

Abstract
Rett syndrome (RTT) is associated with mutations in the transcriptional repressor gene MeCP2. Although the clinical and neuropathological signs of RTT suggest disrupted synaptic function, the specific role of MeCP2 in postmitotic neurons remains relatively unknown. We examined whether MeCP2 deficiency in central neurons contributes to the neuropathogenesis in RTT. Primary cerebellar granule neuronal cultures from wildtype (WT) and MeCP2−/− mice were exposed to NMDA and AMPA-induced excitotoxicity and hypoxic-ischemic insult. The magnitude of cell death in MeCP2−/− cells after excitotoxicity and hypoxia was greater than in the WT littermate control cultures and occurred after shorter exposures that usually, in the WT, would not cause cell death. Pretreatment with the growth factor fibroblast growth factor 1 (FGF-1) under conditions at which WT cells showed complete neuroprotection, only partially protected MeCP2−/−cells. To elucidate specifically the effects of MeCP2 knockout (KO) on cell death, we examined two death cascade pathways. MeCP2−/− neurons exposed to 6 h of hypoxia exhibited enhanced activation of the proapoptotic caspase-3 and increased mitochondrial release of AIF compared to WT neurons, which did not show significant changes. However, pretreatment with the caspase inhibitor ZVAD-FMK had little or no effect on AIF release and its subcellular translocation to the nucleus, suggesting caspase-independent AIF release and their independent contribution to hypoxia-induced cell death. Reintroduction of intact MeCP2 gene in MeCP2−/− cells or MeCP2 gene silencing by MeCP2siRNA in WT cells further confirmed the differential sensitivity of the WT and MeCP2−/− cells and suggest a direct role of MeCP2 in cell death. These results clearly demonstrate increased cell death occurred in neurons lacking MeCP2 expression via both caspase- and AIF-dependent apoptotic mechanisms. Our findings suggest a novel, yet unknown, role for MeCP2 in central neurons in the control of neuronal response to cell death.
Keywords: apoptosis, apoptosis inducing factor, cell death, cerebellar granule neurons, hypoxia, excitotoxicity, caspase-3, MeCP2 knockout
Introduction
Rett Syndrome (RTT) is a progressive neurodevelopmental disorder in females caused by loss-of-function mutations in the X-linked MeCP2 gene (Amir et al., 1999). RTT is characterized by arrested neurological development and subsequent cognitive decline, motor impairments and frequent seizures (Hagberg et al., 1983, Naidu, 1997). Examination of the RTT brain reveals profound microencephaly due, at least in part, to smaller and more densely packed neurons. Other abnormalities include a reduction in dendritic arborization (Kriaucionis and Bird, 2003, Zoghbi, 2003). Several studies have shown that MeCP2 is abundantly expressed within the CNS almost exclusively in neurons where its expression is highly enriched in postmitotic neuronal nuclei (Akbarian et al., 2001, Jung et al., 2003, Mullaney et al., 2004). However, recent reports have also localized MeCP2 outside of the nucleus (Mnatzakanian et al., 2004) and in the post-synaptic compartment (Aber et al., 2003), suggesting that MeCP2 may have a role outside of the nucleus including synaptic function (Aber et al., 2003, Nelson et al., 2006). However, there is little understanding of the extranuclear function of MeCP2 in the CNS.
The MeCP2 gene encodes a protein called the methyl-CpG binding protein 2 (MeCP2)-a member of the methyl-binding protein (MBD) family, which is a key factor in epigenetic transcriptional regulation. MeCP2 functions as a transcriptional repressor that specifically binds to methylated “CpG” dinucleotides preferentially located in the promoter region of genes regulated by methylation (Lewis et al., 1992). Once bound to methylated DNA, MeCP2 is reported to suppress downstream gene expression by virtue of its interaction with histone deacetylase/Sin3 complex (Nan et al., 1997, Jones et al., 1998). Although biochemical evidence is consistent with MeCP2 functioning as a transcriptional repressor (Nan et al., 1997), many recent studies have failed to detect significant gene derepression in MeCP2 mutant mouse (Tudor et al., 2002). The strong expression of MeCP2 in mature neurons and the neuronal phenotype of RTT patients raised the possibility that MeCP2 may be involved as a selective regulator of, as yet unknown, neuronal functions.
The synaptic neurotransmitter abnormalities reported in RTT appear to be associated with high levels of glutamate neurotransmission and higher densities of NMDA and AMPA receptors in younger females, suggesting an enhanced excitatory glutamatergic function in the CNS of younger RTT subjects (Lappalainen et al., 1996, Blue et al., 1999). Several reports have indicated that NMDA receptor activation is required for synaptic pruning and that blocking of NMDA receptors prevents this process (Rabacchi et al., 1992, Luthi et al., 2001). Furthermore, the excitatory neurotransmitter glutamate, which has a trophic effect in the developing brain (McDonald and Johnston, 1990, Johnston, 2001, Johnston et al., 2001), can induce neurotoxicity and cell death following prolonged and excessive activation of NMDA and AMPA subtypes of glutamate receptors (Choi, 1988). Previously, we found that in vitro exposure of primary cerebellar granule neurons to NMDA caused significant neuronal death and the growth and neurotrophic factor fibroblast growth factor 1 (FGF-1) protected against NMDA-induced excitotoxicity and death (Hossain et al., 2002). In the present study, we investigated the effects of insufficient expression of the transcriptional repressor MeCP2 on neuronal functions specific to injury and survival in cultured primary cerebellar granule neurons isolated from postnatal day 6 (P6) old wildtype (WT) and MeCP2 knockout mouse brain. The cerebellum is an important area of interest both clinically and anatomically because of its involvement in motor functions, its prolonged period of postnatal development and delayed expression of MeCP2 in granule cells relative to that in purkinje cells (Naidu et al., 2003, Mullaney et al., 2004). Furthermore, the cerebellum appears to be preferentially affected in RTT from the standpoint of both neuropathology and clinical signs of tremor and ataxia (Oldfors et al., 1990, Murakami et al., 1992). Here, we examined whether MeCP2 deficiency (i) directly contributes to the neuronal response to injury/death mechanism triggered by exposure to excitotoxicity and hypoxia, and (ii) alters the response to neuroprotection by growth and neurotrophic factors.
EXPERIMENTAL PROCEDURES
Cerebellar granule neuronal cultures
The Johns Hopkins University Institutional Animal Care and Use Committee approved all animal protocols used; they complied with the US NIH guide for the Care and Use of Laboratory Animals. All efforts were made to minimize the number of animals used and any pain or suffering. Mecp2tm1.1Bird mice (Jackson Laboratory, Bar Harbor, ME, USA) on a C57BL/6 background (heterozygote backcrossed with C57BL/6 males for at least nine generations) were used for all experimental procedures (Metcalf et al., 2006). The MeCP2 null mouse used in this study provides an excellent animal model for human RTT with the potential for understanding its molecular mechanisms (Guy et al., 2001, Kriaucionis and Bird, 2003). Primary cultures of cerebellar granule cells (CGC) were prepared from postnatal day 6 (P6) old WT and MeCP2 null mice cerebella according to methods described previously (Hossain et al., 2002). Cells were resuspended in a K25+S medium consisting of 10 % FBS (Gemini Bioproducts, Calabasas, CA), 25 mM KCl, Gentamycin (50 μg/ml) and L-glutamine (2 mM) in Basal Medium Eagle (Life Technologies, Inc., Rockville, MD) and were seeded at a density of 2.5 × 105 cells/cm2 area in multi-well plates or in dishes (Corning, Inc., Corning, NY) precoated with poly-l-lysine (100 μg/ml; Sigma). The cells were maintained at 37 °C in the presence of 5 % CO2/95 % air in a humidified incubator. Cytosine arabinofuranoside (AraC, 5 μM; Sigma) was added to cultures 24 h after plating to arrest the growth of non-neuronal cells. With this protocol, 95 to 99% of the cultured cells were granule neurons (Dudek et al., 1997, Li et al., 2000, Hossain et al., 2002). The cells were used for experiments after 10 days in culture (DIV 10).
Induction of excitotoxicity and hypoxia
Experiments were conducted at DIV 10, when cultures consisted primarily of neurons (>95% MAP-2 immunoreactive cells) (MAP-2; Chemicon, Temecula, CA). At least 2 h before excitotoxicity or hypoxia was induced, the K25+S medium for the CGCs was replaced with a serum-free medium containing 25 mM KCl (K25-S) (Hossain et al., 2002). Serum-free medium was used in all experiments to isolate the survival-promoting effect of FGF-1 alone, since in granule cells, serum provides a survival-promoting activity of unknown origin (Timothy and Johnson, 1996). The KCl (25 mM) was included in the medium (K25+S) to ensure normal neuronal development and survival in culture (D’Mello et al., 1993) and to minimize neuronal death from causes other than excitotoxicity.
Neurotoxicity was induced by adding N-methyl-D-aspartate (NMDA; 0–500 μM) or α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA; 0–150 μM) directly to the K25-S medium; and CGCs were incubated for ~20 h at 37°C in a humidified incubator (5 % CO2/95 % air). Immediately after the period of exposure, cell toxicity was measured in the CGCs.
Hypoxia was induced by exposing cells to humidified 95% N2/5% CO2 at 37 °C using a modular incubator chambers (Billups-Rothenberg, Del Mar, CA) for various time periods (0–16 h) as described previously (Hossain et al., 2004a). Control cultures were exposed to humidified 95 % air/5% CO2 at 37 °C for the same duration. Subsets of parallel cultures were pretreated with human recombinant FGF-1 (40 ng/ml; Sigma) for 2 h before exposure to hypoxia (10 h). Cells were placed in a serum-free medium containing 25 mM KCl (K25-S) 2 h before exposure to hypoxia. Control cultures were also in K25-S medium and received an equal volume of BSA (0.1 mg/ml), which had no detectable responses on cell survival or in the biochemical assays (Dudek et al., 1997).
Assessment of cell viability/toxicity
Immediately after the period of exposure to excitotoxicity or hypoxia, cell viability and/or death was determined by independent and complementary methods.
MTT assay
Mitochondrial dehydrogenase activity cleaves 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT; Sigma) and is a biochemical index for cellular viability. A quantitative colorimetric assay of MTT (Nonaka et al., 1998) used to determine cell survival. The tetrazolium ring of MTT is cleaved by various dehydrogenase enzymes in active mitochondria, forming a blue-colored insoluble product, formazan. Cerebellar granule cells were incubated with MTT (125 μg/ml) added to the growth medium for 1 h at 37 °C. The medium was then aspirated and the formazan product was dissolved in 500 ml dimethylsulfoxide and quantified spectrophotometrically at 540 nm using a Spectra MAX 340pc plate reader (Molecular Devices, Sunnyvale, CA) as described previously (Hossain et al., 2002, Hossain et al., 2004a). The results were expressed as a percentage of control culture viability.
LDH assay
Lactate dehydrogenase (LDH) activity released in the media after hypoxic exposure was measured using the CytoTox96 Non-radioactive Cytotoxicity Assay kit (Promega, Madison, WI) as described previously (Hossain et al., 2004a). Percent cell death was determined using the formula: % cytotoxicity = hypoxic LDH release (OD490)/maximum LDH release (OD490) after correcting for baseline absorbance of LDH release at 490 nm.
TUNEL staining
The DeadEnd Fluorometric TUNEL System (Promega) was used to detect cell death in cultured cerebellar granule neurons exposed to hypoxia (10–12 h) as described previously (Hossain et al., 2004a). Fluorescence was visualized in a fluorescence microscope (Carl Zeiss Axioplan 1) with an excitation at 485 nm and an emission at 535 nm. DAPI fluorescence (blue) was visualized with an excitation and emission filters at 365 nm and 450 nm, respectively.
Immunofluorescence
Double immunofluorescence staining of primary CGC cultures (grown on cover slips) was conducted using primary antibodies specific for MeCP2 (Upstate; rabbit polyclonal) and SNAP25, a neuron specific marker, (kindly supplied by Dr. Jonathan Pevsner, Kennedy Krieger Institute, Baltimore) to determine MeCP2 protein expression in neuronal cells. Cerebellar granule cells at DIV 10 were fixed with 3.7% formaldehyde and permeabilized cells were incubated with MeCP2 (1:200) and SNAP25 (1:250) antibodies for overnight at 4 °C according to our previously described method (Hossain et al., 2004b). For negative controls appropriate non-immune IgG was used instead of primary antibodies. Cells were washed and stained with multiple fluorochrome-conjugated secondary antibodies (Texas red-conjugated goat anti-mouse for MeCP2; red, and FITC-conjugated goat anti-rabbit for SNAP-25; green) and 4,6-diamino-2-phenylindole (DAPI; blue) that stain nuclei for 1 h at RT. DAPI stains all cell nuclei with blue fluorescence regardless of viability. Slides were coverslipped with Prolong mounting medium (Molecular Probes, Eugene, OR). MeCP2-and SNAP25-specific immunofluorescence was visualized using a fluorescence microscope (Carl Zeiss Axioplan 1 microscope fitted with AxioVision 3.0 software) at 100 X magnifications.
Subcellular fractionation
Subcellular fractionation was performed as described by Shou et al (2002) (Shou et al., 2002). Briefly, cells were harvested in ice cold PBS and centrifuged at 3600 × g for 5 min. Cell pellets were resuspended in isotonic mitochondrial buffer (MB) consisting of 210 mM mannitol, 70 mM sucrose, 1 mM EDTA and 10 mM HEPES, pH 7.5, supplemented with protease and phosphatase inhibitor cocktails (Calbiochem, San Diego, CA). Cells were homogenized with a Dounce homogenizer for 50 strokes and were centrifuged at 500 × g for 5 min. The resulting supernatant was centrifuged further at 10,000 × g for 30 min at 4°C. The pellet contained the mitochondrial fraction and was suspended in 100 μl MB buffer containing 0.1% triton X-100 to break up the mitochondria; the supernatant was used as the cytosolic fraction.
Transient transfection
Primary cerebellar granule neurons were isolated from mice cerebella and transfected by nucleofection system using Rat Neuron Nucleofector kit (Amaxa, Inc., Cat No. VPG-1003) according to the manufacturer’s instructions (Krauss et al., 2003). Isolated primary cells in suspension containing 12 × 106 cells was mixed with 2 μg of plasmid DNA for nucleofection and seeded at a density of 2.5 × 105 cells per cm2 area in different cell cultures plates depending on the nature of experiments. Using this technique, we achieved >65% transfection efficiencies in primary cortical cultures as determined by green fluorescent protein (GFP) expression.
Commercially available control siRNA (Santa Cruz, Cat No. sc-37007) and MeCP2 siRNA (Santa Cruz, Cat No. sc-35893) was transfected into cells using LipofectAMINE RNAiMAx (Invitrogen) according to manufacturer’s instructions. Briefly, cells were grown on coverslips in 12-well plates at a density of 2.5 × 105/cm2 area and placed in fresh medium without antibiotic. Control scramble siRNA or MeCP2 siRNA (0.3–0.4 nmol) per well and Lipofectamine™ RNAiMAX complex was prepared in a total volume of 100 μl according to manufacturer’s instruction. One hundred microliter of the siRNA-lipofectamine RNAiMAX complex was then added to each well and cells were incubated at 37°C in a 5% CO2 cell culture incubator for 5 h. The transfection medium was then aspirated; cells were washed with fresh transfection medium, and then replenished with previously saved conditioned medium (Li et al., 2000). Experimental treatments were initiated ~ 48 h after transfection. With this protocol, we have achieved >80% reduction in MeCP2 protein levels compared to control siRNA as determined by Western blot and immunofluorescence analyses.
SDS-PAGE and Western blot analyses
SDS-PAGE and immunoblotting were performed according to the method of Laemmli (Laemmli, 1970) with modifications as described previously (Hossain et al., 2002). Briefly, cerebellar granule cell extracts were prepared using 100 μl of ice-cold lysis buffer RIPA (PBS 1X, 1% IGEPAL CA-630, 0.5 % sodium deoxycholate, 0.1% SDS) containing 1 X protease cocktail inhibitor set 1 (Calbiochem, #539131), sodium vanadate (1 mM), sodium pyrophosphate (2 mM) and sodium β-glycerophosphate (1 mM) and stored at −70 °C. Protein concentrations were determined using the Coomasie protein assay (Pierce, Rockford, IL) according to the manufacturer’s instructions.
Total proteins (10–20 μg) were diluted in Laemmli buffer containing β-mercaptoethanol, heated to 100 °C for 5 min, separated on a 4–20% gradient Tris-Glycine pre-cast gel (Invitrogen, Carlsbad, CA) at 140 V for 1 h, and then immunoblotted with specific primary antibody to MeCP2 (1:500; Upstate, Lake Placid, NY), total and cleaved caspase-3 and -9 (1:500; Cell signaling, Baverly, MA), AIF (1:500; Santa Cruz Biotechnology) and actin (1:1000). Horseradish peroxidase (HRP)-conjugated secondary antibodies (Jackson Immonoresearch Laboratories, west Grove, PA) were used at 1:1,000 dilutions for 1 h at RT. HRP reaction product was then visualized by enhanced chemiluminescence using an ECL western blotting detection kit (Amersham-Pharmacia). Digitized images were quantified by densitometry (Molecular Dynamics).
Statistical analysis
Statistics were performed using StatView 5.0 program. Comparisons involving multiple groups were done by ANOVA, followed by Bonferroni/Dunn post-hoc tests where appropriate.
RESULTS
MeCP2 expression in cerebral granule neuronal cultures
To elucidate the effects of MeCP2 deficiency in neuronal response to injury and death, we first examined MeCP2 protein expression in a well-characterized culture system of primary cerebellar granule neurons. Dissociated primary neuronal cultures allow examination of specific neuronal function independent of potential general alterations in brain homeostasis, thereby enabling a distinction between cell-autonomous defects and global systemic dysfunction. Immunofluorescence analysis of MeCP2 expression using a MeCP2-specific antibody showed intense MeCP2-specific immunofluorescence (red) in the nuclear region of wild type neurons (Fig. 1A). In contrast, MeCP2 −/− cerebellar granule cells showed complete absence of MeCP2 proteins (Fig. 1C). Double immunofluorescence staining with MeCP2 and SNAP25, a specific neuronal marker, revealed MeCP2 expression exclusively in neurons (Fig. 1B). No MeCP2- and SNAP25-specific immunofluorescence was observed in cultures incubated with nonimmune IgG (negative controls, Fig. 1E, 1F). We found MeCP2 colocalized with DAPI, a dye that binds nucleic acid and stain nuclei, in SNAP25 positive cells (Fig. 1B). These results indicated that all MeCP2 positive cells were neurons and that MeCP2 staining was within the nucleus.
Fig. 1.
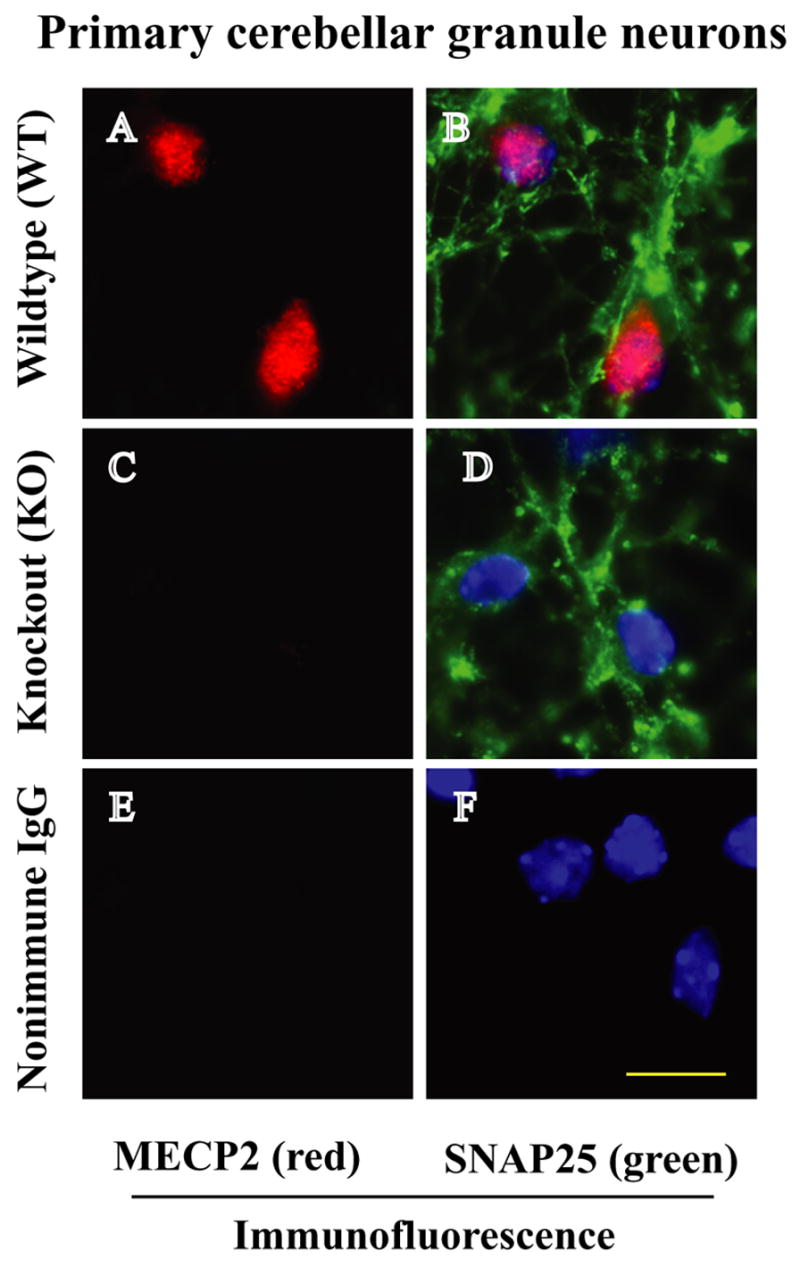
Images of immunofluorescence staining of MeCP2 in WT and MeCP2 KO cerebellar granule neurons. A) MeCP2 is expressed (red staining) in WT cells. B and D) SNAP25 (green staining) is a neuron specific marker that stains the cerebellar granule neurons. Moreover, the same cells were stained with DAPI, a dye that binds nucleic acid and labels cell nuclei. Digitized individual and overlapped images of MeCP2, SNAP25 and DAPI in B indicate that MeCP2 is localized in the nucleus of cerebellar granule neurons. Panel (C) shows complete absence of MeCP2 proteins in MeCP2−/− cerebellar granule neurons (D). Non-immune IgG staining shows no evidence of MeCP2 or SNAP25 (E, F). Scale bar 10 μm.
Effects of NMDA and AMPA induced excitotoxicity in MeCP2−/− cerebellar granule cultures
To determine whether MeCP2 deficiency contributes to neuronal pathogenesis, we exposed primary CGC cultures isolated from MeCP2 null and WT mouse brains to excitotoxic or hypoxic conditions. Primary CGC cultures (DIV 10) were exposed to NMDA (0–500 μM) (Fig. 2A) or AMPA (0–150 μM) (Fig. 2B) for ~20 h. Quantification of cell viability by MTT assay revealed that 200 μM of NMDA and 100 μM of AMPA resulted in ~ 40–50% cell death (p<0.01) compared to respective littermate controls. At each concentration of NMDA or AMPA, percent cell death was significantly higher in MeCP2−/− granule neurons (p<0.01) compared to that in WT cultures (Fig. 2). Since in RTT, girls are heterozygous for the MeCP2 gene, we also examined excitotoxicity in CGC cultures from heterozygous (+/−) mice. Exposure of heterozygous (+/−) CGC cultures to NMDA and AMPA (not shown) showed a similar pattern of cell death (Fig. 2C) to that from MeCP2−/− cultures (Fig. 2A & 2B). Thus, the percentage of NMDA and AMPA-induced neuronal death in MeCP2 protein deficient or absent cells is increased relative to that in WT littermate cerebellar granule neurons.
Fig. 2.

Effects of NMDA and AMPA-induced excitotoxicity on WT and MeCP2−/−primary cerebellar granule neurons. CGC cultures (DIV 10) were exposed to increasing concentrations of NMDA (A, C) and AMPA (B) for ~20 h at 37°C. Quantification of neuronal viability by MTT reduction colorimetric assays revealed that 200 μM of NMDA (A) and 100 μM of AMPA (B) resulted in ~ 50% cell death in MeCP2−/− cells (p<0.01) compared to WT controls. MeCP2 heterozygous cells also had a significantly higher percentage of cell death than WT controls. Values are shown as mean ± SEM of % cell viability relative to controls (100%) from 6 independent observations (n=6). *p<0.01
Hypoxia-induced cell death in MeCP2−/− cerebellar granule neurons
Since glutamate receptor mediated excitotoxicity is a major contributor to neuronal death in hypoxic-ischemic brain injury (Johnston, 2001), we next examined whether MeCP2 deficiency altered the neuronal response to hypoxia-induced cell death. Hypoxia (0–16 h) was induced in DIV 10 cultured cells. MTT quantification showed a significantly lower cell viability (50%; p<0.01) occurring in MeCP2−/− granule cells after 8 h of hypoxia than that observed in WT cells (~75%) under identical conditions (Fig. 3A). These results indicate that WT and MeCP2−/− CGC cultures have a differential sensitivity to hypoxia-induced cell death. Our results also revealed that the onset of significant neuronal death in MECP2−/− cells occurred earlier (5h) than in the WT cultures (p<0.01) (Fig. 3A). Pretreatment of CGC cultures with FGF-1 (40 ng/ml) was neuroprotective (p<0.01) against hypoxia (10 h) -induced death both in MeCP2−/− and WT neurons (Fig. 3B). However, the magnitude of FGF-1-mediated neuroprotection was less in MeCP2 null cells (p<0.05) than in the WT (Fig. 3B). Collectively, the results demonstrate that MeCP2 deficiency in cerebellar granule neurons produced a higher degree of cell death when MeCP2−/− neurons were exposed to excitotoxicity or hypoxia, and that MeCP2 deficiency altered the neuronal response to the FGF-1 mediated neuroprotection against cell death.
Fig. 3.
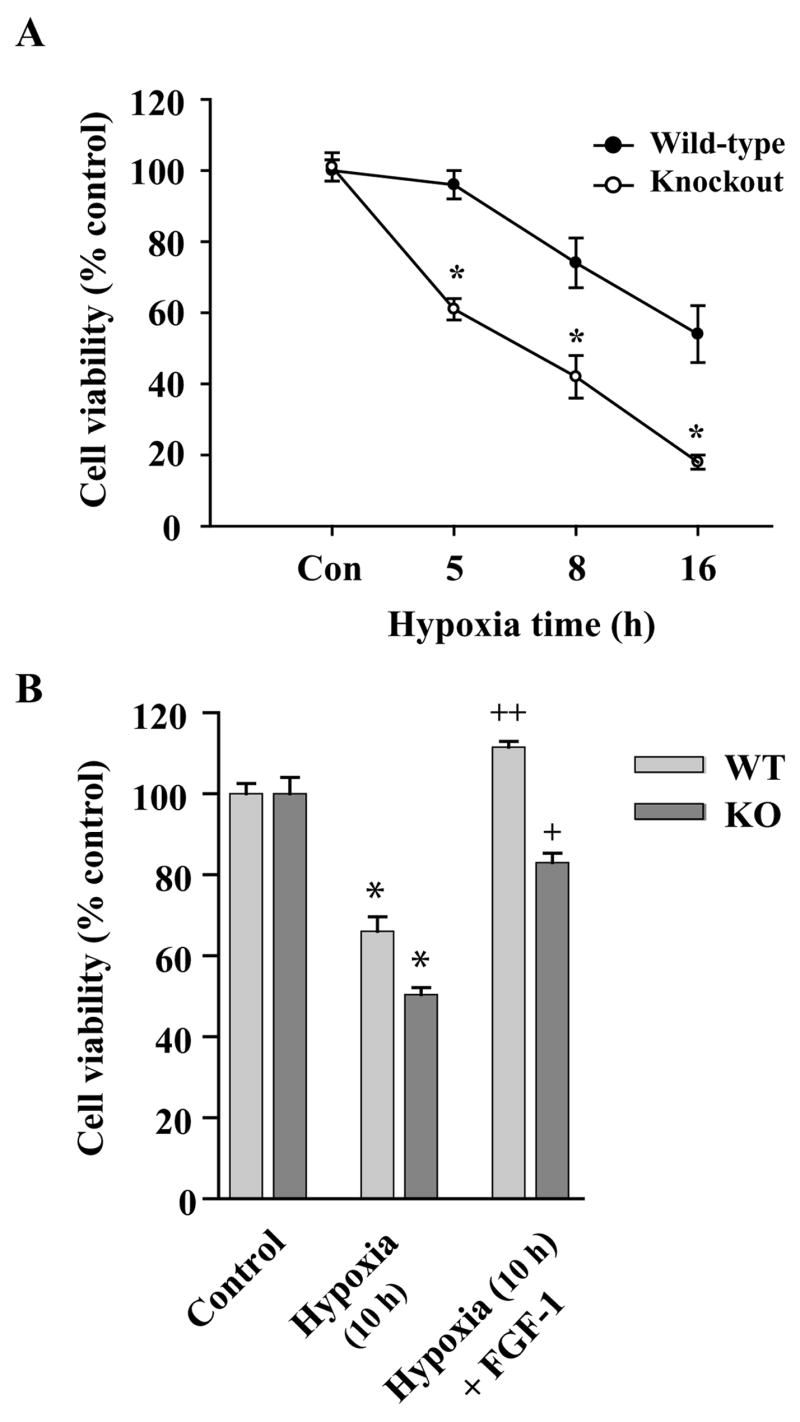
MeCP2−/− cerebellar granule neurons exposed to hypoxia show increased neuronal death. Primary CGC cultures at DIV 10 were exposed to hypoxia for 5, 8 and 16 hours. A) After 5 hours of hypoxia, a MTT cell viability assay revealed a ~40% increase in cell death in compared to WT neurons, which showed relatively little cell death at that time point. At 8 and 16 h exposure times, the MeCP2−/− cells also showed significantly greater cell death than the WT cells. B) MTT assays of CGCs pretreated with FGF-1 (40 ng/ml) 2 h prior to 10 h of hypoxia showed complete neuroprotection of WT neurons, while MeCP2−/− cells were only partially protected by the FGF−1 pretreatment. Data is expressed as percent control of normoxia cells (mean ± SEM, n=8; *p< 0.01 vs. normoxia controls; ++ p<0.01 and +p<0.05 vs. respective hypoxia group).
MeCP2 deficiency in neurons enhances proapoptotic death pathways
Mitochondria release proteins that propagate both caspase-dependent and caspase-independent cell death pathways. Caspase-3 is a terminator protein that triggers the execution phase of apoptosis. We performed a Western blot analysis of total cellular extracts from WT and MeCP2−/− cells (DIV 10 cultures) under normoxia or after 6 h exposure to hypoxia. Densitometric quantification of cleaved caspase-3 specific immunoreactive protein bands (~17 kDa; normalized to actin) revealed a 2-fold increase in the MeCP2−/− cells, (p<0.01) in levels of activated caspase-3 after 6h of hypoxia, compared to WT and KO normoxia controls and WT hypoxia controls (100%) (Fig. 4). This result suggests that the greater magnitude of cell death in MeCP2−/− cells is due the enhanced activation of caspase-3 proteins.
Fig. 4.

Hypoxia induced increases in cleaved caspase-3. Primary cerebellar granule cultures at DIV 10 were exposed to hypoxia for 6 h. Total cellular proteins (20 μg/lane) were analyzed by Western blot analysis using an antibody specific for cleaved caspase-3, normalizing the density to that for actin. After 6 hr of hypoxia, MeCP2−/− cerebellar granule neurons exhibited a 2-fold increase in cleaved caspase-3 protein levels compared to WT cells under normoxic or hypoxic conditions (*p<0.01). Values represent mean ± SEM (n=3) relative to controls. Representative blots are shown.
We next determined whether caspase-independent pathways also contribute to the increased cell death in MeCP2−/− neurons after hypoxia, by examining the role of apoptosis inducing factor (AIF), a caspase-independent death regulator. Cell fractionation experiments isolated mitochondrial and cytoplasmic fractions, which were then examined by Western blot analysis. AIF was released and translocated to the cytoplasm in response to hypoxia. Under normoxia conditions, AIF-specific intense immunoreactive protein band was detected (67 kDa) in the mitochondrial fraction from both WT and MeCP2−/− cells (Fig. 5A). After exposure to hypoxia for 6h, mitochondrial AIF release increased in MeCP2−/− cells. Evidence for this release was the concurrent decrease in AIF protein levels (>50%; p<0.01) in the mitochondrial fraction (normalized to mitochondrial specific marker prohibitin) along with the simultaneous increase of AIF in the cytoplasmic fraction (p<0.01) (Fig. 5A). AIF levels in mitochondrial and cytosolic fractions of WT cells did not change after exposure to hypoxia (Fig. 5A). The results suggest that increases in AIF release may contribute to the enhanced cell death observed in MeCP2−/− cerebral granule cells after hypoxia. Thus, both caspase- and AIF-dependent cell death pathways contributed to enhanced cell death after hypoxia in MeCP2−/− cells.
Fig. 5.
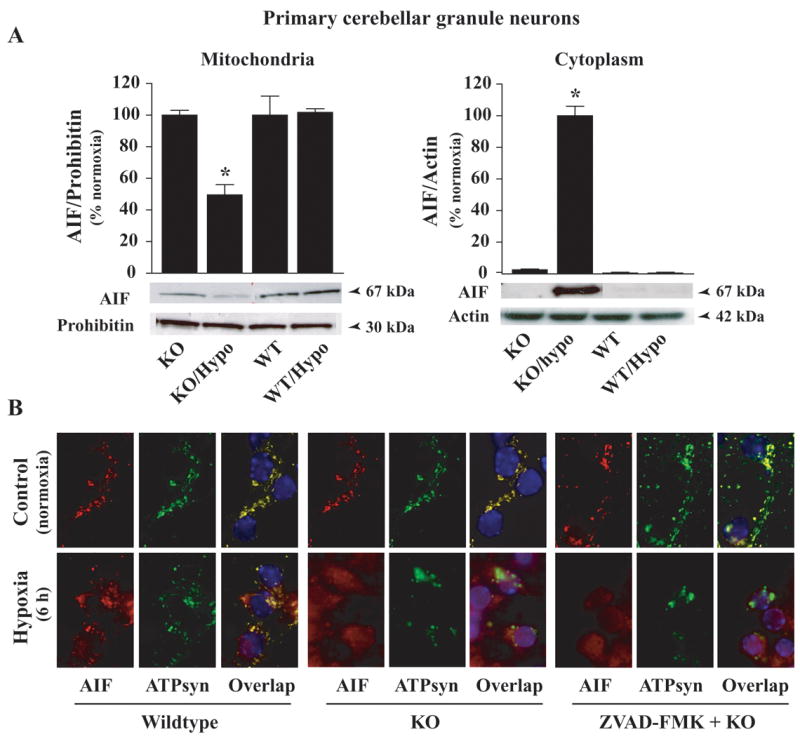
Increased mitochondrial release of AIF in MeCP2−/− cerebellar granule neurons after hypoxia. AIF release form mitochondrial to cytosolic fractions was analyzed by (A) Western blot and (B) immunofluorescence analyses using AIF-specific primary antibody (1:500) (PharMingen). Quantification of AIF-specific protein bands showed that 6 h of hypoxia lead to an increase in AIF specific protein bands in the cytosolic fraction with concomitant decrease in the bands in the mitochondrial fraction from MeCP2−/− cells compared to WT cells. Values represent mean ± SEM (n=3–4) relative to controls (*p<0.01). Representative blots are shown. B) WT and MeCP2−/− primary cerebellar granule neurons were immunostained with AIF (1:200; red) and ATP synthase (1:300; green); DAPI stained the nucleus of cells blue. Under normoxia conditions, AIF and ATP Synthase are colocalized (yellow) in both WT and MeCP2−/− cells. After hypoxia (6 h), most AIF-specific immunofluorescence was localized in the cytoplasm and nucleus (purple) of MeCP2−/− cells, it remained localized in mitochondria in WT cells. The caspase inhibitor ZVAD-FMK had little or only marginally affected AIF release and nuclear translocation. No AIF or ATP synthase specific immunofluorescence was observed in cells incubated with nonimmune IgG (−ve controls).
To more specifically determine whether hypoxia-induced AIF release occurs via a caspase-dependent or independent manner, we pretreated WT and MeCP2−/− cells with the general caspase inhibitor ZVAD-FMK (100 μM) (BD Pharmingen) 1 h before exposure to hypoxia (6 h). Cells were double immunostained with AIF (Santa Cruz) and mitochondrial specific marker ATP synthase (Chemicon) and coverslipped with mounting medium that contained DAPI, a dye that binds and stains the nucleus (Fig. 5B). Overlapping digitized images of mitochondrial proteins AIF (red) and ATP synthase (green) staining showed overlapping images (yellow), which were completely absent in the nucleus (blue) (Fig. 5B; upper panels). In contrast, after 6 h of hypoxia, the majority of the AIF-specific immunofluorescence (red) was localized in the cytoplasm and nucleus (purple) of MeCP2−/− cells (bottom panels) compared to mitochondrial localization in the WT cells. Pretreatment with the caspase inhibitor ZVAD-FMK had no effects or only marginally reduced the magnitude of AIF translocation to the cytoplasm and nucleus in MeCP2−/− cells (Fig. 5B). Our results suggest that AIF release from mitochondria in the MeCP2−/− cells after hypoxia occurred via a caspase-independent pathway.
Specificity of MeCP2 deficiency in cell death mechanism
To examine the mechanistic role of MeCP2 deficiency in the cell death response, we used a strategy of overexpressing the intact MeCP2 gene in MeCP2 null cerebellar granule cells. MeCP2−/− and WT cells were transfected with control plasmid DNA or the pTAU-MeCP2pAneo expression vector DNA (kindly provided by Dr. Rudolf Jaenisch) (Luikenhuis et al., 2004) as described and 48 h later exposed to hypoxia (8 h). Hypoxia-induced neuronal death was assessed independently by LDH release and MTT cell viability assays. Hypoxia exposure caused >50% cell death in MeCP2−/− cells compared to WT cells (Fig. 6). However, reintroduction of MeCP2 gene into null cells significantly reduced cell death (p<0.001) after hypoxia (Fig. 6).
Fig. 6.

Reintroduction of the MeCP2 gene in null cells reduces hypoxia-induced death in MeCP2−/− cerebellar granule neurons. Hypoxia-induced neuronal death was assessed independently by complementary LDH release (A) and MTT cell viability (B) assays. After 8 h hypoxia, >50% of MeCP2−/− cells died. Introduction of MeCP2 into null cells significantly reduced cell death after hypoxia compared to that in the hypoxia only group. Data are expressed as percent of WT control cells under normoxia conditions (mean ± SEM, n=6; *p< 0.001 vs. normoxia controls; +p<0.01 vs. respective hypoxia group).
We next used TUNEL staining in the CGC cultures as an independent complementary method for examining neuronal death in MeCP2−/− cells (Fig. 7). Intense TUNEL (+) cells were detected both in the WT and MeCP2−/−cultures after 8 h of hypoxia compared to respective normoxia controls. However, the number of TUNEL (+)-cells was significantly higher (~5-fold; p<0.01) in MeCP2−/− cultures than that in the MeCP2 WT cells under identical conditions (Fig. 7A). Over expression of MeCP2 in MeCP2 null neurons lead to a significant decrease in the number of TUNEL (+) cells compared to untransfected null neurons (Fig. 7B). Next, we used MeCP2 gene silencing strategy in WT CGC cultures using MeCP2 siRNA (Santa Cruz) to examine the functional requirement of MeCP2 for the observed effects. WT cerebellar granule cells at DIV 8 were transfected with MeCP2 siRNA or control siRNA as indicated and 48 h after transfection were exposed to 8 h hypoxia (Fig. 7C). TUNEL staining and quantification of TUNEL (+) cells revealed a significant increase in cell death (p<0.01) in MeCP2siRNA transfected cells after exposure to hypoxia relative to control siRNA transfected cells (Fig. 7C). These results together demonstrate a direct role of MeCP2 in neuronal death/survival mechanism, and that insufficient expression of MeCP2 is directly involved in the greater magnitude of cell death in MeCP2−/− cerebellar granule cells.
Fig. 7.
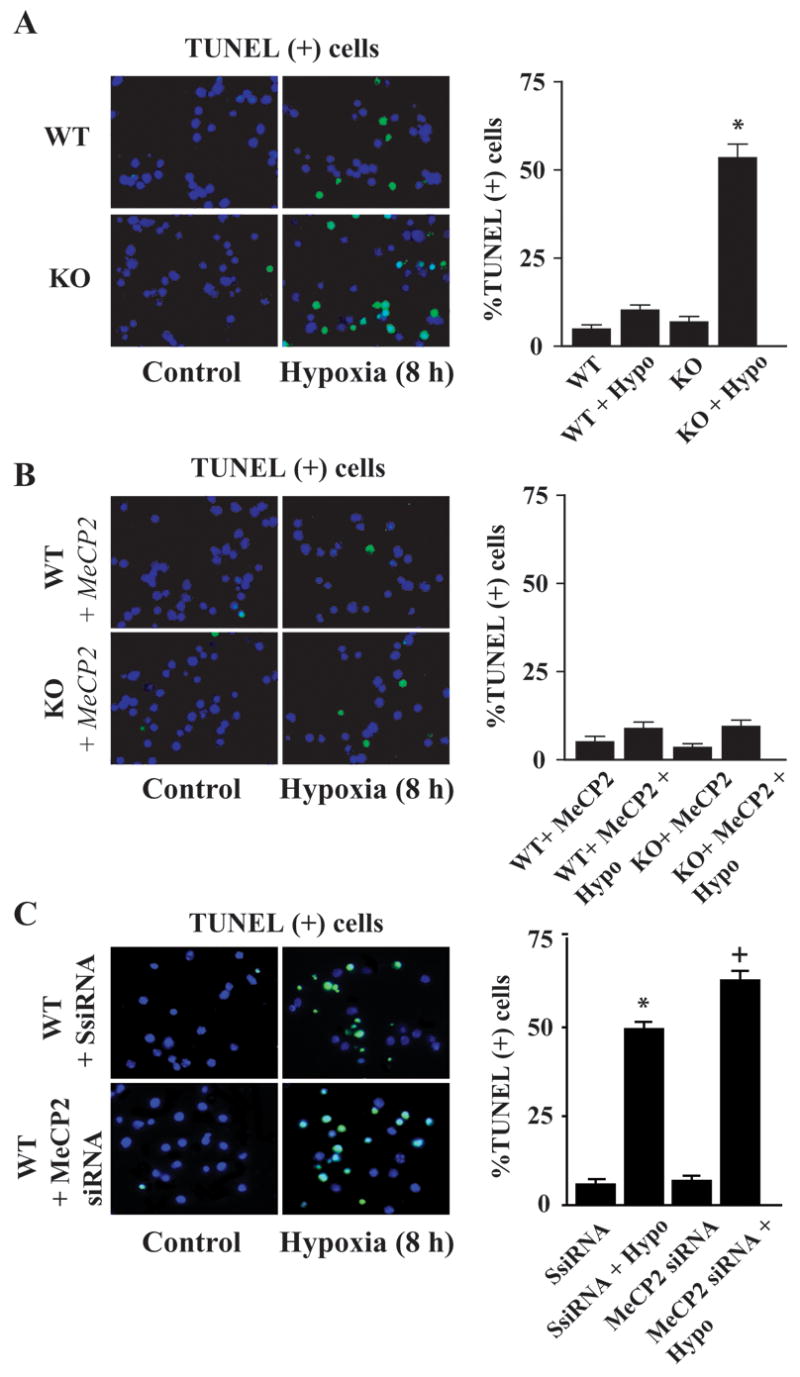
Hypoxia caused increased apoptotic cell death in MeCP2−/− cerebellar granule neurons. WT and MeCP2−/− primary cerebellar granule neurons were exposed to hypoxia (8 h). A) The number of TUNEL (+) cells (green) with darkly stained chromatin was increased in MeCP2−/− cell cultures compared to that in WT controls. B) Introduction of the intact MeCP2 gene into MeCP2 null cells reduced the number of hypoxia-induced TUNEL (+) cells. C) MeCP2 gene silencing in WT CGC by transfection with 3 target-specific siRNA (20–25 nt) (Santa Cruz) increased the number of TUNEL (+) cells after hypoxia compared to control scramble siRNA transfected cells. Quantification of TUNEL (+) cells over total cells (DAPI staining; blue) revealed >5-fold more cell death in MeCP2siRNA transfected cells compared to that in wild type cells exposed to hypoxia. Values represent mean ± SEM (n=4) relative to controls (*p<0.01 vs. control SsiRNA; +p<0.01 vs. normoxia WT + MeCP2 siRNA group).
DISCUSSION
The present study provides the first evidence that the loss of functional MeCP2 expression in central neurons leads to greater cell injury/death after exposure to different pathological conditions such as excitotoxicity and hypoxic-ischemic insults. We also found shorter durations of exposure, which caused no or relatively little cell death in WT neurons, lead to significant cell death in MeCP2−/− CGC cultures. Particularly interesting was the finding that CGC cultures from MeCP2 heterozygous mice also showed cell death with a magnitude comparable to that in MeCP2 null cells. The specific action of the MeCP2 deficit on the cell death mechanism was further demonstrated by MeCP2 gain/loss-of-function strategies using MeCP2−/− and WT cells. After hypoxia, activation of the proapoptotic death effector caspase-3 was enhanced and AIF release from the mitochondria and translocation to the nucleus was increased in MeCP2−/− cells. Furthermore, the caspase-3 activation and increased AIF release was observed at a shorter duration of exposure in MeCP2−/− cells compared to that in WT cells. Our findings suggest that these proapoptotic death cascade pathways could account for the enhanced cell death observed in MeCP2−/− cerebellar granule neurons.
Modeling Rett syndrome in primary neuronal cultures
The cerebellum is an important area of interest for RTT research; it is an area with distinct changes in neuropathology and cerebellar deficits may underlie the clinical signs of tremor and ataxia (Oldfors et al., 1990, Murakami et al., 1992, Naidu et al., 2003). In the present study, we used well-characterized primary neuronal cultures of cerebellar granule neurons obtained from MeCP2 null mice cerebella as an in vitro RTT model. Primary neuronal cultures provide an excellent model for investigating the molecular and intracellular signaling events that regulate neuronal death and survival. The high degree of cellular homogeneity (D’Mello et al., 1993, Dudek et al., 1997) of primary neuronal cultures, the predictable expression of a neuronal phenotype in vitro, including an extensive neuritic network and the expression of multiple glutamate receptor subtypes (Cox et al., 1990), make it possible to distinguish between cell-autonomous defects and global systemic dysfunction. We examined the effects of MeCP2 loss on cell death mechanisms in this model of cultured cerebellar granule cells to determine whether MeCP2 deficiency in central neurons contributes to neuronal pathogenesis observed in RTT.
The MeCP2 null mouse used for this study is an established animal model of human RTT (Guy et al., 2001, Kriaucionis and Bird, 2003). Studies using MeCP2 null and heterozygous mice suggest that MeCP2 heterozygous females, which are genetically most comparable to human RTT patients display symptoms similar to MeCP2 null mice but at an older age (Guy et al., 2001, Kriaucionis and Bird, 2003). We observed that MeCP2 deficiency in cerebellar granule neurons leads to a greater degree of cell death when MeCP2−/− neurons were exposed to excitotoxicity and hypoxic conditions. We also found that cerebellar granule neurons from MeCP2 (+/−) heterozygous females were more sensitive to NMDA-mediated excitotoxicity than cells from MeCP2 wild type mice.
The role of MeCP2 in enhanced cell death
The NMDA and AMPA subtypes of glutamate receptors play a critical role in mediating cytotoxicity in hypoxic-ischemic brain damage (McDonald and Johnston, 1990, Johnston, 2001, Johnston et al., 2001). Our previous studies showed that NMDA receptor density was higher in young girls with RTT (Blue et al., 1999). Our hypothesis was that enhanced excitatory neurotransmission during the early course of RTT and in encephalopathic phase could worsen the disorder by impairing cortical plasticity, synaptogenesis and pruning, and behavior (Lappalainen et al., 1996, Blue et al., 1999) (Mullaney et al., 2004). Thus, the higher degree of cell death observed in MeCP2 null and heterozygous cells is possibly the result of enhanced excitotoxicity triggered by NMDA subtype of glutamate receptors and hypoxia.
The importance of MeCP2 in neuronal function is also evident from our findings that reintroduction of the intact MeCP2 gene into MeCP2−/− cells significantly reduced their vulnerability to hypoxia. The direct involvement of MeCP2 is further supported by MeCP2 loss-of-function experiments. MeCP2 gene silencing using MeCP2 siRNA in WT cerebellar granule cells significantly increased the number of TUNEL (+) cells comparable to that observed in MeCP2−/− cells. These results suggest a correlation between MeCP2 function and the cell viability after exposure to hypoxia. The specific importance of functional MeCP2 role in neuropathology is also evident from our neurotrophic factor FGF-1 pretreatment findings. Under conditions that we previously found to provide almost complete neuroprotection (Hossain et al., 2002, Russell et al., 2006) that the FGF-1 pretreatment only partially attenuated hypoxia-induced neuronal death in MeCP2−/− cells compared to wild type control cells. Taken together, these results suggest a direct role for MeCP2 function in neuronal injury and death triggered by enhanced excitatory neurotransmission (Lappalainen et al., 1996, Blue et al., 1999, Nelson et al., 2006).
Caspase dependent and independent mechanisms for cell death
Many human disorders affecting the brain are due to mitochondrial dysfunction (Dotti et al., 1993, Armstrong, 2005), sometimes resulting in increased oxidative stress or induction of neuronal apoptosis (Penninger and Kroemer, 2003). More recently, mitochondrial dysfunction due to physiological changes has been shown to contribute to the pathology in the MeCP2-null mouse (Kriaucionis et al., 2006). Activation of the caspase cascade and AIF release are mitochondria-mediated events that lead to apoptotic cell death under various cytotoxic insults (Susin et al., 1999, Penninger and Kroemer, 2003). Mitochondrial membrane permeablization leads to caspase activation (Kroemer and Reed, 2000) and release of AIF from mitochondria into cytoplasm and nucleus (Susin et al., 1999, Cande et al., 2004). MeCP2−/− cells showed increased activation of caspase-3 under hypoxic conditions. This caspase-dependent apoptotic (programmed cell death) mechanism is more active in the cerebellum at this time point due to its prolonged period of development (Naidu et al., 2003, Mullaney et al., 2004).
AIF is a key player in caspase-independent apoptosis, which after its release from mitochondria translocates to nucleus in response to specific death signals and causes high molecular weight DNA fragmentation and cell death (Susin et al., 1999, Daugas et al., 2000, Yu et al., 2003, Cheung et al., 2005). We found that AIF release was increased in MeCP2−/− cells after hypoxia. The caspase inhibitor ZVAD-FMK had little or no effect on the AIF release and its nuclear translocation, suggesting that the AIF-mediated contribution to cell death was caspase-independent (Susin et al., 1999, Cande et al., 2004). Taken together these results indicate that both caspase-dependent and caspase-independent AIF-mediated mechanisms contribute to cell death under excitotoxic and hypoxic conditions.
In summary, the loss of MeCP2 function or complete knockdown of MeCP2 induces a death program at an earlier time period than in wild type mice after hypoxia and excitotoxicity in cerebellar granule neurons. The neuronal death was significantly greater in cerebellar granule cells lacking functional MeCP2 protein expression compared to wild types, and occurred via both caspase- and AIF-dependent manner. In view of the fact that MeCP2 inactivation derepresses gene expression or transcriptional dysregulation (Chen et al., 2003), it is possible that overexpression of prodeath component molecules or their enhanced levels of activation in central neurons makes neurons more vulnerable to injury and death. Our findings clearly suggest a novel, yet unknown, role for MeCP2 in the CNS in the control of neuronal response to cell death.
Acknowledgments
This work was supported by grants from the NICHD program project HD24448 (SN) and the NIH RO1 NS046030 (MAH).
Abbreviations
- AIF
apoptosis inducing factor
- AraC
cytosine arabinofuranoside
- CGC
cerebellar granule cells
- DIV
days in vitro
- KO
knock out
- FGF-1
fibroblast growth factor 1
- MeCP2
methyl-CpG binding protein 2
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- RTT
Rett syndrome
- TUNEL
terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling
- WT
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aber KM, Nori P, MacDonald SM, Bibat G, Jarrar MH, Kaufmann WE. Methyl-CpG-binding protein 2 is localized in the postsynaptic compartment: an immunochemical study of subcellular fractions. Neuroscience. 2003;116:77–80. doi: 10.1016/s0306-4522(02)00586-9. [DOI] [PubMed] [Google Scholar]
- Akbarian S, Chen RZ, Gribnau J, Rasmussen TP, Fong H, Jaenisch R, Jones EG. Expression pattern of the Rett syndrome gene MeCP2 in primate prefrontal cortex. Neurobiol Dis. 2001;8:784–791. doi: 10.1006/nbdi.2001.0420. [DOI] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Armstrong DD. Neuropathology of Rett syndrome. J Child Neurol. 2005;20:747–753. doi: 10.1177/08830738050200090901. [DOI] [PubMed] [Google Scholar]
- Blue ME, Naidu S, Johnston MV. Development of amino acid receptors in frontal cortex from girls with Rett syndrome. Ann Neurol. 1999;45:541–545. doi: 10.1002/1531-8249(199904)45:4<541::aid-ana21>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Cande C, Vahsen N, Metivier D, Tourriere H, Chebli K, Garrido C, Tazi J, Kroemer G. Regulation of cytoplasmic stress granules by apoptosis-inducing factor. J Cell Sci. 2004;117:4461–4468. doi: 10.1242/jcs.01356. [DOI] [PubMed] [Google Scholar]
- Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- Cheung EC, Melanson-Drapeau L, Cregan SP, Vanderluit JL, Ferguson KL, McIntosh WC, Park DS, Bennett SA, Slack RS. Apoptosis-inducing factor is a key factor in neuronal cell death propagated by BAX-dependent and BAX-independent mechanisms. J Neurosci. 2005;25:1324–1334. doi: 10.1523/JNEUROSCI.4261-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- Cox JA, Felder CC, Hennebury RC. Differential expression of excitatory amino acid receptor subtypes in cultured cerebellar neurons. Neuron. 1990;4:941–947. doi: 10.1016/0896-6273(90)90147-8. [DOI] [PubMed] [Google Scholar]
- D’Mello SR, Galli C, Ciotti T, Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci U S A. 1993;90:10989–10993. doi: 10.1073/pnas.90.23.10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugas E, Nochy D, Ravagnan L, Loeffler M, Susin SA, Zamzami N, Kroemer G. Apoptosis-inducing factor (AIF): a ubiquitous mitochondrial oxidoreductase involved in apoptosis. FEBS Lett. 2000;476:118–123. doi: 10.1016/s0014-5793(00)01731-2. [DOI] [PubMed] [Google Scholar]
- Dotti MT, Manneschi L, Malandrini A, De Stefano N, Caznerale F, Federico A. Mitochondrial dysfunction in Rett syndrome. An ultrastructural and biochemical study. Brain Dev. 1993;15:103–106. doi: 10.1016/0387-7604(93)90045-a. [DOI] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: report of 35 cases. Ann Neurol. 1983;14:471–479. doi: 10.1002/ana.410140412. [DOI] [PubMed] [Google Scholar]
- Hossain MA, Bailone JC, Gomez R, Laterra J. Neuroprotection by scatter factor/hepatocyte growth factor and FGF-1 in cerebellar granule neurons is phosphatidylinositol 3-kinase/Akt-dependent and MAPK/CREB-independent. J Neurochem. 2002;81:365–378. doi: 10.1046/j.1471-4159.2002.00837.x. [DOI] [PubMed] [Google Scholar]
- Hossain MA, Russell JC, Miknyoczki S, Ruggeri B, Lal B, Laterra J. Vascular endothelial growth factor mediates vasogenic edema in acute lead encephalopathy. Ann Neurol. 2004a;55:660–667. doi: 10.1002/ana.20065. [DOI] [PubMed] [Google Scholar]
- Hossain MA, Russell JC, O’Brien R, Laterra J. Neuronal pentraxin 1: a novel mediator of hypoxic-ischemic injury in neonatal brain. J Neurosci. 2004b;24:4187–4196. doi: 10.1523/JNEUROSCI.0347-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston MV. Excitotoxicity in neonatal hypoxia. Ment Retard Dev Disabil Res Rev. 2001;7:229–234. doi: 10.1002/mrdd.1032. [DOI] [PubMed] [Google Scholar]
- Johnston MV, Trescher WH, Ishida A, Nakajima W. Neurobiology of hypoxic-ischemic injury in the developing brain. Pediatr Res. 2001;49:735–741. doi: 10.1203/00006450-200106000-00003. [DOI] [PubMed] [Google Scholar]
- Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- Jung BP, Jugloff DG, Zhang G, Logan R, Brown S, Eubanks JH. The expression of methyl CpG binding factor MeCP2 correlates with cellular differentiation in the developing rat brain and in cultured cells. J Neurobiol. 2003;55:86–96. doi: 10.1002/neu.10201. [DOI] [PubMed] [Google Scholar]
- Krauss M, Kinuta M, Wenk MR, De Camilli P, Takei K, Haucke V. ARF6 stimulates clathrin/AP-2 recruitment to synaptic membranes by activating phosphatidylinositol phosphate kinase type Igamma. J Cell Biol. 2003;162:113–124. doi: 10.1083/jcb.200301006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriaucionis S, Bird A. DNA methylation and Rett syndrome. Hum Mol Genet. 2003;12(Spec No 2):R221–227. doi: 10.1093/hmg/ddg286. [DOI] [PubMed] [Google Scholar]
- Kriaucionis S, Paterson A, Curtis J, Guy J, Macleod N, Bird A. Gene expression analysis exposes mitochondrial abnormalities in a mouse model of Rett syndrome. Mol Cell Biol. 2006;26:5033–5042. doi: 10.1128/MCB.01665-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lappalainen R, Lindholm D, Riikonen R. Low levels of nerve growth factor in cerebrospinal fluid of children with Rett syndrome. J Child Neurol. 1996;11:296–300. doi: 10.1177/088307389601100407. [DOI] [PubMed] [Google Scholar]
- Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, Klein F, Bird A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell. 1992;69:905–914. doi: 10.1016/0092-8674(92)90610-o. [DOI] [PubMed] [Google Scholar]
- Li M, Wang X, Meintzer MK, Laessig T, Birnbaum MJ, Heidenreich KA. Cyclic AMP promotes neuronal survival by phosphorylation of glycogen synthase kinase 3beta. Mol Cell Biol. 2000;20:9356–9363. doi: 10.1128/mcb.20.24.9356-9363.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikenhuis S, Giacometti E, Beard CF, Jaenisch R. Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc Natl Acad Sci U S A. 2004;101:6033–6038. doi: 10.1073/pnas.0401626101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthi A, Schwyzer L, Mateos JM, Gahwiler BH, McKinney RA. NMDA receptor activation limits the number of synaptic connections during hippocampal development. Nat Neurosci. 2001;4:1102–1107. doi: 10.1038/nn744. [DOI] [PubMed] [Google Scholar]
- McDonald JW, Johnston MV. Physiological and pathophysiological roles of excitatory amino acids during central nervous system development. Brain Res Brain Res Rev. 1990;15:41–70. doi: 10.1016/0165-0173(90)90011-c. [DOI] [PubMed] [Google Scholar]
- Metcalf BM, Mullaney BC, Johnston MV, Blue ME. Temporal shift in methyl-CpG binding protein 2 expression in a mouse model of Rett syndrome. Neuroscience. 2006;139:1449–1460. doi: 10.1016/j.neuroscience.2006.01.060. [DOI] [PubMed] [Google Scholar]
- Mnatzakanian GN, Lohi H, Munteanu I, Alfred SE, Yamada T, MacLeod PJ, Jones JR, Scherer SW, Schanen NC, Friez MJ, Vincent JB, Minassian BA. A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome. Nat Genet. 2004;36:339–341. doi: 10.1038/ng1327. [DOI] [PubMed] [Google Scholar]
- Mullaney BC, Johnston MV, Blue ME. Developmental expression of methyl-CpG binding protein 2 is dynamically regulated in the rodent brain. Neuroscience. 2004;123:939–949. doi: 10.1016/j.neuroscience.2003.11.025. [DOI] [PubMed] [Google Scholar]
- Murakami JW, Courchesne E, Haas RH, Press GA, Yeung-Courchesne R. Cerebellar and cerebral abnormalities in Rett syndrome: a quantitative MR analysis. AJR Am J Roentgenol. 1992;159:177–183. doi: 10.2214/ajr.159.1.1609693. [DOI] [PubMed] [Google Scholar]
- Naidu S, Bibat G, Kratz L, Kelley RI, Pevsner J, Hoffman E, Cuffari C, Rohde C, Blue ME, Johnston MV. Clinical variability in Rett syndrome. J Child Neurol. 2003;18:662–668. doi: 10.1177/08830738030180100801. [DOI] [PubMed] [Google Scholar]
- Naidu SB. Rett syndrome. Indian J Pediatr. 1997;64:651–659. doi: 10.1007/BF02726119. [DOI] [PubMed] [Google Scholar]
- Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88:471–481. doi: 10.1016/s0092-8674(00)81887-5. [DOI] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, Monteggia LM. MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr Biol. 2006;16:710–716. doi: 10.1016/j.cub.2006.02.062. [DOI] [PubMed] [Google Scholar]
- Nonaka S, Hough CJ, Chuang DM. Chronic lithium treatment robustly protects neurons in the central nervous system against excitotoxicity by inhibiting N-methyl-D- aspartate receptor-mediated calcium influx. Proc Natl Acad Sci U S A. 1998;95:2642–2647. doi: 10.1073/pnas.95.5.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfors A, Sourander P, Armstrong DL, Percy AK, Witt-Engerstrom I, Hagberg BA. Rett syndrome: cerebellar pathology. Pediatr Neurol. 1990;6:310–314. doi: 10.1016/0887-8994(90)90022-s. [DOI] [PubMed] [Google Scholar]
- Penninger JM, Kroemer G. Mitochondria, AIF and caspases--rivaling for cell death execution. Nat Cell Biol. 2003;5:97–99. doi: 10.1038/ncb0203-97. [DOI] [PubMed] [Google Scholar]
- Rabacchi S, Bailly Y, Delhaye-Bouchaud N, Mariani J. Involvement of the N-methyl D-aspartate (NMDA) receptor in synapse elimination during cerebellar development. Science. 1992;256:1823–1825. doi: 10.1126/science.1352066. [DOI] [PubMed] [Google Scholar]
- Russell JC, Szuflita N, Khatri R, Laterra J, Hossain MA. Transgenic expression of human FGF-1 protects against hypoxic-ischemic injury in perinatal brain by intervening at caspase-XIAP signaling cascades. Neurobiol Dis. 2006;22:677–690. doi: 10.1016/j.nbd.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Shou Y, Li N, Li L, Borowitz JL, Isom GE. NF-kappaB-mediated up-regulation of Bcl-X(S) and Bax contributes to cytochrome c release in cyanide-induced apoptosis. J Neurochem. 2002;81:842–852. doi: 10.1046/j.1471-4159.2002.00880.x. [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Timothy MM, Johnson EM. Metabolic and genetic analyses of apoptosis in potassium/serum-deprived rat cerebellar granule neurons. J Neurosci. 1996;16:7487–7495. doi: 10.1523/JNEUROSCI.16-23-07487.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tudor M, Akbarian S, Chen RZ, Jaenisch R. Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proc Natl Acad Sci U S A. 2002;99:15536–15541. doi: 10.1073/pnas.242566899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SW, Wang H, Dawson TM, Dawson VL. Poly(ADP-ribose) polymerase-1 and apoptosis inducing factor in neurotoxicity. Neurobiol Dis. 2003;14:303–317. doi: 10.1016/j.nbd.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]