisomycin (32). To study whether the histone H3 phosphorylatable motif at Ser10 or Ser28 specifically induces cell transformation promoted by EGF, we replaced Ser10 of histone H3 with alanine (S10A), Ser28 with alanine (S28A) or both Ser10 and Ser28 with alanine (S10/28A), and then subcloned these constructs into the pcDNA3.1/V5-His vector (Fig. 3A). These plasmids were then introduced into JB6 Cl41 cells. Expression of constructs was confirmed with an anti-V5 antibody against V5-histone H3 (Fig. 3B). Stable transfectant H3 WT or mutants, S10A, S28A, or S10/28A, were then employed to determine their susceptibility to cell transformation promoted by EGF. Cells were treated with EGF (1 ng/ml) in a soft agar matrix and incubated at 37 °C in a 5% CO2 incubator for 10 days. Results indicated that colony numbers promoted by EGF were greatly decreased in S10A, but not S28A, mutant JB6 Cl41 cells compared with cell transformation in JB6 Cl41 cells overexpressing wildtype histone H3 (Fig. 3C). These data indicate that serine 10 of histone H3 is most likely a critical site for mediating neoplastic cell transformation promoted by EGF.
Figure 3.
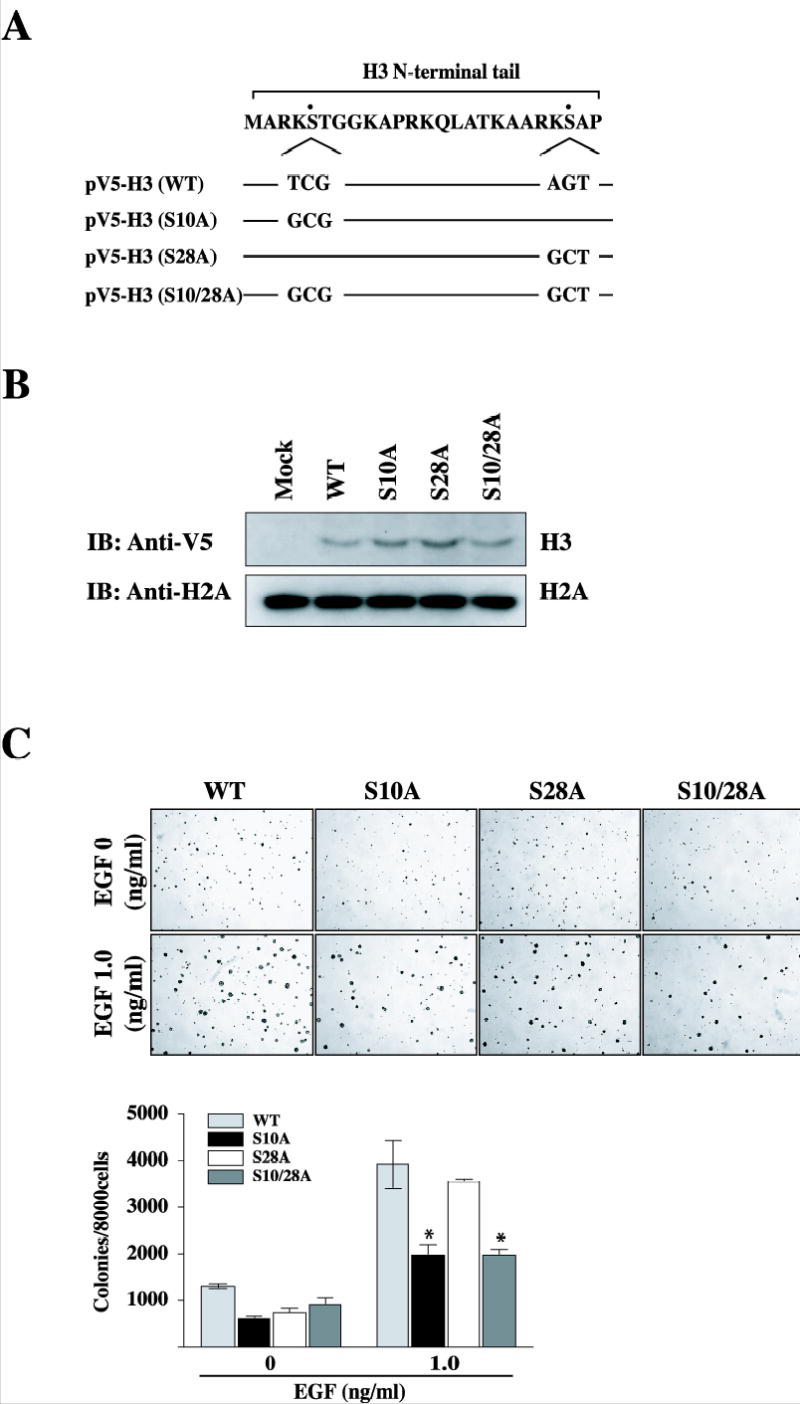
Mutants of histone H3 (S10A) suppress neoplastic cell transformation promoted by EGF. (A) Schematics of histone H3 mutation of Ser10 to alanine (S10A), mutation of Ser28 to alanine (S28A), and both mutations of Ser10 and Ser28 (S10/28A). (B) The JB6 Cl41 cells were transfected with mock, pV5-H3, pV5-H3 S10A, S28A, or S10/28A, selected with 600 μg/ml G418 for 10 days, and then pooled. pV5-H3 WT, S10A, S28A, and S10/28A expression were detected with an antibody against the V5 epitope. For visualizing equal loading of protein, total histone H2A was detected by western blotting. Mock denotes the pcDNA3.1/V5 transfected control cell line. IB: immunoblotting. (C) pV5-H3, pV5-H3 S10A, pV5-H3 S28A, or pV5-H3 S10/28A transfected cells were subjected to a soft agar assay in the presence of EGF. Cells (8 × 103/ml) were exposed to EGF (1 ng/ml) in 1 ml of 0.3 basal medium Eagle agar containing 10% FBS. The culture was maintained at 37 °C in a 5% CO2 atmosphere for 10 days. The average colony number was photographed and calculated from three separate experiments. Significant differences were evaluated using the Student’s t test (*: p < 0.05); significant decrease in EGF-induced cell transformation in pV5-H3 S10A or S10/28A cells compared to pV5-H3 cells.
Induction of the c-jun and c-fos Promoter is Critical for Neoplastic Cell Transformation Elicited by H3.
c-Jun and c-Fos are nuclear proto-oncoproteins whose expression is stimulated by a variety of growth-promoting agents and activated oncogenes (9). Phospho-acetylated histone H3 occurs on nucleosomes associated with the active, but not the inactive, c-fos and c-jun proto-oncogene promoters under physiological conditions using EGF as a stimulus (33). To investigate whether cell transformation elicited by overexpression of H3 results from direct stimulation of the c-jun or c-fos promoter, we took advantage of the availability of the reporter plasmid carrying the luc gene under the control of the murine c-jun or c-fos promoter (34, 35). Twenty-four h after transfection with these reporters in Mock or H3 wildtype overexpressing JB6 Cl41 cells, cells were starved for another 24 h by incubating in serum-deprived MEM at 37 °C in a 5% CO2 atmosphere. Cells were then treated or not treated with EGF (1, 10, 30, 50, and 100 ng/ml). As shown in Fig. 4, A and B, EGF-promoted c-jun or c-fos transcriptional activity was significantly induced in JB6 Cl41 cells overexpressing wildtype histone H3 (H3 WT) compared with cells expressing pcDNA3.1/V5 (Mock). These data support our notion that induction of the c-jun or c-fos promoter by EGF is one of the mechanisms explaining the increased anchorage-independent growth of JB6 Cl41 cells overexpressing wildtype histone H3. In contrast, the histone H3 mutant, S10A (H3 S10A), but not S28A (H3 S28A), suppressed EGF-promoted c-jun or c-fos gene induction, compared with H3 WT cells (Fig. 4C). Cotransfection of the reporters with the histone H3 cDNA in JB6 Cl41 cells revealed that histone H3 can strongly induce the activity of the c-jun or c-fos promoter (Fig. 4D). These data provide evidence that the induction of the c-jun and c-fos genes is involved in neoplastic cell transformation induced by overexpression of the wildtype histone H3 protein.
Figure 4.
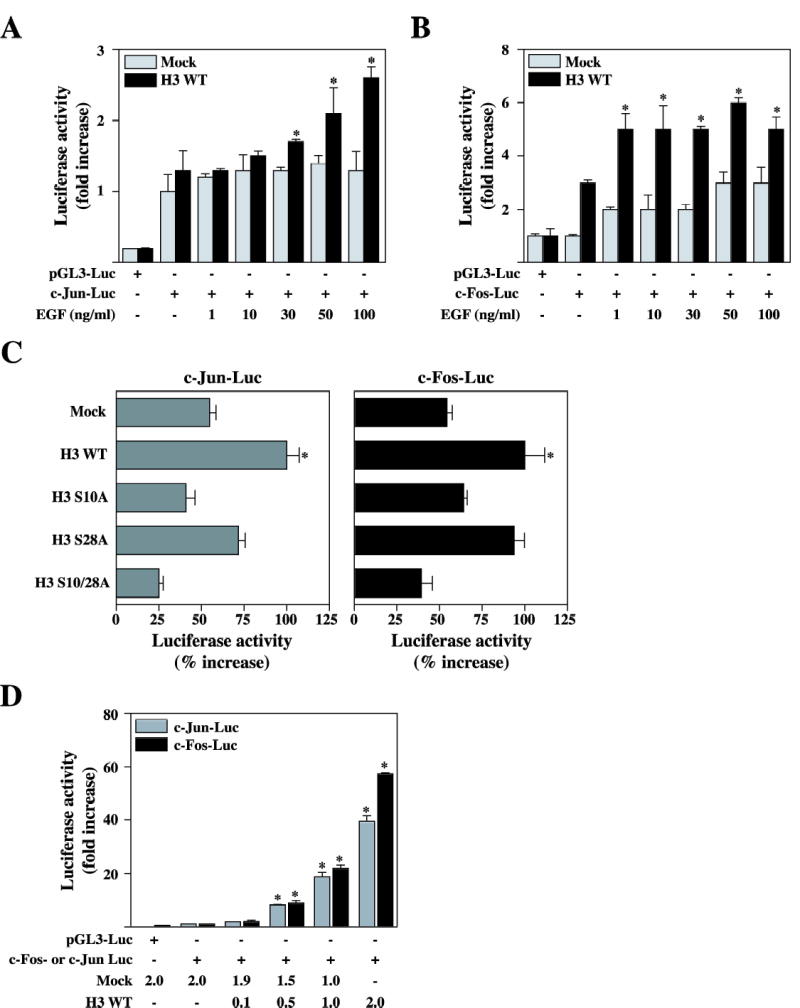
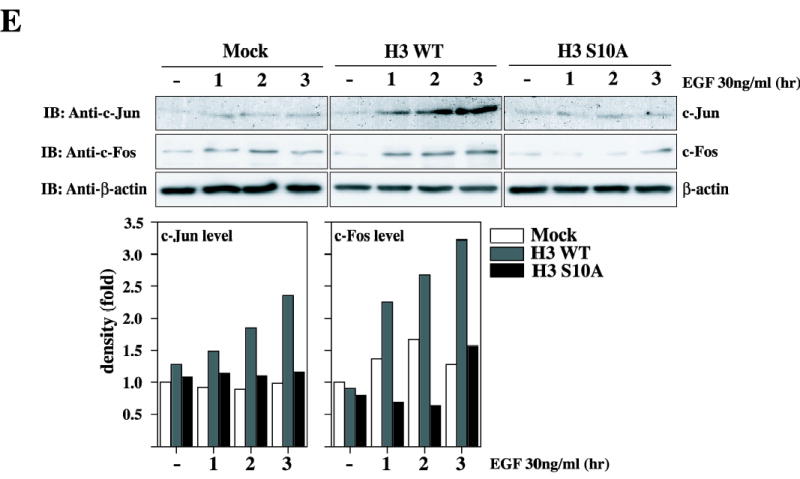
Histone H3 upregulates c-fos and c-jun promoter activity, and increase endogeneous c-Jun and c-Fos protein level. (A and B) Mock and pV5-H3 WT stably transfected JB6 Cl41 cells were transfected with a plasmid mixture containing the c-fos-luciferase reporter gene (0.5 μg, A) or the c-jun-luciferase reporter gene (0.5 μg, B), each with the pRL-SV40 gene (0.1 μg). At 24 h post-transfection, cells were starved for 24 h by incubating in serum-deprived MEM at 37 °C in a 5% CO2 atmosphere, and then incubated in the presence or absence of EGF (1, 10, 30, 50, and 100 ng/ml) for 3h. The firefly luciferase activity was determined in cell lysates and normalized against renilla luciferase activity. (C) Mock, pV5-H3 WT, S10A, S28A, and S10/28A JB6 Cl41 cells were transfected with a plasmid mixture containing the c-fos-luciferase reporter gene (0.5 μg, left panel) or the c-jun-luciferase reporter gene (0.5 μg, right panel), each with the pRL-SV40 gene (0.1 μg). At 24 h post-transfection, cells were starved for 24 h by incubating in serum-deprived MEM at 37 °C in a 5% CO2 atmosphere, and then incubated in the presence or absence of 30 ng/ml EGF for 3 h. The firefly luciferase activity was determined in cell lysates and normalized against renilla luciferase activity. (D) JB6 Cl41 cells were co-transfected with a plasmid mixture containing the c-fos-luciferase reporter gene (0.5 μg) or the c-jun-luciferase reporter gene (0.5 μg), each with the pV5-H3 WT (0.1, 0.5, 1.0, and 2.0 μg) or mock (1.0–2.0 μg) with pRL-SV40 gene (0.1 μg). At 36 h post-transfection, the firefly luciferase activity was determined in cell lysates and normalized against renilla luciferase activity. All experiments were performed at least twice with triplicate samples and depicted as means ± S.E. Data were recorded as relative luciferase activity (fold or %) using a Luminoskan Ascent (Thermo Electron Corp., Helsinki, Finland). Significant differences were evaluated using the Student’s t test (*: p < 0.05); significant increase in EGF-induced c-Jun or c-Fos activity in pV5-H3 cells compared to mock cells. Mock denotes the pcDNA3.1/V5 transfected control cell line. (E) Mock, H3WT, or H3 S10A transfected cells were analyzed to detect an endogenous c-Jun or c-Fos protein levels in the presence of EGF. Cells were starved for 24 hr by incubating in serum-deprived MEM at 37°C in a 5% CO2. atmosphere, and then incubated in the presence or absence of EGF 30 ng/ml for in a time-dependent way. Protein levels of c-Jun or c-Fos in H3 WT cells or H3 S10A cells was quantitated compared to Mock cells.
We next decided to assess the effect of histone H3 on the expression of the endogenous c-Jun or c-Fos proteins. For these experiments, mock, H3 WT, or H3 S10A cells were treated with EGF (30 ng/ml) in a time-dependent way (1, 2, or 3 hr) and then the endogenous c-Jun or c-Fos protein level was analyzed by immunoblotting using a specific antibody against c-Jun or c-Fos, respectively. As shown in Fig. 4E, only H3 WT (middle panels), but not H3 S10A cells, dramatically increased endogenous c-Jun and c-Fos protein levels induced by EGF compared with mock cells. Taken together, these data therefore indicate that histone H3 can regulate the expression of c-Jun and c-Fos, and in turn, that c-Jun and c-Fos represents potential candidates to mediate the neoplastic cell transformation elicited by histone H3.
AP-1 Activity is Enhanced in H3 Overexpressing Cells.
The AP-1 transcription factor is a dimeric complex that comprises members of the JUN, FOS, ATF (activating transcription factor) and MAF (musculoaponeurotic fibro sarcoma) protein families (36). The regulation of cell proliferation by AP-1 might be of crucial importance for the multi-stage development of tumors (37, 38). AP-1 is induced by several external stimuli, such as EGF, that increase MAPK activity (39). To determine whether the overexpression of wildtype histone H3 in JB6 Cl41 cells is responsible for the AP-1 activation response to EGF, we next co-transfected the AP-1 luciferase reporter plasmid and the pRL-SV40 gene into wildtype histone H3 overexpressing or control JB6 Cl41 cells. At 24 h post-transfection, cells were starved for an additional 24 h by incubating in serum-deprived MEM at 37 °C in a 5% CO2 atmosphere. They were then treated or not treated with EGF (1, 10, 30, 50, or 100 ng/ml). The EGF-induced AP-1 activation response in JB6 Cl41 cells was much more pronounced in wildtype histone H3 overexpressing cells (pV5-H3 WT) than mock vector control cells (Mock) (Fig. 5A). In contrast, no significant gains of AP-1 transactivation were observed in histone H3 mutant S10A (pV5-H3 S10A) or S10/28A (pV5-H3 S10/28A) overexpressing JB6 Cl41 cells (Fig. 5B). When wildtype histone H3 was transiently overexpressed in JB6 Cl41 cells, AP-1 luciferase activity was increased in a dose-dependent way by overexpression of wildtype histone H3 (pV5-H3 WT), similar to that observed for c-jun or c-fos activity (Fig. 5C).
Figure 5.
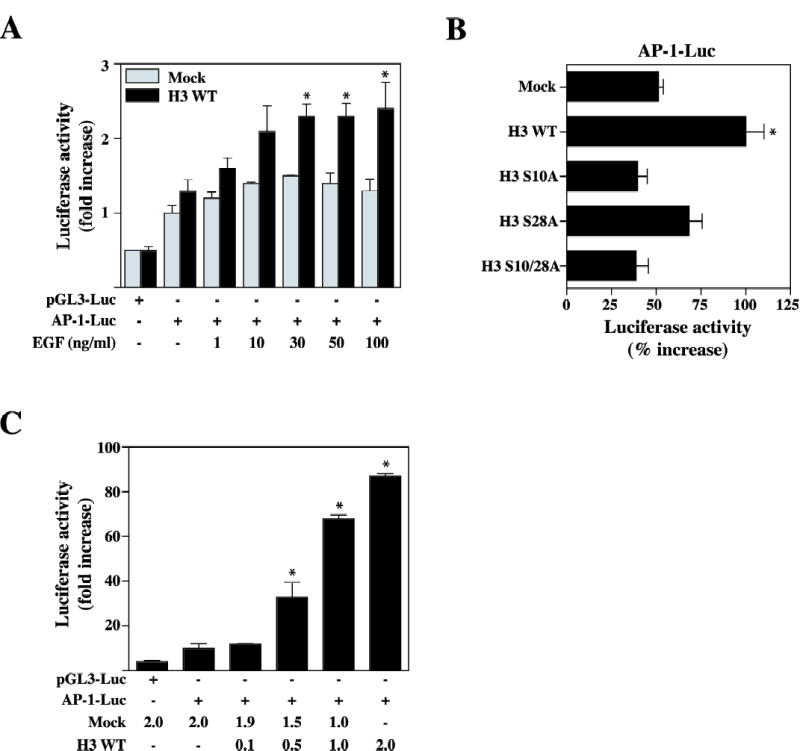
Wildtype (WT) Histone H3 is associated with AP-1 transcriptional activation. (A) Mock and pV5-H3 WT stably transfected JB6 Cl41 cells were co-transfected with a plasmid mixture containing the AP-1 luciferase reporter gene (1 μg) with the pRL-SV40 gene (0.1 μg). At 24 h post-transfection, cells were starved for 24 h by incubating in serum-deprived MEM at 37 °C in a 5% CO2 atmosphere, and then incubated in the presence or absence of EGF (1, 10, 30, 50, and 100 ng/ml) for 3 h. The firefly luciferase activity was determined in cell lysates and normalized against renilla luciferase activity. (B) Mock, pV5-H3 WT, S10A, S28A, and S10/28A JB6 Cl41 cells were co-transfected with a plasmid mixture containing the AP-1 luciferase reporter gene (1 μg) with the pRL-SV40 gene (0.1 μg). At 24 h post-transfection, cells were starved for 24 h by incubating in serum-deprived MEM at 37 °C in a 5% CO2 atmosphere, and then incubated in the presence or absence of EGF (1, 10, 30, 50, and 100 ng/ml) for 3 h. The firefly luciferase activity was determined in cell lysates and normalized against renilla luciferase activity. (C) JB6 Cl41 cells were transfected with a plasmid mixture containing the AP-1 luciferase reporter gene (1 μg) with the pV5-H3 (0.1, 0.5, 1.0, and 2.0 μg) or mock (1.0–2.0 μg) with pRL-SV40 gene (0.1 μg). At 36 h post-transfection, the firefly luciferase activity was determined in cell lysates and normalized against renilla luciferase activity. Data were recorded as relative luciferase activity (fold or %) using a Luminoskan Ascent. All experiments were performed at least twice with triplicate samples and depicted as means ± S.E. Significant differences were evaluated using the Student’s t test (*: p < 0.05); significant increase in EGF-induced AP-1 activity in pV5-H3 WT cells compared to mock cells.
A Knockdown of c-Jun and c-Fos Inhibits H3-Induced Cell Transformation.
To evaluate the function of c-Jun and c-Fos in neoplastic cell transformation induced by overexpression of histone H3, we tested whether siRNA-mediated knockdown of c-Jun or c-Fos in pV5-H3 WT cells would inhibit the cell transforming activity. Immunoblot analysis of c-Jun or c-Fos siRNA- transfected pV5-H3 cells revealed that a maximal protein knockdown of up to 90% or 70%, respectively, was induced by c-Jun- or c-Fos-specific siRNA but had no effect on β-actin protein expression (Fig. 6, A and B). We next examined whether knockdown of c-Jun or c-Fos affected the ability of histone H3 to form colonies in soft agar. Mock or pV5-H3 WT cells were treated with 100 pmol each of control siRNA, c-Jun-, or c-Fos-specific siRNA and plated in soft agar with/without EGF. Cells were incubated at 37 °C in a 5% CO2 incubator for only 4 days. The overexpression of histone H3 caused a dramatic increase in cell transformation with addition of EGF (Fig. 6C). In contrast, both c-Jun- and c-Fos-specific siRNAs almost totally inhibited the number of colonies formed in the H3-enhanced cell transformation by EGF (Fig. 6C). Finally, to determine whether c-Jun- or c-Fos specific siRNA inhibits cell transformation in the histone overexpressing cells through the AP-1 transactivation activity, we treated cells with 100 pmol each of control siRNA, c-Jun-, or c-Fos-specific siRNA. After 24h treatment of siRNAs, we transfected AP-1-luciferase reporter plasmind in the mock or pV5-H3 cells and used these transfectants for analysis of AP-1 transactivation activities. At 24 h post-transfection, cells were starved for an additional 24 h by incubating in serum-deprived MEM at 37 °C in a 5% CO2 atmosphere. They were then treated or not treated with EGF (30 ng/ml) for 3 hr. The EGF-induced AP-1 activation response in JB6 Cl41 cells was much more pronounced in wildtype histone H3 overexpressing cells (pV5-H3 WT) than mock vector control cells (Mock) (Fig. 6D). In contrast, no significant gains of AP-1 transactivation were observed in c-Jun- or c-Fos-siRNA transfected pV5-H3 cells (Fig. 6D).
Figure 6.
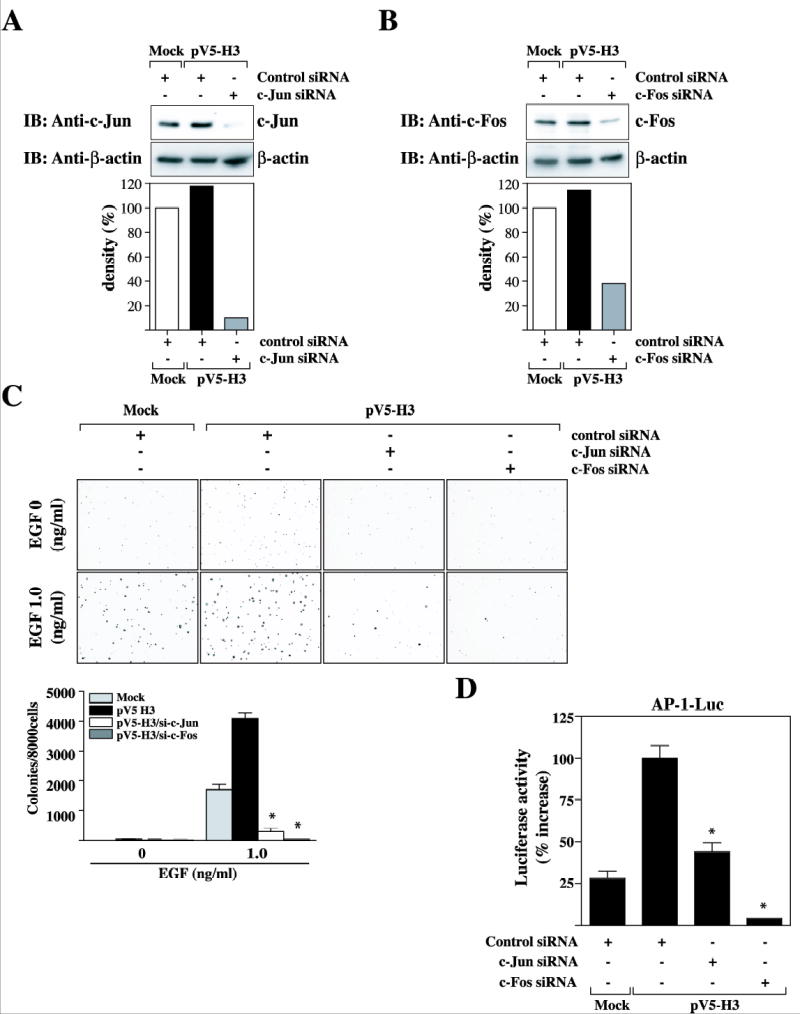
Knockdown of c-Jun or c-Fos expression in H3 overexpressing cells inhibited neoplastic cell transformation and AP-1 transactivation activity elicited by H3. (A and B) Mock or pV5-H3 cells were transfected with control siRNA, c-Jun siRNA, or c-Fos siRNA duplex. After 48 hr siRNA treatment, the expression of c-Jun or c-Fos was analyzed with a specific antibody against c-Jun or c-Fos, respectively. Knockdown of c-Jun or c-Fos in pV5-H3/si-c-Jun or pV5-H3/si-c-Fos cells was quantitated compared to mock cells. (C) Control siRNA, c-Jun-, or c-Fos-specific siRNA-treated Mock or pV5-H3 cells, pV5-H3/si-c-Jun, or pV5-H3/si-c-Fos cells were used to assess cell transformation in a soft agar assay. Cells (8 × 103 cells/ml) treated with control siRNA, c-Jun-siRNA, or c-Fos-siRNA were exposed to EGF (0 or 1 ng/ml) in 1 ml of 0.3 basal medium Eagle agar containing 10% FBS. The culture was maintained at 37°C in a 0.5% CO2 atmosphere for only 4 days and then colonies were counted automatically. The average colony number was calculated and photographed from three separate experiments. Significant differences were evaluated using the student’s t test (**: p < 0.05); significant decrease in EGF-induced cell transformation in pV5-H3/si-c-Jun or pV5-H3/si-c-Fos cells compared to pV5-H3 cells. (D) Control siRNA, c-Jun-, or c-Fos-specific siRNA-treated Mock, pV5-H3 WT, S10A, S28A, and S10/28A JB6 Cl41 cells were co-transfected with a plasmid mixture containing the AP-1 luciferase reporter gene (1 μg) with the pRL-SV40 gene (0.1 μg). At 24 h post-transfection, cells were starved for 24 h by incubating in serum-deprived MEM at 37 °C in a 5% CO2 atmosphere, and then incubated in the presence or absence of EGF (30 ng/ml) for 3 h. The firefly luciferase activity was determined in cell lysates and normalized against renilla luciferase activity.
Discussion
Mitotic phosphorylation of histone H3 at Ser10 is critical for proper chromosome condensation and segregation (40). The phosphorylation status of histone H3 at Ser10 in turn influences methylation at lysine 9 of histone H3 by the SUV39H1 methylase (41). Methylation of histone H3 at lysine 9 by SUV39H1 has also been implicated in recruiting heterochromatin protein 1 to heterochromatin sites, where it mediates gene silencing (42–44). This observation has led us to believe that chromatin-based regulatory mechanisms may be involved in epigenetic alteration of cancer cells, leading to tumor establishment and progression. However, the intriguing question is whether the modification in the amino-terminal tails of histone H3 directly affects neoplastic cell transformation.
In this study, we found that overexpression of histone H3 induced neoplastic cell transformation and cell proliferation in JB6 Cl41 cells. In contrast, knockdown of histone H3 expression using the siRNA technique suppressed cell transformation and cell proliferation. Surprisingly, mutation of serine 10 of histone H3 with alanine suppressed cell transformation promoted by EGF. Numerous oncogenes act by mimicking normal growth signaling and many of the cell surface receptors that transduce growth stimulatory signals into the cell interior are themselves targets of deregulation during tumor pathogenesis. For example, the EGF receptor is upregulated in stomach, brain, and breast tumors (45) and the SOS-Ras-Raf-MAPK cascade plays a central role in tumorigenesis stimulated by growth factors, such as EGF (46). In about 25% of human tumors, Ras proteins are present in structurally altered forms that enable them to release a flux of mitogenic signals into cells, without stimulation by their normal upstream regulators (47). Constitutive activation of the Ras-MAPK signaling pathway in mouse fibroblasts transformed with oncogenes (e.g., H-ras) elevates the level of phosphorylated H3 (6). The phosphorylation of histone H3 at serine 10 is essential for maintenance of proper chromosome dynamics during mitosis (40). As indicated earlier, the elevation of phosphorylated H3 (Ser10) levels may contribute to the aberrant gene expression observed in the oncogene-transformed cells, resulting from persistent activation of the Ras-MAPK pathway and MSK1 (48). Increased mitotic H3 (Ser10) phosphorylation was also observed in various colorectal tumor cells with high AIM-1 expression levels (19). AIM-1, a mammalian lpl1/aurora kinase involved in H3 phosphorylation, is overexpressed in many tumor cell lines, including human colorectal tumors of advanced grade and stage. This accumulated evidence suggested that the post-translational modification of histone H3 at the N-terminal tail might not only regulate gene expression, chromosomal condensation and mitosis, but also have oncogenic effects. These previous studies strongly supported our finding that histone H3, especially the serine 10 motif, might have an important role in carcinogenesis, resulting from the EGF-induced activation of MSK1 and the Ras-Raf-MAPK pathway.
For signaling pathway-activated genes such as c-fos and c-jun, the coupling of histone phosphorylation at specific loci may contribute to mechanisms that allow them to be rapidly activated in response to external stimuli (28). Overexpression of the Fos and Jun proteins was found to correlate with a positive effect on cell transformation (33). c-Jun is believed to be primarily a positive regulator of cell proliferation because c-Jun-deficient fibroblasts have a marked proliferation defect in vitro (49). To fully promote cell-cycle progression, the c-Jun protein is activated by c-Jun amino-terminal kinases (JNKs) (50). Subsequently, the activated c-Jun-containing AP-1 complex induces the transcription of positive regulators of cell-cycle progression, including cyclin D1, or represses negative regulators, including the tumor suppressor p53 and the cyclin-dependent kinase INK4A (36). c-Fos might have a more important role than c-Jun during late-stage tumorigenesis (51). The importance of c-Fos in tumor invasion has been supported in vivo, as the progression of chemically induced papillomas to invasive squamous-cell carcinomas is impaired in c-Fos-deficient mice (52).
In this study, even though many factors have been shown to interact with and activate histone H3, we found that the c-fos or c-jun gene is a common target of histone H3 leading to induction of AP-1 activity, which may include its transactivating potential, DNA-binding capacity and the stability of AP-1 components. The phosphorylation of the ternary complex factor (TCF) by the Ras-Raf-MEK-ERK signal cascade (12) and the stimulation of the activity of the c-fos SRE by histone H3 phosphorylation (Ser10) (28) might promote c-Fos expression, and thereby stabilize the c-Fos/c-Jun heterodimer leading to expression of AP-1-regulated genes (53). By linking histone H3 phosphorylation to AP-1 transactivating potential, these finding provide an attractive explanation of how post-translational modification of histone H3 can be related to neoplastic cell transformation. Thus these results suggest that histone H3 may be a crucial target for cancer chemotherapy or genetic therapy in the future.
Acknowledgments
Grant support: This work was supported in part by The Hormel Foundation and National Institutes of Health grants CA77646 and CA91064
We are grateful to Dr. Ron Prywes (Columbia University, New York) for the c-Jun-Luc (pJC6 CL3) and c-Fos-Luc (c-Fos WT CL3) promoter.
References
- 1.Gurley LR, D'Anna JA, Barham SS, Deaven LL, Tobey RA. Histone phosphorylation and chromatin structure during mitosis in Chinese hamster cells. Eur J Biochem. 1978;84:1–15. doi: 10.1111/j.1432-1033.1978.tb12135.x. [DOI] [PubMed] [Google Scholar]
- 2.Wei Y, Mizzen CA, Cook RG, Gorovsky MA, Allis CD. Phosphorylation of histone H3 at serine 10 is correlated with chromosome condensation during mitosis and meiosis in Tetrahymena. Proc Natl Acad Sci U S A. 1998;95:7480–4. doi: 10.1073/pnas.95.13.7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goto H, Tomono Y, Ajiro K, et al. Identification of a novel phosphorylation site on histone H3 coupled with mitotic chromosome condensation. J Biol Chem. 1999;274:25543–9. doi: 10.1074/jbc.274.36.25543. [DOI] [PubMed] [Google Scholar]
- 4.Hendzel MJ, Wei Y, Mancini MA, et al. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997;106:348–60. doi: 10.1007/s004120050256. [DOI] [PubMed] [Google Scholar]
- 5.Van Hooser A, Goodrich DW, Allis CD, Brinkley BR, Mancini MA. Histone H3 phosphorylation is required for the initiation, but not maintenance, of mammalian chromosome condensation. J Cell Sci. 1998;111 ( Pt 23):3497–506. doi: 10.1242/jcs.111.23.3497. [DOI] [PubMed] [Google Scholar]
- 6.Chadee DN, Hendzel MJ, Tylipski CP, et al. Increased Ser-10 phosphorylation of histone H3 in mitogen-stimulated and oncogene-transformed mouse fibroblasts. J Biol Chem. 1999;274:24914–20. doi: 10.1074/jbc.274.35.24914. [DOI] [PubMed] [Google Scholar]
- 7.Mahadevan LC, Willis AC, Barratt MJ. Rapid histone H3 phosphorylation in response to growth factors, phorbol esters, okadaic acid, and protein synthesis inhibitors. Cell. 1991;65:775–83. doi: 10.1016/0092-8674(91)90385-c. [DOI] [PubMed] [Google Scholar]
- 8.Thomson S, Clayton AL, Hazzalin CA, Rose S, Barratt MJ, Mahadevan LC. The nucleosomal response associated with immediate-early gene induction is mediated via alternative MAP kinase cascades: MSK1 as a potential histone H3/HMG-14 kinase. Embo J. 1999;18:4779–93. doi: 10.1093/emboj/18.17.4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herschman HR. Primary response genes induced by growth factors and tumor promoters. Annu Rev Biochem. 1991;60:281–319. doi: 10.1146/annurev.bi.60.070191.001433. [DOI] [PubMed] [Google Scholar]
- 10.McMahon SB, Monroe JG. Role of primary response genes in generating cellular responses to growth factors. Faseb J. 1992;6:2707–15. doi: 10.1096/fasebj.6.9.1612295. [DOI] [PubMed] [Google Scholar]
- 11.Hunter T, Karin M. The regulation of transcription by phosphorylation. Cell. 1992;70:375–87. doi: 10.1016/0092-8674(92)90162-6. [DOI] [PubMed] [Google Scholar]
- 12.Sachsenmaier C, Radler-Pohl A, Zinck R, Nordheim A, Herrlich P, Rahmsdorf HJ. Involvement of growth factor receptors in the mammalian UVC response. Cell. 1994;78:963–72. doi: 10.1016/0092-8674(94)90272-0. [DOI] [PubMed] [Google Scholar]
- 13.Ben-Ari ET, Bernstein LR, Colburn NH. Differential c-jun expression in response to tumor promoters in JB6 cells sensitive or resistant to neoplastic transformation. Mol Carcinog. 1992;5:62–74. doi: 10.1002/mc.2940050111. [DOI] [PubMed] [Google Scholar]
- 14.Dong Z, Birrer MJ, Watts RG, Matrisian LM, Colburn NH. Blocking of tumor promoter-induced AP-1 activity inhibits induced transformation in JB6 mouse epidermal cells. Proc Natl Acad Sci U S A. 1994;91:609–13. doi: 10.1073/pnas.91.2.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sassone-Corsi P, Mizzen CA, Cheung P, et al. Requirement of Rsk-2 for epidermal growth factor-activated phosphorylation of histone H3. Science. 1999;285:886–91. doi: 10.1126/science.285.5429.886. [DOI] [PubMed] [Google Scholar]
- 16.Hsu JY, Sun ZW, Li X, et al. Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell. 2000;102:279–91. doi: 10.1016/s0092-8674(00)00034-9. [DOI] [PubMed] [Google Scholar]
- 17.Giet R, Glover DM. Drosophila aurora B kinase is required for histone H3 phosphorylation and condensin recruitment during chromosome condensation and to organize the central spindle during cytokinesis. J Cell Biol. 2001;152:669–82. doi: 10.1083/jcb.152.4.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adams RR, Maiato H, Earnshaw WC, Carmena M. Essential roles of Drosophila inner centromere protein (INCENP) and aurora B in histone H3 phosphorylation, metaphase chromosome alignment, kinetochore disjunction, and chromosome segregation. J Cell Biol. 2001;153:865–80. doi: 10.1083/jcb.153.4.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ota T, Suto S, Katayama H, et al. Increased mitotic phosphorylation of histone H3 attributable to AIM-1/Aurora-B overexpression contributes to chromosome number instability. Cancer Res. 2002;62:5168–77. [PubMed] [Google Scholar]
- 20.Graber MW, Schweinfest CW, Reed CE, Papas TS, Baron PL. Isolation of differentially expressed genes in carcinoma of the esophagus. Ann Surg Oncol. 1996;3:192–7. doi: 10.1007/BF02305800. [DOI] [PubMed] [Google Scholar]
- 21.Yu JY, DeRuiter SL, Turner DL. RNA interference by expression of short-interfering RNAs and hairpin RNAs in mammalian cells. Proc Natl Acad Sci U S A. 2002;99:6047–52. doi: 10.1073/pnas.092143499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Colburn NH, Wendel EJ, Abruzzo G. Dissociation of mitogenesis and late-stage promotion of tumor cell phenotype by phorbol esters: mitogen-resistant variants are sensitive to promotion. Proc Natl Acad Sci U S A. 1981;78:6912–6. doi: 10.1073/pnas.78.11.6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clark GJ, Cox AD, Graham SM, Der CJ. Biological assays for Ras transformation. Methods Enzymol. 1995;255:395–412. doi: 10.1016/s0076-6879(95)55042-9. [DOI] [PubMed] [Google Scholar]
- 24.Huang C, Ma WY, Dong Z. Requirement for phosphatidylinositol 3-kinase in epidermal growth factor-induced AP-1 transactivation and transformation in JB6 P+ cells. Mol Cell Biol. 1996;16:6427–35. doi: 10.1128/mcb.16.11.6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li JJ, Dong Z, Dawson MI, Colburn NH. Inhibition of tumor promoter-induced transformation by retinoids that transrepress AP-1 without transactivating retinoic acid response element. Cancer Res. 1996;56:483–9. [PubMed] [Google Scholar]
- 26.Dong Z, Cmarik JL. Harvesting cells under anchorage-independent cell transformation conditions for biochemical analyses. Sci STKE. 2002. p. PL7. [DOI] [PubMed]
- 27.Carpenter G, Cohen S. Epidermal growth factor. Annu Rev Biochem. 1979;48:193–216. doi: 10.1146/annurev.bi.48.070179.001205. [DOI] [PubMed] [Google Scholar]
- 28.Cheung P, Tanner KG, Cheung WL, Sassone-Corsi P, Denu JM, Allis CD. Synergistic coupling of histone H3 phosphorylation and acetylation in response to epidermal growth factor stimulation. Mol Cell. 2000;5:905–15. doi: 10.1016/s1097-2765(00)80256-7. [DOI] [PubMed] [Google Scholar]
- 29.Clayton AL, Mahadevan LC. MAP kinase-mediated phosphoacetylation of histone H3 and inducible gene regulation. FEBS Lett. 2003;546:51–8. doi: 10.1016/s0014-5793(03)00451-4. [DOI] [PubMed] [Google Scholar]
- 30.Zhong S, Jansen C, She QB, et al. Ultraviolet B-induced phosphorylation of histone H3 at serine 28 is mediated by MSK1. J Biol Chem. 2001;276:33213–9. doi: 10.1074/jbc.M103973200. [DOI] [PubMed] [Google Scholar]
- 31.Zhong S, Zhang Y, Jansen C, Goto H, Inagaki M, Dong Z. MAP kinases mediate UVB-induced phosphorylation of histone H3 at serine 28. J Biol Chem. 2001;276:12932–7. doi: 10.1074/jbc.M010931200. [DOI] [PubMed] [Google Scholar]
- 32.Soloaga A, Thomson S, Wiggin GR, et al. MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14. Embo J. 2003;22:2788–97. doi: 10.1093/emboj/cdg273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clayton AL, Rose S, Barratt MJ, Mahadevan LC. Phosphoacetylation of histone H3 on c-fos- and c-jun-associated nucleosomes upon gene activation. Embo J. 2000;19:3714–26. doi: 10.1093/emboj/19.14.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gupta P, Prywes R. ATF1 phosphorylation by the ERK MAPK pathway is required for epidermal growth factor-induced c-jun expression. J Biol Chem. 2002;277:50550–6. doi: 10.1074/jbc.M209799200. [DOI] [PubMed] [Google Scholar]
- 35.Zhu C, Johansen FE, Prywes R. Interaction of ATF6 and serum response factor. Mol Cell Biol. 1997;17:4957–66. doi: 10.1128/mcb.17.9.4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859–68. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- 37.Liu Y, Ludes-Meyers J, Zhang Y, et al. Inhibition of AP-1 transcription factor causes blockade of multiple signal transduction pathways and inhibits breast cancer growth. Oncogene. 2002;21:7680–9. doi: 10.1038/sj.onc.1205883. [DOI] [PubMed] [Google Scholar]
- 38.Park YG, Nesterova M, Agrawal S, Cho-Chung YS. Dual blockade of cyclic AMP response element- (CRE) and AP-1-directed transcription by CRE-transcription factor decoy oligonucleotide. gene-specific inhibition of tumor growth. J Biol Chem. 1999;274:1573–80. doi: 10.1074/jbc.274.3.1573. [DOI] [PubMed] [Google Scholar]
- 39.Tyagi A, Agarwal R, Agarwal C. Grape seed extract inhibits EGF-induced and constitutively active mitogenic signaling but activates JNK in human prostate carcinoma DU145 cells: possible role in antiproliferation and apoptosis. Oncogene. 2003;22:1302–16. doi: 10.1038/sj.onc.1206265. [DOI] [PubMed] [Google Scholar]
- 40.de la Barre AE, Gerson V, Gout S, Creaven M, Allis CD, Dimitrov S. Core histone N-termini play an essential role in mitotic chromosome condensation. Embo J. 2000;19:379–91. doi: 10.1093/emboj/19.3.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rea S, Eisenhaber F, O'Carroll D, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–9. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 42.Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–20. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- 43.Bannister AJ, Zegerman P, Partridge JF, et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–4. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 44.Nielsen SJ, Schneider R, Bauer UM, et al. Rb targets histone H3 methylation and HP1 to promoters. Nature. 2001;412:561–5. doi: 10.1038/35087620. [DOI] [PubMed] [Google Scholar]
- 45.Yarden Y, Ullrich A. Growth factor receptor tyrosine kinases. Annu Rev Biochem. 1988;57:443–78. doi: 10.1146/annurev.bi.57.070188.002303. [DOI] [PubMed] [Google Scholar]
- 46.Clarke N, Arenzana N, Hai T, Minden A, Prywes R. Epidermal growth factor induction of the c-jun promoter by a Rac pathway. Mol Cell Biol. 1998;18:1065–73. doi: 10.1128/mcb.18.2.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neri A, Murphy JP, Cro L, et al. Ras oncogene mutation in multiple myeloma. J Exp Med. 1989;170:1715–25. doi: 10.1084/jem.170.5.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strelkov IS, Davie JR. Ser-10 phosphorylation of histone H3 and immediate early gene expression in oncogene-transformed mouse fibroblasts. Cancer Res. 2002;62:75–8. [PubMed] [Google Scholar]
- 49.Schreiber M, Kolbus A, Piu F, et al. Control of cell cycle progression by c-Jun is p53 dependent. Genes Dev. 1999;13:607–19. doi: 10.1101/gad.13.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Behrens A, Sibilia M, Wagner EF. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat Genet. 1999;21:326–9. doi: 10.1038/6854. [DOI] [PubMed] [Google Scholar]
- 51.Reichmann E, Schwarz H, Deiner EM, et al. Activation of an inducible c-FosER fusion protein causes loss of epithelial polarity and triggers epithelial-fibroblastoid cell conversion. Cell. 1992;71:1103–16. doi: 10.1016/s0092-8674(05)80060-1. [DOI] [PubMed] [Google Scholar]
- 52.Saez E, Rutberg SE, Mueller E, et al. c-fos is required for malignant progression of skin tumors. Cell. 1995;82:721–32. doi: 10.1016/0092-8674(95)90469-7. [DOI] [PubMed] [Google Scholar]
- 53.Chiu R, Boyle WJ, Meek J, Smeal T, Hunter T, Karin M. The c-Fos protein interacts with c-Jun/AP-1 to stimulate transcription of AP-1 responsive genes. Cell. 1988;54:541–52. doi: 10.1016/0092-8674(88)90076-1. [DOI] [PubMed] [Google Scholar]