

Abstract
We introduce and analyze a general model of a population evolving over a network of selectively neutral genotypes. We show that the population’s limit distribution on the neutral network is solely determined by the network topology and given by the principal eigenvector of the network’s adjacency matrix. Moreover, the average number of neutral mutant neighbors per individual is given by the matrix spectral radius. These results quantify the extent to which populations evolve mutational robustness—the insensitivity of the phenotype to mutations—and thus reduce genetic load. Because the average neutrality is independent of evolutionary parameters—such as mutation rate, population size, and selective advantage—one can infer global statistics of neutral network topology by using simple population data available from in vitro or in vivo evolution. Populations evolving on neutral networks of RNA secondary structures show excellent agreement with our theoretical predictions.
Kimura’s (1) contention that a majority of genotypic change in evolution is selectively neutral has gained renewed attention with the recent analysis of evolutionary optimization methods (2, 3) and the discovery of neutral networks in genotype-phenotype models for RNA secondary structure (4–6) and protein structure (7). It was found that collections of mutually neutral genotypes, which are connected via single mutational steps, form extended networks that permeate large regions of genotype space. Intuitively, a large degeneracy in genotype-phenotype maps, when combined with the high connectivity of (high-dimensional) genotype spaces, readily leads to such extended neutral networks. This intuition is now supported by recent theoretical results (8, 9).
In evolution of ribozymes in vitro, mutations responsible for an increase in fitness are only a small minority of the total number of accepted mutations (10). This fact indicates that, even in adaptive evolution, the majority of point mutations is neutral. The fact that only a minority of loci is conserved in sequences evolved from a single ancestor similarly indicates a high degeneracy in ribozymal genotype-phenotype maps (11). Neutrality is also implicated in experiments where RNA sequences evolve a given structure starting from a range of different initial genotypes (12). More generally, neutrality in RNA and protein genotype-phenotype maps is indicated by the observation that their structures are much better conserved during evolution than their sequences (13, 14).
Given the presence of neutral networks that preserve structure or function in sequence space, one asks, how does an evolving population distribute itself over a neutral network? Can we detect and analyze structural properties of neutral networks from data on biological or in vitro populations? To what extent does a population evolve toward highly connected parts of the network, resulting in sequences that are relatively insensitive to mutations? Such mutational robustness has been observed in biological RNA structures (15) and in simulations of the evolution of RNA secondary structure (16). However, an analytical understanding of the phenomenon, the underlying mechanisms, and their dependence on evolutionary parameters—such as mutation rate, population size, selection advantage, and the topology of the neutral network—has up to now not been available.
Here, we develop a model for the evolution of populations on neutral networks and show analytically that, for biologically relevant population sizes and mutation rates, a population’s distribution over a neutral network is determined solely by the network’s topology. Consequently, one can infer important structural information about neutral networks from data on evolving populations, even without specific knowledge of the evolutionary parameters. Simulations of the evolution of a population of RNA sequences, evolving on a neutral network defined with respect to secondary structure, confirm our theoretical predictions and illustrate their application to inferring network topology.
Modeling Neutrality
We assume that genotype space contains a neutral network of high, but equal fitness, genotypes on which the majority of a population is concentrated and that the neighboring parts of genotype space consist of genotypes with markedly lower fitness. The genotype space consists of all sequences of length L over a finite alphabet 𝒜 of A symbols. The neutral network on which the population moves can be most naturally regarded as a graph G embedded in this genotype space. The vertex set of G consists of all genotypes that are on the neutral network; its size is denoted by |G|. Two vertices are connected by an edge if and only if they differ by a single point mutation.
We will investigate the dynamics of a population evolving on this neutral network and analyze the dependence of several population statistics on the topology of the graph G. With these results, we will then show how measuring various population statistics enables one to infer the structural properties of G.
For the evolutionary process, we assume a discrete-generation selection-mutation dynamics with constant population size M. Individuals on the neutral network G have a fitness of σ. Individuals outside the neutral network have fitnesses that are considerably smaller than σ. With the approximations we use, the exact fitness values for genotypes off G turn out to be immaterial. Each generation, M individuals are selected with replacement and with probability proportional to fitness and then mutated with probability μ. These individuals form the next generation.
This dynamical system is a discrete-time version of Eigen’s molecular-evolution model (17). Our analysis can be translated directly to the continuous-time equations for the Eigen model. The results remain essentially unchanged.
Although our analysis can be extended to more complicated mutation schemes, we will assume that only single point mutations can occur at each reproduction of an individual. With probability μ, one of the L symbols is chosen with uniform probability and is mutated to one of the A − 1 other symbols. Thus, with a mutation, a genotype s moves with uniform probability to one of the L(A − 1) neighboring points in genotype space.
For the results presented below to hold, it is not necessary that all genotypes in G have exactly the same fitness. As in any model of neutral evolution (1, 18), it is sufficient to assume that the fitness differentials between distinct genotypes in G are smaller than the reciprocal 1/M of the population size. Additionally, we assume that the fitness differentials between genotypes in G and genotypes outside G are much larger than 1/M. These assumptions break down when there is a continuum of fitness differentials between genotypes or in the case of very small population size, which readily allows the spreading of mildly deleterious mutations (19).
Infinite-Population Solution
The first step is to solve for the asymptotic distribution of the population over the neutral network G in the limit of very large population size.
Once the (infinite) population has come to equilibrium, there will be a constant proportion P of the population located on the network G and a constant average fitness 〈f〉 in the population. Under selection, the proportion of individuals on the neutral network increases from P to σP/〈f〉. Under mutation, a proportion 〈ν〉 of these individuals remains on the network, whereas a proportion 1−〈ν〉 falls off the neutral network to lower fitness. At the same time, a proportion Q of individuals located outside G mutate onto the network so that an equal proportion P ends up on G in the next generation. Thus, at equilibrium, we have a balance equation:
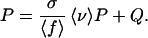 |
1 |
In general, the contribution of Q is negligible. As mentioned above, we assume that the fitness σ of the network genotypes is substantially larger than the fitnesses of those off the neutral network and that the mutation rate is small enough so that the bulk of the population is located on the neutral network. Moreover, because their fitnesses are smaller than the average fitness 〈f〉, only a fraction of the individuals outside the network G produces offspring for the next generation. Of this fraction, only a small fraction mutates onto the neutral network G. Therefore, we neglect the term Q in Eq. 1 and obtain
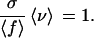 |
2 |
This equation expresses the balance between selection expanding the population on the network and deleterious mutations reducing it by moving individuals off.
Eq. 2 can also be phrased in terms of the genetic load ℒ, defined as the relative distance of the average fitness from the optimum fitness in the population: ℒ = (σ −〈f〉)/σ. ℒ measures the selection pressure that the population is experiencing. According to Eq. 2, in the presence of neutrality, ℒ is simply equal to the proportion 1 −〈ν〉 of offspring that falls off the network G. Thus, Eq. 2 states that ℒ is equal to the proportion of deleterious mutations per generation, in accordance with Haldane’s original result (20).
Under mutation, an individual located at genotype s of G with vertex degree ds (the number of neutral-mutant neighbors) has probability
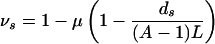 |
3 |
to remain on the neutral network G. If asymptotically a fraction Ps of the population is located at genotype s, then 〈ν〉 is simply the average of νs over the asymptotic distribution on the network 〈ν〉 = Σs∈GνsPs/P. As Eq. 3 shows, the average 〈ν〉 is simply related to the population neutrality 〈d〉 = Σs∈GdsPs/P. Moreover, using Eq. 2, we can directly relate the population neutrality 〈d〉 to the average fitness 〈f〉:
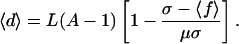 |
4 |
Despite the fact that neither the details of the topology of G nor the fitness values of the genotypes lying off the neutral network are given, one can relate the population neutrality 〈d〉 of the individuals on the neutral network directly to the average fitness 〈f〉 in the population. It may seem surprising that such a simple relation is possible at all. Because the population consists partly of sequences off the neutral network, one expects that the average fitness is determined in part by the fitnesses of these sequences. However, under the assumption that back mutations from low-fitness genotypes off the neutral network onto G are negligible, the fitnesses of sequences outside G influence only the total proportion P of individuals on the network but not the average fitness in the population.
Eq. 4 shows that the population neutrality 〈d〉 can be inferred from the average fitness and other parameters—such as mutation rate. However, as we will now show, the population neutrality 〈d〉 can also be obtained independently from knowledge of the topology of G alone.
The asymptotic equilibrium proportions {Ps} of the population at network nodes s are the solutions of the simultaneous equations:
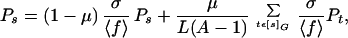 |
5 |
where [s]G is the set of neighbors of s that are also on the network G. By using Eq. 4, Eq. 5 can be rewritten as an eigenvector equation:
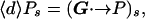 |
6 |
where G is the adjacency matrix of the graph G:
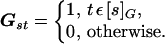 |
7 |
Because G is nonnegative and the neutral network G is connected, the adjacency matrix is irreducible. Therefore, the theorems of Frobenius–Perron for nonnegative irreducible matrices apply (21). These imply that the proportions Psof the limit distribution on the network are given by the principal eigenvector of the graph adjacency matrix G. Moreover, the population neutrality is equal to G’s spectral radius ρ: 〈d〉 = ρ. In this way, one concludes that, asymptotically, the population neutrality 〈d〉 is independent of evolutionary parameters (μ, L, σ) and of the fitness values of the genotypes outside the neutral network. It is a function only of the neutral network topology as determined by the adjacency matrix G.
In genetic load terminology, our results imply that
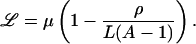 |
8 |
We see that Haldane’s result (ℒ = μ; ref. 20) is recovered in the absence of neutrality (ρ = 0). In the presence of neutrality, the genetic load is reduced by a factor that depends only on the spectral radius ρ of the network’s adjacency matrix.
The fortunate circumstance that the population neutrality depends only on the topology of G allows us to consider several practical consequences. Note that knowledge of μ, σ, and 〈f〉 allows one to infer a dominant feature of the topology of G, namely, the spectral radius ρ of its adjacency matrix. In evolution experiments in vitro in which biomolecules have evolved, say, to bind a particular ligand (22), by measuring the proportion 〈ν〉 of molecules that remain functional after mutation, we can now infer the spectral radius ρ of their neutral network. In other situations, such as in the bacterial evolution experiments described in ref. 23, it might be more natural to measure the average fitness 〈f〉 of an evolving population and then use Eq. 4 to infer the population neutrality 〈d〉 of viable genotypes in sequence space.
Blind and Myopic Random Neutral Walks
Above, we solved for the asymptotic average neutrality 〈d〉 of an infinite population under selection and mutation dynamics and showed that it was uniquely determined by the topology of the neutral network G. To put this result in perspective, we now compare the population neutrality 〈d〉 with the effective neutralities observed under two different kinds of random walks over G. The results illustrate informative extremes of how network topology determines the population dynamics on neutral networks and affects the evolution of robustness.
The first kind of random walk that we consider is generally referred to as a “blind ant” random walk. An ant starts out on a randomly chosen node of G. At each time step, it chooses one of its L(A − 1) neighbors at random. If the chosen neighbor is on G, the ant steps to this node; otherwise, it remains at the current node for another time step. It is easy to show that this random walk asymptotically spends equal amounts of time at all of the nodes of G (24). Therefore, the network neutrality d̄ of the nodes visited under this type of random walk is simply given by
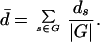 |
9 |
It is instructive to compare this result with the effective neutrality observed under another random walk, called the “myopic ant”. An ant again starts at a random node s∈G. At each time step, the ant determines the set [s]G of network neighbors of s and then steps to one at random. Under this random walk, the asymptotic proportion Ps of time spent at node s is proportional to the node degree ds (24). It turns out that the myopic neutrality d̂ seen by this ant can be expressed in terms of the mean d̄ and variance Var(d) of node degrees over G:
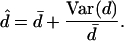 |
10 |
The network and myopic neutralities d̄ and d̂ are thus directly given in terms of local statistics of the distribution of vertex degrees, whereas the population neutrality 〈d〉 is given by ρ, the spectral radius of the adjacency matrix of G. The latter is an essentially global property of G.
Mutational Robustness
With these cases in mind, we now consider how different network topologies are reflected by these neutralities. In prototype models of populations evolving on neutral networks, the networks are often assumed to be or are approximated as regular graphs (3, 9, 25–27). If the graph G is, in fact, regular, each node has the same degree d and, obviously, one has 〈d〉 = d̄ = d̂ = d.
In more realistic neutral networks, one expects the neutralities of G to vary over the network. When this occurs, the population neutrality is typically larger than the network neutrality: 〈d〉 = ρ > d̄. Their difference precisely quantifies the extent to which a population seeks out the most connected areas of the neutral network. Thus, a population will evolve a mutational robustness that is larger than if the population were to spread uniformly over the neutral network. Additionally, the mutational robustness tends to increase during the transient phase in which the population relaxes toward its asymptotic distribution.
Assume, for instance, that initially the population is located entirely off the neutral network G at lower-fitness sequences. At some time, a genotype s∈G is discovered by the population. To a rough approximation, one can assume that the probability of genotype s being discovered first is proportional to the number of neighbors, L(A − 1) − ds, that s has outside the neutral network. Therefore, the population neutrality 〈d0〉 when the population first enters the neutral network G is approximately given by
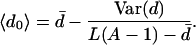 |
11 |
We define the excess robustness r to be the relative increase in neutrality between this initial neutrality and the (asymptotic) population neutrality
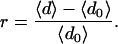 |
12 |
For networks that are sparse, i.e., d̄ ≪ L(A − 1), Eq. 12 is well approximated by r ≈ (〈d〉 − d̄)/d̄. Note that, although r is defined in terms of population statistics, the preceding results have shown that this robustness is only a function of the topology of G and should thus be considered a property of the network.
Finite-Population Effects
Our analysis of the population distribution on the neutral network G assumed an infinite population. For finite populations, it is well known that sampling fluctuations cause a population to converge, which raises a question: to what extent does the asymptotic distribution Ps still describe the distribution over the network for small populations? As a finite population diffuses over a neutral network (28), one might hope that the time average of the distribution over G is still given by Ps. Indeed, the simulation results shown below indicate that for moderately large population sizes, this approximation is the case. However, a simple argument shows that it cannot be true for arbitrarily small populations.
Assume that the population size M is so small that the product of mutation rate and population size is much smaller than one; i.e., Mμ ≪ 1. In this limit, the population, at any point in time, is converged completely onto a single genotype s on the neutral network G. With probability Mμ, a single mutant will be produced at each generation. Such a mutant is equally likely to be one of the L(A − 1) neighbors of s. If this mutant is not on G, it will quickly disappear because of selection. However, if the mutant is on the neutral network, there is a probability 1/M that it will take over the population. When this happens, the population effectively will have taken a random-walk step on the network, of exactly the kind followed by the blind ant. Therefore, for Mμ ≪ 1, the population neutrality will be equal to the network neutrality: 〈ν〉 = d̄. In this regime, r ≈ 0, and excess mutational robustness will not emerge through evolution.
The extent to which the population neutrality 〈d〉 approaches its infinite population value ρ is determined by the extent to which the population is not converged by sampling fluctuations. In neutral evolution, population convergence is generally only a function of the product Mμ (29–31). Thus, as the product Mμ ranges from values much smaller than one to values much larger than one, we predict that the population neutrality 〈d〉 shifts from the network neutrality d̄ to the infinite-population neutrality, given by G’s spectral radius ρ.
RNA Evolution on Structurally Neutral Networks
The evolution of RNA molecules in a simulated flow reactor provides an excellent arena in which to test the theoretical predictions of evolved mutational robustness. The replication rates (fitnesses) were chosen to be a function only of the secondary structures of the RNA molecules. The secondary structure of RNA is an essential aspect of its phenotype, as documented by its conservation in evolution (13) and the convergent in vitro evolution toward a similar secondary structure when selecting for a specific function (12). RNA secondary structure prediction based on free-energy minimization is a standard tool in experimental biology and has been shown to be reliable, especially when the minimum free-energy structure is thermodynamically well defined (32). RNA secondary structures were determined with the vienna package (33), which uses the free energies described in ref. 34. Free energies of dangling ends were set to zero.
The neutral network G on which the population evolves consists of all RNA molecules of length L = 18 that fold into a particular target structure. A target structure (Fig. 1) was selected that contains sufficient variation in its neutrality to test the theory, but is not so large as to preclude an exhaustive analysis of its neutral network topology.
Figure 1.

The target RNA secondary structure.
By using only single point mutations per replication, purine–pyrimidine base pairs (G–C, G–U, A–U) can mutate into each other, but not into pyrimidine–purine (C–G, U–G, U–A) base pairs. The target structure contains 6 base pairs that can each be taken from one or the other of these two sets. Thus, the approximately 2 × 108 sequences that are consistent with the target’s base pairs separate into 26 = 64 disjoint sets. Of these, we analyzed the set in which all base pairs were of the purine–pyrimidine type and found that the set contains two neutral networks of 51,028 and 5,169 sequences that fold into the target structure. Simulations were performed on the largest of the two. The exhaustive enumeration of this network showed that it has a network neutrality of d̄ ≈ 12.0 with a standard deviation of 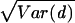 ≈ 3.4, a maximum neutrality of ds = 24, and a minimum of ds = 1. The spectral radius of the network’s 51,028 × 51,028 adjacency matrix is ρ ≈ 15.7. The theory predicts that, when Mμ ≫ 1, the population neutrality should converge to this value.
≈ 3.4, a maximum neutrality of ds = 24, and a minimum of ds = 1. The spectral radius of the network’s 51,028 × 51,028 adjacency matrix is ρ ≈ 15.7. The theory predicts that, when Mμ ≫ 1, the population neutrality should converge to this value.
The simulated flow reactor contains a population of replicating and mutating RNA sequences (17, 35). The replication rate of a molecule depends on whether its calculated minimum free-energy structure equals that of the target: sequences that fold into the target structure replicate on average once per time unit, whereas all other sequences replicate once per 104 time units on average. During replication, the progeny of a sequence has probability μ of a single point mutation. Selection pressure in the flow reactor is induced by an adaptive dilution flux that keeps the total RNA population fluctuating around a constant capacity M.
Evolution was seeded from various starting sequences with either a relatively high or a relatively low neutrality. Independent of the starting point, the population neutrality converged to the predicted value, as shown in Fig. 2.
Figure 2.

The evolution of RNA mutational robustness: convergence of the population’s average neutrality to the theoretical value, 〈d〉 = ρ ≈ 15.7, predicted by the spectral radius of G (upper dashed line). The network’s average neutrality d̄ is represented by the lower dashed line. Simulations used a population size of M = 104 and mutation rates of μ = 0.5 and μ = 0.1 per sequence. Simulations were started at sequences with either a relatively large number of neutral neighbors (ds = 24) (upper curves for each mutation rate) or a small number (ds = 5) (lower curves).
Subsequently, we tested the finite-population effects on the population’s average neutrality at several different mutation rates. Fig. 3 shows the dependence of the asymptotic average population neutrality on population size M and mutation rate μ. As expected, the population neutrality depends on only the product Mμ of population size and mutation rate. For small Mμ, the population neutrality increases with increasing Mμ, until Mμ ≈ 500, where it saturates at the predicted value of 〈d〉 ≈ 15.7. Because small populations do not form a stationary distribution over the neutral net but diffuse over it (28), the average population neutrality at each generation may fluctuate considerably for small populations. Theoretically, sampling fluctuations in the proportions of individuals at different nodes of the network scale inversely with the square root of the population size. We therefore expect the fluctuations in population neutrality to scale as the inverse square root of the population size as well. This prediction was indeed observed in our simulations.
Figure 3.

Dependence of the population neutrality on mutation rate μ and population size M. Simulations used three mutation rates, μ∈ {0.5, 0.1, 0.01}, and a range of population sizes, M∈ {10,000, 5,000, 1,000, 500, 250, 100, 50, 20}. The results show that the evolved neutrality in the population depends on the product Mμ of population size and mutation rate. Neutrality increases with increasing Mμ and saturates when Mμ > 500. When Mμ < 1 population neutrality approximates G’s average neutrality d̄ ≈ 12.0. When Mμ > 500, population neutrality converges to the spectral radius of the network’s adjacency matrix, ρ ≈ 15.7.
Finally, the fact that r ≈ 0.31 for this neutral network shows that, under selection and mutation, a population will evolve a mutational robustness that is 31% higher than if it were to spread randomly over the network.
Conclusions
We have shown that, under neutral evolution, a population does not move over a neutral network in an entirely random fashion but tends to concentrate at highly connected parts of the network, resulting in phenotypes that are relatively robust against mutations. Moreover, the average number of point mutations that leave the phenotype unaltered is given by the spectral radius of the neutral network’s adjacency matrix. Thus, our theory provides an analytical foundation for the intuitive notion that evolution selects genotypes that are mutationally robust and that reduce genetic load.
Perhaps surprisingly, the tendency to evolve toward highly connected parts of the network is independent of evolutionary parameters—such as mutation rate, selection advantage, and population size (as long as Mμ ≫ 1)—and is solely determined by the network’s topology. One consequence is that one can infer properties of the neutral network’s topology from simple population statistics.
Simulations with neutral networks of RNA secondary structures confirm the theoretical results and show that, even for moderate population sizes, the population neutrality converges to the infinite-population prediction. Typical sizes of in vitro populations are such that the data obtained from experiments are expected to accord with the infinite-population results derived here. It seems possible then to devise in vitro experiments that, by using the results outlined above, would allow one to obtain information about the topological structure of neutral networks of biomolecules with similar functionality.
Finally, we focused only on the asymptotic distribution of the population on the neutral network, but how did the population attain this equilibrium? The transient relaxation dynamics, such as those shown in Fig. 2, can be analyzed in terms of the nonprincipal eigenvectors and eigenvalues of the adjacency matrix G. Because the topology of a graph is almost entirely determined by the eigensystem of its adjacency matrix, one should, in principle, be able to infer the complete structure of the neutral network from accurate measurements of the transient population dynamics.
Acknowledgments
We thank Sergey Gavrilets for pointing us to the connections of our results with genetic load and the participants of the Santa Fe Institute Workshop on Evolutionary Dynamics for stimulating this work, which was supported in part at the Santa Fe Institute by National Science Foundation Grant IRI-9705830, by Sandia National Laboratory Contract BE-7463, and by Keck Foundation Grant 98-1677. M.H. gratefully acknowledges support from a fellowship of the Royal Netherlands Academy of Arts and Sciences.
References
- 1.Kimura M. The Neutral Theory of Molecular Evolution. Cambridge, U.K.: Cambridge Univ. Press; 1983. [Google Scholar]
- 2.Crutchfield J P, Mitchell M. Proc Natl Acad Sci USA. 1995;92:10742–10746. doi: 10.1073/pnas.92.23.10742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Nimwegen E, Crutchfield J P, Mitchell M. Phys Lett A. 1997;229:144–150. [Google Scholar]
- 4.Fontana W, Stadler P F, Bornberg-Bauer E G, Griesmacher T, Hofacker I L, Tacker M, Tarazona P, Weinberger E D, Schuster P. Phys Rev E. 1992;47:2083–2099. doi: 10.1103/physreve.47.2083. [DOI] [PubMed] [Google Scholar]
- 5.Grüner W, Giegerich R, Strothmann D, Reidys C M, Weber J, Hofacker I L, Stadler P F, Schuster P. Monatsh Chem. 1996;127:355–389. [Google Scholar]
- 6.Schuster P, Fontana W, Stadler P F, Hofacker I. Proc R Soc London B. 1994;255:279–284. doi: 10.1098/rspb.1994.0040. [DOI] [PubMed] [Google Scholar]
- 7.Babajide A, Hofacker I L, Sippl M J, Stadler P F. Fold Des. 1997;2:261–269. doi: 10.1016/S1359-0278(97)00037-0. [DOI] [PubMed] [Google Scholar]
- 8.Gavrilets S, Gravner J. J Theor Biol. 1997;184:51–64. doi: 10.1006/jtbi.1996.0242. [DOI] [PubMed] [Google Scholar]
- 9.Reidys C M, Stadler P F, Schuster P. Bull Math Biol. 1997;59:339–397. doi: 10.1007/BF02462007. [DOI] [PubMed] [Google Scholar]
- 10.Wright M C, Joyce G F. Science. 1997;276:614–617. doi: 10.1126/science.276.5312.614. [DOI] [PubMed] [Google Scholar]
- 11.Landweber L F, Pokrovskaya I D. Proc Natl Acad Sci USA. 1999;96:173–178. doi: 10.1073/pnas.96.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ekland E H, Bartel D P. Nature (London) 1996;382:373–376. doi: 10.1038/382373a0. [DOI] [PubMed] [Google Scholar]
- 13.Gutell R, Power A, Hertz G Z, Putz E J, Stormo G D. Nucleic Acids Res. 1993;20:5785–5795. doi: 10.1093/nar/20.21.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huynen M A, Doerks T, Eisenhaber F, Orengo C, Sunyaev S, Yuan Y, Bork P. J Mol Biol. 1998;280:323–326. doi: 10.1006/jmbi.1998.1884. [DOI] [PubMed] [Google Scholar]
- 15.Huynen M A, Konings D, Hogeweg P. J Theor Biol. 1993;165:251–267. doi: 10.1006/jtbi.1993.1188. [DOI] [PubMed] [Google Scholar]
- 16.Huynen M A, Hogeweg P. J Mol Evol. 1994;39:71–79. doi: 10.1007/BF00178251. [DOI] [PubMed] [Google Scholar]
- 17.Eigen M. Naturwissenschaften. 1971;58:465–523. doi: 10.1007/BF00623322. [DOI] [PubMed] [Google Scholar]
- 18.Gillespie J H. The Causes of Molecular Evolution. New York: Oxford Univ. Press; 1991. [Google Scholar]
- 19.Moran N A. Proc Natl Acad Sci USA. 1996;93:2873–2878. doi: 10.1073/pnas.93.7.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haldane J B S. Am Nat. 1937;71:337–349. [Google Scholar]
- 21.Gantmacher F R. Applications of the Theory of Matrices. New York: Interscience; 1959. [Google Scholar]
- 22.Türk C. Methods Mol Biol. 1997;67:219–230. doi: 10.1385/0-89603-483-6:219. [DOI] [PubMed] [Google Scholar]
- 23.Elena S F, Cooper V S, Lenski R E. Science. 1996;272:1802–1804. doi: 10.1126/science.272.5269.1802. [DOI] [PubMed] [Google Scholar]
- 24.Hughes B D. Random Walks and Random Environments. Vol. 2. Oxford: Clarendon; 1996. [Google Scholar]
- 25.Forst C V, Reidys C M, Weber J. In: Advances in Artificial Life, Lecture Notes in Artificial Intelligence. Moran F, Moreno A, Merelo J, Chacon P, editors. Vol. 929. Springer; 1995. pp. 128–147. [Google Scholar]
- 26.Reidys C M. Ph. D. thesis. Jena, Germany: Friedrich Schiller Universität; 1995. [Google Scholar]
- 27.van Nimwegen, E., Crutchfield, J. P. & Mitchell, M. (1999) Theor. Comput. Sci., in press.
- 28.Huynen M A, Stadler P F, Fontana W. Proc Natl Acad Sci USA. 1996;93:397–401. doi: 10.1073/pnas.93.1.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Derrida B, Peliti L. Bull Math Biol. 1991;53(3):355–382. [Google Scholar]
- 30.Kimura M. J Appl Probl. 1964;1:177–232. [Google Scholar]
- 31.Wright S. Genetics. 1931;16:97–159. doi: 10.1093/genetics/16.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huynen M A, Gutell R, Konings D. J Mol Biol. 1997;267:1104–1112. doi: 10.1006/jmbi.1997.0889. [DOI] [PubMed] [Google Scholar]
- 33.Hofacker I L, Fontana W, Stadler P F, Bonhoeffer S, Tacker M, Schuster P. Monatsh Chem. 1994;125:167–188. [Google Scholar]
- 34.Walter A E, Turner D H, Kim J, Lyttle M H, Muller P, Mathews D H, Zuker M. Proc Natl Acad Sci USA. 1994;91:9218–9222. doi: 10.1073/pnas.91.20.9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fontana W, Schnabl W, Schuster P. Phys Rev A. 1989;40:3301–3321. doi: 10.1103/physreva.40.3301. [DOI] [PubMed] [Google Scholar]