

Abstract
Whole-genome analysis using high-density single-nucleotide–polymorphism oligonucleotide arrays allows identification of microdeletions, microduplications, and uniparental disomies. We studied 67 children with unexplained mental retardation with normal karyotypes, as assessed by G-banded chromosome analyses. Their DNAs were analyzed with Affymetrix 100K arrays. We detected 11 copy-number variations that most likely are causative of mental retardation, because they either arose de novo (9 cases) and/or overlapped with known microdeletions (2 cases). The eight deletions and three duplications varied in size from 200 kb to 7.5 Mb. Of the 11 copy-number variations, 5 were flanked by low-copy repeats. Two of those, on chromosomes 15q25.2 and Xp22.31, have not been described before and have a high probability of being causative of new deletion and duplication syndromes, respectively. In one patient, we found a deletion affecting only a single gene, MBD5, which codes for the methyl-CpG-binding domain protein 5. In addition to the 67 children, we investigated 4 mentally retarded children with apparent balanced translocations and detected four deletions at breakpoint regions ranging in size from 1.1 to 14 Mb.
Mental retardation (MR) has a prevalence of ∼2%–3%.1 Whereas the frequencies of mild MR differ among studies, most authors agree that severe MR, defined as an intelligence quotient (IQ) of <50, has a prevalence of 0.3%–0.4%.2 Aside from trisomy 21, which accounts for ∼5%–15% of MR cases,2 chromosome abnormalities are detected in not more than 5% of cases by cytogenetic analysis of chromosomes prepared from peripheral-blood lymphocytes.3,4 The resolution of cytogenetic techniques is typically 5–10 Mb. Smaller rearrangements in the subtelomeric regions have been detected in ∼5% of affected children by regional analysis using FISH or muliplex ligation-dependent probe amplification.5,6 It has further been shown that another 10%–20% of rearrangements can be found by array-based comparative genomic hybridization (array CGH).7–10 Recently, high-density SNP microarrays have been evaluated for this purpose.11,12 Special efforts have been undertaken to analyze regions that are flanked by segmental duplication, since these are known to predispose to recurrent rearrangements in genomic disorders.13 Array CGH and SNP microarrays have also revealed the presence of several thousand copy-number variations (CNVs) in the general population,14 complicating the interpretation of the findings in disease cases.
Here, we used Affymetrix GeneChip Human Mapping 100K arrays (100K arrays) to analyze DNA from 67 children with unexplained MR. Gains or losses that are likely to be causative of the disease were present in 11 (16%) of the children. We also show that the resolution of this array is sufficient to detect single gene deletions in regions with low gene content and that the determination of the breakpoints is precise enough to amplify the junction fragments after narrowing the breakpoint with few quantitative PCR (qPCR) products, unless the breakpoints are flanked by repetitive sequences or the rearrangement is complex.15
Material and Methods
Patients
The 67 children with unexplained MR were ascertained mainly in a single human genetics practice (that of S.S. and B.K.). We classified MR as mild or severe on the basis of clinical criteria and the competence of adaptive behavior. If available, an IQ of <50 on a standardized IQ test was used to classify MR as severe. According to these criteria, 61 cases were classified as severe, and 3 cases as mild. Most of the children had additional but often mild symptoms (table A1 in appendix A). Children with brain malformations were excluded from the study. All children had normal G-banded chromosomes (banding level 500–550). In 29 of the cases, cytogenetic analysis was performed in two different laboratories, with identical results. FISH with subtelomeric probes and metabolic investigations were inconspicuous in 42 and 11 children, respectively. DNA samples were available from the parents of 44 children, which allowed us to investigate whether CNVs originated de novo and to check which parental allele was lost, in cases of de novo deletions. In addition to the children with normal G-banded chromosomes, we analyzed DNA from four mentally retarded children in whom de novo balanced reciprocal translocations had been detected by cytogenetic analysis. DNA samples from 415 children referred for molecular diagnostics of the FMR1 gene but for whom no mutation was found were used for sequence analysis of the MBD5 gene. The study was approved by the Ethics Committee of the Medical Department of the Technical University Munich.
Array CGH
Genomic DNA was isolated from peripheral-blood leukocytes by use of a modified salting-out procedure.16 DNA concentrations were measured with a NanoDrop spectrophotometer (ND-1000 V.3.1.2). The 100K arrays consist of two arrays, the Xba240 and the Hind240 array, which together include 116,204 SNPs with an average spacing of 23.6 kb. DNA was processed in accordance with the manufacturer′s instructions. In brief, 250 ng of total genomic DNA was digested with XbaI or HindIII and then was ligated to adaptors. A generic primer that recognizes the adaptor sequence was used to amplify the adaptor-ligated DNA fragments in a GeneAmp PCR System 9700 (Applied Biosystems). After purification with the Macherey-Nagel NucleoFast 96 PCR Clean-Up ultrafiltration technology, a total of 40 μg of PCR product was fragmented and labeled with biotin. Hybridization was performed in the Affymetrix GeneChip Hybridization Oven 640. Arrays were washed and stained with the Affymetrix GeneChip Fluidics Station 450 and were scanned with the Affymetrix GeneChip Scanner 3000 7G. Image processing was performed with GCOS 1.4, and genotypes were called with GTYPE 4.0 software by use of the default call threshold of 0.25.
Data Analysis
To account for experimental variations, we hierarchically clustered the euclidean distance matrix generated from the binary logarithm of the sum of the median-normalized intensity values of both alleles. The arrays were then ordered into groups with a similar intensity profile. For each group, copy numbers were calculated as follows. We first determined the raw intensity values at each SNP locus by calculating separately the mean of the perfect-match probes for the A and B alleles. The raw intensity values were then median normalized. The log2 ratio of the intensity values were calculated separately for the three genotypes (AA, AB, and BB) and the no-calls by dividing the normalized intensity values of the test array by the median values of all arrays with the same genotype for each SNP locus. One can show that the noise is lower when genotype-specific intensity values are used than when the sum of the intensity of both alleles for all genotypes is used. To make intensity values from male and female X chromosomes comparable, the mean dosage of the male SNPs on the X chromosome was adjusted to the mean dosage of the autosomal SNPs.
In many dosage plots, we observed that the mean dosage level depended on the length of the restriction fragments. Thus, we corrected for this dependence by using quadratic regression as described elsewhere.17 This increased the signal-to-noise ratio (SNR) further.
We used two measures for the assessment of data quality. First, we calculated the SD and the median absolute deviation (MAD) of the final log2 intensity ratios. Second, we calculated an SNR in male DNA samples by substracting the median log2 intensity ratio of the X-chromosomal SNPs from the median log2 ratio of the autosomal SNPs. This difference was then divided by half the sum of the MAD of the log2 intensity ratio of the autosomal and X-chromosomal SNPs,
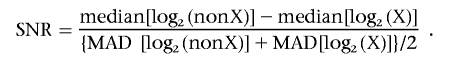 |
Note that the numerator measures the separation between the log2 intensity levels of autosomal (“nonX”) and X-chromosomal SNPs in males. Because of technological nonlinearities, this difference is <1. We found an average difference of 0.63 in 95 HindIII arrays and of 0.70 in 98 XbaI arrays. The denominator estimates the scale of the variation, putting equal emphasis on the autosomal and X-chromosomal SNPs. All characteristics are estimated robustly, to safeguard against deviations from the standard normal model and against outliers.
The minimal number of consecutive lowly expressed SNPs that significantly indicates a deletion was calculated with the expression
 |
where sep1copy,2copies is the median log2 intensity ratio of SNPs with 1 copy or 2 copies, respectively; P denotes probabilities for CN, the measured copy number of a SNP with real copy number 2 under a normal model; and Φ is the distribution function of the standard normal distribution. The factor 67×105 indicates the Bonferroni correction for multiple testing.
We also implemented tools to select regions conspicuous for gains and losses and to detect loss of parental alleles. To select CNVs, we took into account the array-specific SD. The number of consecutive SNPs had to be larger when the SD was higher. As a minimum, we used the median of five consecutive SNPs to define a CNV. Conspicuous regions were compared with known CNVs, as provided by the Database of Genomic Variants and the DECIPHER database. All analysis tools were implemented as R or Perl scripts (available at Scripts Web site).
qPCR
We designed 1–3 amplicons for validation of each CNV (table A2 in appendix A). qPCR was performed on a 7900HT real-time PCR system (Applied Biosystems) by use of SYBR Green I for detection. Reaction mixtures contained 0.2 μM of each primer and 10 μl of 2 × Power SYBR Green PCR Master Mix (Applied Biosystems). Each assay included a no-template control, two male and two female control DNAs, and the patient DNA at a final concentration of 2.5 ng/μl in duplicate, in a total volume of 20 μl. We used the same cycling conditions for all reactions: initial step at 50°C for 2 min, denaturation at 95°C for 10 min, then 40 cycles at 95°C for 15 s, and a combined annealing and extension at 58°C for 60 s. To exclude the presence of unspecific products, a melting-curve analysis of the products was performed after completion of the amplification. To control for differences in DNA concentration, reaction efficiency, and threshold cycles (Ct), the Ct values were normalized using the Ct value of a reference gene (BNC1) for each DNA sample. Analysis was performed as relative quantification by use of the comparative Ct method.
Breakpoint Analysis
We used qPCR to narrow the interval of the deletion breakpoints to 2–5 kb and generated junction fragments spanning the breakpoints by long-range PCR (table A2). Junction fragments were cloned into the pGEM-T vector (Promega) and were sequenced using BigDye v3.1 cycle sequencing (Applied Biosystems).
Mutation Analysis
Dye-binding/high-resolution DNA melting analysis was used to screen for single-nucleotide variations in MBD5. Unlabeled primers flanking each coding exon were designed with the ExonPrimer software (table A2). Genomic DNA (∼10 ng) was subjected to PCR amplification performed in 5 μl total volume containing 1× Thermo-Start High Performance Buffer (ABgene), MgCl2 (1.25 mM), 100 μM of each deoxynucleotide triphosphate, 0.25 U of Thermo-Start DNA polymerase (ABgene), primers (0.4 μM each), and the dye LCGreen PLUS (Idaho Technology) at 1× final concentration. After PCR, the samples were heated to 94°C for 30 s and then were cooled to 20°C before melting. Melting acquisition was performed on a LightScanner HR I 384 instrument (Idaho Technology) in accordance with the manufacturer′s standard procedures. Melting-curve data were analyzed with the standard software provided by Idaho Technology. Abnormal melting profiles were confirmed or excluded by sequencing of independent PCR products.
X-Chromosome Inactivation Analysis
For the investigation of the X-chromosome pattern, we used the trinucleotide repeat in the first exon of the androgen-receptor gene.18 A total of 2 μg of DNA was digested with 20 units of the methylation-sensitive restriction enzymes HhaI and HpaII (New England Biolabs). After overnight digestion, an aliquot of 2 μl was amplified by PCR by use of the FAM-labeled primer 5′-TCCAGAATCTGTTCCAGAGCGTGC-3′ and the unlabeled primer 5′-GCTGTGAAGGTTGCTGTTCCTCAT-3′. The PCR products were separated on an ABI Prism 3730 DNA sequencer and were analyzed with the GeneMapper software (Applied Biosystems).
Results
Array Data Analysis
DNA from 67 patients with MR and normal G-banded chromosomes and 4 patients with MR and a balanced translocation diagnosis were analyzed with 100K arrays. These arrays use a one-color technique; therefore, the normalized intensity values of a test chip have to be compared with one or more reference chips. We first assessed the data quality by using the SD of the copy-number values of all SNPs. Preliminary analysis showed that using the arrays processed during the study instead of external reference arrays provided by the manufacturer resulted in better data quality. We used hierarchical clustering to group arrays with similar intensity profiles and observed that the SNR could be increased when arrays with similar intensity profiles were analyzed together. The HindIII and XbaI arrays showed at least four and three groups, respectively, with clearly distinctive intensity profiles (fig. 1). Differences in the intensity profiles are most likely caused by DNA and experimental variations. Notably, arrays processed in a single experiment tended to show similar profiles. We eventually determined genotype-specific log2 intensity ratios for each SNP locus within each group. To increase the SNR further, we corrected for the dependence of the log2 intensity ratio on the fragment length by using quadratic regression. In total, we analyzed 169 array sets. The data quality was assessed by the SD and the MAD of the log2 intensity ratio of all autosomal probes; for the HindIII arrays, the mean SD was 0.19 and the mean MAD was 0.16, and, for the XbaI arrays, the mean SD was 0.18 and the mean MAD was 0.16. These values are in accordance with previously published reports.14,17
Figure 1. .

Heat map of the median normalized intensity values of 169 HindIII arrays. The log2 intensity values of 1,000 SNPs are displayed after hierarchical clustering. The sum of the intensity values of alleles A and B was used for this calculation. The columns contain the different arrays; the rows contain the different SNPs. The banner across the top of the heat map shows a color code for the four different time points at which the arrays were hybridized. Although the copy-number profile should be almost the same for each chip, it shows at least four clearly separated groups.
Different array types and platforms show different rates of the increase and decrease for the log2 intensities because of duplications and deletions, respectively. Thus, the MAD or SD alone is not appropriate for the comparison of different array types and platforms. To render our results comparable across different platforms, we estimated the SNR by using the difference of the log2 intensity ratios of the autosomal and the X-chromosomal SNPs of male samples. The HindIII arrays showed a mean SNR of 4.6, and the XbaI arrays showed a mean SNR of 5.14. These values mean that, on average, any six SNPs for HindIII arrays and five consecutive SNPs for XbaI arrays indicate a deletion at a 95% significance level over all 67 arrays. In our real data, the individual arrays had different SDs, and the log2 intensity ratios were not normally distributed. We therefore adjusted the number of consecutive SNPs that define a candidate region, depending on the SD, so that we obtained at most eight CNVs per sample.
CNVs
By applying our rules for CNV detection, we obtained 27 candidate regions in 24 patients (table 1). All regions that considerably overlapped with known CNVs provided by the Database of Genomic Variants had previously been excluded. The 27 candidate regions were evaluated by qPCR. The 14 regions that were defined by >20 SNPs could be confirmed by qPCR. Of the 13 regions defined by 5–20 SNPs, 5 were determined to be false-positive findings. In summary, we could confirm 22 CNVs (table 1). They varied in size from 10 kb to 7.5 Mb and contained up to 49 genes. Genotype information revealed that four CNVs originated on the paternal chromosome and two CNVs on the maternal chromosome. From these 22 confirmed CNVs, we further excluded five regions that were inherited from one parent and six regions that did not contain known coding regions or that could not be tested to determine whether they occurred de novo, because we did not have parental DNA available.
Table 1. .
qPCR[Note]
| SNP |
Overlap(%) |
||||||||||||
| Patient ID |
Chromosome | Gain or Loss | Start | End | No. of SNPs | Region Length (Mb) |
qPCRa | No. of qPCRs | Status of Inheritance | No. of Genes | CNVb | DGVc | Primer(s) |
| 27384 | 1q31.1-q31.3 | Loss | rs10494585 | rs10494695 | 464 | 7.75 | Yes | 1 | De novo | 17 | 10 | 100 | 8920 |
| 30437 | 2p25.3-p25.1 | Loss | rs2313466 | rs1964092 | 193 | 3.79 | Yes | 1 | De novo | 4 | 5 | 100 | 8930, 9069, and 9070 |
| 29922 | 8p23.1 | Loss | rs2945251 | rs2466115 | 177 | 3.9 | Yes | 3 | NA | 34 | 6 | 30 | 8926, 9061, and 9062 |
| 30375 | 3p25.3-p25.2 | Loss | rs10510400 | rs10510422 | 98 | 2.83 | Yes | 3 | De novo | 39 | 4 | 100 | 8928, 9065, and 9066 |
| 28735 | 12p13.33 | Loss | rs953385 | rs2283285 | 62 | 2.01 | Yes | 3 | De novo | 18 | 27 | 100 | 8924, 9077, and 9078 |
| 30428 | 17q21.31 | Loss | rs436667 | rs1918798 | 38 | 0.48 | Yes | 1 | De novo | 7 | 6 | 9 | 8929, 9067, and 9068 |
| 28283 | 7q21.13 | Loss | rs10487988 | rs10488004 | 36 | 0.27 | Yes | 3 | Paternal | 1 | 0 | 0 | 9651, 9652, and 9653 |
| 29836 | Xp22.31 | Gain | rs719632 | rs10521669 | 31 | 1.42 | Yes | 4 | Maternal | 5 | 47 | 100 | 9820, 9821, 9822, and 9060 |
| 27737 | 17p11.2 | Gain | rs4073940 | rs1373147 | 29 | 3.22 | Yes | 3 | NA | 49 | 11 | 100 | 8921, 9054, and 9055 |
| 28430 | 15q25.2 | Loss | rs17158372 | rs10520569 | 27 | 1.37 | Yes | 3 | De novo | 11 | 11 | 100 | 8923, 9058, and 9059 |
| 27581 | 9p23 | Loss | rs10511570 | rs1900218 | 23 | 0.28 | Yes | 1 | NA | 0 | 85 | 63 | 8934 |
| 28701 | 13q12.11 | Gain | rs9315234 | rs4570685 | 23 | 0.53 | Yes | 3 | De novo | 5 | 18 | 46 | 8922, 9056, and 9057 |
| 29945 | 3p13 | Loss | rs10511008 | rs10511014 | 22 | 0.44 | Yes | 5 | NA | 1 | 0 | 0 | 8939, 9063, and 9064 |
| 27581 | 1q25.2 | Gain | rs10494517 | rs7555418 | 20 | 0.59 | No | 3 | … | … | … | … | 8932, 9073, and 9074 |
| 27831 | 3q24 | Loss | rs2140300 | rs10513274 | 19 | 0.19 | Yes | 1 | NA | 0 | 24 | 100 | 8935 |
| 27526 | Xp22.31 | Gain | rs10521668 | rs5934414 | 15 | 0.34 | No | 4 | … | … | … | … | 9820, 9821, 9822, and 9060 |
| 29608 | 4q28.3 | Gain | rs1495265 | rs10518595 | 14 | 0.22 | Yes | 1 | NA | 0 | 100 | 34 | 8937 |
| 27733 | 11q14.1 | Gain | rs870066 | rs10501436 | 13 | 0.11 | No | 1 | … | … | … | … | 8941 |
| 29195 | 2q23.1 | Loss | rs2890919 | rs10497034 | 13 | 0.2 | Yes | 2 | De novo | … | 0 | 0 | 8936 and 8948 |
| 27733 | 9p23 | Loss | rs372412 | rs983282 | 11 | 0.14 | Yes | 1 | NA | 0 | 37 | 13 | 8940 |
| 29996 | 5p15.2 | Loss | rs31953 | rs26152 | 10 | 0.03 | Yes | 1 | NA | 1 | 0 | 0 | 8944 |
| 30227 | 16p13.12 | Loss | rs190013 | rs9452 | 8 | 0.05 | Yes | 1 | Maternal | 1 | 2 | 2 | 8946 |
| 29700 | 13q31.2 | Loss | rs221022 | rs452708 | 7 | 0.03 | Yes | 4 | Paternal | 0 | 100 | 7 | 8938, 9655, 9656, and 9657 |
| 29945 | 3p14.2 | Loss | rs9311853 | rs10510892 | 7 | 0.05 | No | … | … | … | … | … | 8939 |
| 28283 | 8q22.1 | Loss | rs6996243 | rs1378125 | 6 | 0.66 | No | 3 | … | … | … | … | 9648, 9649, and 9650 |
| 29199 | 6q22.33 | Loss | rs3778130 | rs265353 | 6 | 0.01 | Yes | 1 | Maternal | 1 | 0 | 0 | 8942 |
| 30221 | 1q23.3 | Gain | rs9330294 | rs10494355 | 6 | 0.06 | Yes | 2 | Paternal | 1 | 46 | 11 | 8945 and 9076 |
Note.— The breakpoint regions were sequenced in individuals 29195 and 29945 (GenBank accession numbers EF504248 and EF504249). NA = parental DNA not available.
Confirmation by qPCR.
Percentage of the CNV detected in this study that overlaps with one or more CNVs listed in the Database of Genomic Variants.
Percentage of CNVs listed in the Database of Genomic Variants that overlaps with the CNV detected in this study.
We finally considered 11 CNVs—8 deletions and 3 duplications—to be causative of MR by criteria as follows (fig. 2 and table 2). Eight of the rearrangements were assumed to be causative because they occurred de novo. Two rearrangements, in 8p23.1 and 17p11.2, were assumed to be causative because they overlapped with known deletion or duplication syndromes (e.g., 17p11.2 duplication syndrome [MIM 610883]), although they could not be proved to have occurred de novo, because of missing paternal DNA. At last, a maternally inherited 1.4-Mb duplication in Xp22.31, including the STS gene, in a male patient was thought to be causative because deletions of this region are known to cause MR and because the chromosome carrying the duplication was nonrandomly inactivated in the mother.
Figure 2. .

Genomic profiles showing the log2 intensity ratios of CNVs and of the surrounding genomic regions detected in patients with MR. The length of the CNVs is indicated on the X-axis. log2 Intensity ratios are calculated as described in the “Material and Methods” section. Displayed are the log2 intensity ratios after median smoothing with a window of 9. The dosage values of homozygous and heterozygous SNPs are depicted in black and gray, respectively. The gray horizontal lines are drawn at log2(0.75) and log2(1.25).
Table 2. .
Deletions and Duplications[Note]
| SNP |
LOHa |
|||||||||||
| Patient ID |
Gain or Loss | Chromosome | Start | End | Region Length (Mb) |
No. of SNPs | No. of Genes | Segmental Duplications | Paternal | Maternal | No. of qPCRs | Confirmation by Second Hybridization |
| 27384 | Loss | 1q31.1-31.3 | rs10494585 | rs10494695 | 7.5 | 464 | 17 | No | 48 | 0 | 1 | Yes |
| 29922 | Loss | 8p23.1 | rs2945251 | rs2466115 | 3.9 | 177 | 34 | Yes | NA | 0 | 3 | No |
| 30437 | Loss | 2p25.3-25.1 | rs2313466 | rs1964092 | 3.8 | 193 | 4 | No | 0 | 18 | 1 | No |
| 27737 | Gain | 17p11.2 | rs4073940 | rs1373147 | 3.2 | 29 | 49 | Yes | NA | … | 3 | Yes |
| 30375 | Loss | 3p25.3-25.2 | rs10510400 | rs10510422 | 2.7 | 98 | 39 | No | 8 | 0 | 3 | No |
| 28735 | Loss | 12p13.33 | rs953385 | rs2283285 | 2.0 | 62 | 18 | No | 4 | 0 | 3 | No |
| 28430 | Loss | 15q25.2 | rs17158372 | rs10520569 | 1.4 | 27 | 11 | Yes | 0 | 2 | 3 | Yes |
| 29836 | Gain | Xp22.31 | rs719632 | rs10521669 | 1.4 | 31 | 5 | Yes | … | … | 3 | Yes |
| 30428 | Loss | 17q21.31 | rs436667 | rs1918798 | 0.5 | 38 | 7 | Yes | 26 | 0 | 1 | Yes |
| 28701 | Gain | 13q12.11 | rs9315234 | rs4570685 | 0.5 | 23 | 5 | No | … | … | 3 | Yes |
| 29195 | Loss | 2q23.1 | rs2890919 | rs10497034 | 0.2 | 13 | 1 | No | 0 | 0 | 2 | Yes |
Note.— NA = not available.
The number of SNPs for which a paternal or maternal allele is missing. LOH = loss of heterozygosity.
In addition, we investigated DNA of four mentally retarded children with de novo translocations—three terminal and one interstitial translocation—containing a total of eight breakpoints. According to GTG-banding analysis, they appeared to be balanced. By microarray analysis, we detected deletions at two of the six breakpoints in the terminal translocations and at two of the breakpoints in the interstitial translocation containing 3–63 genes (table 3).
Table 3. .
Reciprocal Translocations
| SNP |
|||||
| Patient ID, GTG Banding,a and Chromosomeb |
Start | End | Region Length (Mb) |
No. of SNPs | No. of Genes |
| 31922, 46,XY,t(12;13)(q22;q32): | |||||
| 13q33.2-33.3 | rs2149144 | rs9301245 | 1.8 | 146 | 3 |
| 28526, 46,XY,t(1;10)(p13.1;p13): | |||||
| 10p14 | rs2259442 | rs1243963 | 1.1 | 57 | 8 |
| 28181, 46,XY,t(4;6)(q28.3q31.1;q23.1q22.2): | |||||
| 4q28.3-31.1 | rs10519357 | rs6841039 | 3.9 | 186 | 6 |
| 6q16.1-21 | rs9320518 | rs6568702 | 14.3 | 625 | 63 |
GTG banding (with 500–550 bands) contains results of karyotyping.
Chromosome contains results of microarray experiment.
CNVs Flanked by Low-Copy Repeats
Of the 11 validated CNVs, 5 were flanked by low-copy repeats (LCRs). The first one was a 3.9-Mb deletion in 8p23.1, indicated by 177 SNPs in a child (patient identification [ID] 29922) with mild MR. The mother carried two copies, and the maternal genotypes in the deleted region are consistent with the boy carrying one of the maternal alleles. Thus, it is most likely that the deletion occurred on the paternal chromosome, although we did not have paternal DNA available for investigation. The deletion overlapped with the known 8p23.1 deletion syndrome and included the GATA4 gene, as confirmed by qPCR.19 The syndromic features of our patient included microcephaly, hypospadia, and an atrial septum defect described to be attributable to haploinsufficiency of GATA4.20 The child did not present with facial dysmorphisms, diaphragmatic hernia, or epilepsy.
The second CNV flanked by LCRs was a duplication of 3.2 Mb in 17p11.2, indicated by 29 SNPs in a boy (ID 27737) with mild MR. His mother carried two copies. DNA from the father was not available. As estimated by the dosage values, the duplication coincides with the common 3.7-Mb interstitial duplication of the 17p11.2 duplication syndrome, a region that harbors deletions in Smith-Magenis syndrome (MIM 182290).21,22 The patient we investigated had normal birth weight and length, but feeding was poor, and he failed to thrive postnatally. He had a hypospadia grade I. At age 10 mo, his length was in the 10th percentile, his head circumference was <3rd percentile, and he was found to have hypotonia. Facial features, such as telecanthus, triangular face, and broad forehead, were in accordance with the findings recently described in other children who were given a diagnosis of Potocki-Lupski syndrome (fig. 3A).22 Later, the mother reported that the child started speaking single words only at age 27 mo.
Figure 3. .

Facial features of patients. A, Patient 27737, with a common 17p11.2 duplication, at age 10 mo, showing a telecanthus, triangular face, and broad forehead. B, Patient 29195, with an MBD5 deletion, at age 15 mo, showing a normal facies without dysmorphic features.
The third CNV flanked by LCRs was a 500-kb deletion in 17q21.3 in a girl (ID 30428) with moderate MR. The deletion encompasses a known inversion polymorphism.23 We used the genotype information from 38 SNPs within the deleted region to define the haplotypes in the girl and her parents. The father was homozygous for the H2 haplotype, and the mother was homozygous for the H1 haplotype. The deletion occurred on one of the paternal H2 haplotypes. Deletions of the same region have recently been found in 10 other patients and have defined a new microdeletion syndrome.13,24,25 The deletions reported therein also occurred on the H2 haplotype and may be facilitated by the direct orientation of the repeats on this haplotype.
The fourth CNV flanked by LCRs was a 1.4-Mb deletion on chromosome 15q25.2, which comprises ∼11 genes, in a patient (ID 28430) with mild MR. Both parents carried two copies of the region. The deletion was indicated by the dosage values of 27 SNPs. Two informative SNPs showed that the maternal allele was deleted. The patient was affected by intrauterine growth retardation. She was born in the 38th wk of gestation, with a weight of 1,950 g and a length of 42 cm. She had only very slight dysmorphic signs. The main symptoms were psychomotor retardation, polysplenia, and a hypoproliferative, macrocytic anemia that developed in the 1st year of life and that required blood transfusions until she was age 4 years. Because of short stature and anemia, the diagnosis Diamond-Blackfan anemia (MIM 105650) was considered. At age 11 years, she was referred to the emergency ward with bleeding from esophageal varices. Ultrasound sonography revealed a severe portal vein stenosis, although liver structure was normal.
The fifth CNV flanked by LCRs was a 1.4-Mb duplication in Xp22.31 in a boy (ID 29836) with severe MR. The duplication was located between the VCX3A and VCX2 genes and was indicated by 18 SNPs (fig. 4A). qPCR revealed a single dose for amplicons ∼6 kb distal to VCX3A and 3 kb proximal to VCX2, whereas amplicons 3 kb proximal to VCX3A and 5 kb distal to VCX2 showed a double dose. Therefore, VCX3A or VCX2 is duplicated or an additional fusion product of both genes was generated, depending on where the recombination occurred. In addition, all genes between VCX3A and VCX2 are duplicated (HDHD1A, STS, VCX, and PNPLA4). Investigations of the parental DNAs showed that the father had a single copy of this region, whereas the healthy mother had three copies and therefore is a carrier of the duplication. Studies of the X-inactivation pattern in peripheral-blood DNA of the mother showed a nonrandom X-inactivation, with the chromosome carrying the duplication being inactivated. We therefore conclude that the duplication is most likely causative of the MR in her son. He had normal motor development but showed, in addition to MR, speech and language deficits. At age 9 years, he used only 2-word sentences. He developed postnatal microcephaly, with a head circumference of 50 cm (3rd percentile) at age 9 years. A recent report describes additional patients with MR carrying duplications of this region.26 The same region contains deletions in 80%–90% of patients with X-linked ichthyosis (MIM 308100) caused by deficiency of the steroid sulfatase enzyme, encoded by the STS gene.27 Most patients with deletions are affected only by ichthyosis, but a few also display MR, although the size of the deletion is often very similar to that of the deletion breakpoints situated near the directly orientated genes VCX3A (VCXA) and VCX2 (VCXB), which are embedded in an LCR of ∼9 kb. Data from two studies suggested that, depending on whether VCX-proximal or VCX-distal homologous sequences are used for recombination, VCX3A is deleted and VCX2 is retained, or vice versa.28,29 MR seems to be associated with the deletion of VCX3A,29 although the data are not unequivocal, because patients with Xp;Yq translocations leading to a deletion of VCX3A have been reported to have normal intelligence.
Figure 4. .

Schematic representation of the regions affected in patients 29836 and 29195. A, Duplication in Xp22.31 in patient 29836. Genes are indicated by white boxes. The arrows denote the direction of transcription. The primers used for qPCR are shown above the genomic representation as numbered black bars. Primers 9060 and 9821 showed a single dosage; primers 9822 and 9820, a double dosage. The nonallelic homologous recombination must have occurred within the homologous regions flanked by primers 9060 and 9820 and primers 9822 and 9821. B, Deletion of part of the MBD5 gene in patient 29195. Exons are indicated by numbered black boxes (untranslated sequence) or gray boxes (translated sequence). The extent of the deletion is shown by a black bar below the gene scheme. Exons 1A–1E of MBD5 denote the 5′ untranslated exons established in this study.
CNVs Not Flanked by LCRs
In six patients, de novo CNVs could be identified, with a size between 200 kb and 7.5 Mb. The child (ID 27384) with the largest deletion (7.5 Mb) has psychomotor retardation but did not have additional symptoms besides slight dysmorphic facial features. The most prominent syndromic features in these six patients included hydrocephalus and cleft palate (in patient 28701), microcephaly and adipositas (in patient 30437), and an ostium secundum defect (in patient 30375). The 2.7-Mb deletion in the last boy (ID 30437) overlapped with the proximal part of the 3p syndrome, which has been described to cause atrioventricular septal defects in most of the affected individuals.30 The 3.8-Mb deletion on 2p25.1-25.3 contained only four genes, including SOX11. This gene is expressed transiently during embryonic development in many tissues and causes severe defects in several organ systems in homozygous Sox11-deficient mice, leading to death shortly after birth.31
The smallest CNV suggestive of being causative of the clinical phenotype observed is a 200-kb deletion on chromosome 2q23.1 in a boy (ID 29195) with severe MR. The deletion was indicated by 13 SNPs, which affected the first 7 of 10 exons of the methyl-CpG–binding domain protein 5 gene (MBD5) (fig. 4B). The deletion region was present in the parental DNA but provided no information with regard to the origin of the deletion. Sequencing of the coding region of the remaining allele did not show any deviation from the reference sequence (GenBank accession number NM_018328). The junction fragment was amplified and sequenced after the breakpoints were narrowed by qPCR (GenBank accession number EF504248). The proximal breakpoint lies within an LTR/ERVL repeat (MLT2B5), and the distal breakpoint lies within single-copy sequence in intron 7 of the MBD5 gene. ESTs suggested that the GenBank entry for MBD5 is 5′ incomplete. We extended the mRNA by five noncoding exons, using 5′ rapid amplification of cDNA ends (RACE) and RT-PCR (GenBank accession number EF542797). The boy had a sandal gap between the first and second toe but no facial dysmorphic features (fig. 3B). In addition to MR, motor development was slightly retarded. At age 8 mo, he had febrile seizures. First seizures without fever started at age 16 mo and proved to be drug resistant. The boy is hypoactive, and social interactions are very limited. To provide further evidence of the pathogenicity, we screened 415 DNAs from children with MR. We found four missense variants that were not present in ∼660 controls (table 4). We did not have parental DNAs available to check whether these variants occurred de novo.
Table 4. .
Variations in MBD5
| No. of Control Individuals with Genotype |
||||||
| Variation Type and Sequence Change |
Protein Change | Patient ID(s) | Exon/Intron | 11 | 12 | 22 |
| Nonsynonymous: | ||||||
| c.431C→T | p.T144I | A12 | 4 | CC: 660 | CT: 0 | TT: 0 |
| c.1368G→T | p.S456K | A6 and B135 | 4 | GG: 663 | GT: 1 | TT: 0 |
| c.1382G→A | p.R461H | A47 and B217 | 4 | GG: 649 | GA: 0 | AA: 0 |
| c.1962C→A | p.D654E | C231 | 4 | CC: 655 | CA: 0 | AA: 0 |
| c.1963G→A | p.A655T | 30224a | 4 | GG: 653 | GA: 0 | AA: 0 |
| c.2030G→A | p.S677N | B134, C264, and C281 | 4 | GG: 662 | GA: 3 | AA: 0 |
| c.2569G→A | p.A857T | 31833a | 5 | GG: 670 | GA: 0 | AA: 0 |
| c.3143C→T | p.T1048I | C260 | 7 | CC: 640 | CT: 0 | TT: 0 |
| Synonymous: | ||||||
| c.1638C→T | p.A546A | A109 and B139 | 4 | CC: 658 | CT: 3 | TT: 0 |
| c.2286C→T | p.H762H | B225 | 4 | CC: 338 | CT: 0 | TT: 0 |
| c.3279C→T | p.V1094V | A53 | 7 | TT: 648 | CT: 0 | CC: 0 |
One of the parents carries the same mutation.
MBD5 was originally identified because of sequence homologies to MBD1–MBD4 and MECP2, which is mutated in Rett syndrome.32 It also contains a PWWP motif, which has been shown to bind to DNA and is found in proteins often containing other chromatin-association domains.33
Discussion
Hybridization using synthetic oligonucleotide arrays offers great promise for the detection of submicroscopic deletions and duplications. As the technology evolves, driven mainly by the need for dense SNP arrays in genomewide association studies, the resolution to detect relatively small deletions and duplications with strong effects on rare phenotypes will improve. We used arrays that cover the entire genome, with 100,000 SNPs, to test them against 71 patients with MR. Samples from all 71 patients had previously been subjected to cytogenetic analysis, because they had slight or overt symptoms characteristic of chromosomal abnormalities.
Our study led to the identification of 11 deletions or duplications that were interpreted to be causative, because they could be shown to have occurred de novo or because they corresponded to established disease-associated indel mutations. Our detection rate of 16% is in the same range as that of earlier studies that used 100K arrays12 or whole-genome tiling-path resolution array CGH.7 The patient (ID 29922) with 8p23.1 deletion syndrome and the patient (ID 27737) with 17p11.2 duplication syndrome raise the question of why the clinical picture in these patients did not lead to a specific diagnosis and testing. In both cases, the differential diagnosis did include the mentioned syndromes, but its features were by no means unequivocal. In the case of the 17q21.3 deletion, the genotype-phenotype connection was established only during the course of this study.13,24,25
We identified two CNVs, in 15q25.2 and Xp22.31, with a high probability of being causative of new deletion and duplication syndromes. They were flanked by LCRs, which make it likely that other cases exist, both deletions and duplications. Whereas no other cases have been described so far for 15q25.2, duplications in Xp22.31, which are characterized by MR in combination with autistic behavior, seem to be more frequent.26
The smallest CNV detected and concluded to be causative is a deletion of a single gene, MBD5 (in patient 29195). The deletion measured 200 kb in size. It was detected by 13 consecutive SNPs and deleted one noncoding and seven coding exons of the gene, which codes for the methyl-CpG–binding domain protein 5. Two previous reports and a DECIPHER entry (about patient 1079) have described contiguous gene deletions that also involve the MBD5 gene7,34 (J. Veltman, personal communication). The clinical features of our case patient and two of the previously described patients include epileptic seizures. A mutation screen involving 415 DNAs of children with unexplained MR revealed four missense variants not present in 660 controls, but no deletions or nonsense mutations were found. We could not check whether the missense variants occurred de novo because we did not have parental DNA available. Thus, a confirmation that the MBD5 mutations are pathogenic awaits the screening of a panel enriched for epileptic seizures for which parental DNA is available.
The CNVs suggest that the genes involved cause the related pathology by dosage differences. The most likely group of genes to be considered for such a mechanism is transcription factors.35 With the exception of the MBD5 deletion, the Xp22.31 duplication, and the 17q21.31 deletion, all regions contained at least one gene involved in the transcription process. From the 189 genes contained in the reported deletions and duplications, 129 have a Gene Ontology annotation, and 16 (12%) of those 129 are annotated as transcription factors.
We showed that 100K arrays allow the detection of CNVs with a size between 10 kb and 7.5 Mb, a range that is not reliably accessible by microscopic cytogenetic techniques. The quality of the intensity data obtained from the arrays was variable and depended on the experimental circumstances, restrictions that must be accounted for in the analysis procedure. The detection of CNVs by at least 20 neighboring SNPs was found to be reliable, whereas the detection of CNVs by 5–20 neighboring SNPs had a false-positive rate of 30%. The resolution might improve with new protocols and new generations of arrays that reduce the experimental error and provide a better SNR. The interpretation of the results in children with MR or other diseases is complicated by the presence of thousands of CNVs within the normal population, especially when their size is small. One usually applies the criterion that a CNV has to have occurred de novo for it to be likely pathogenic. However, the frequency of de novo deletions and duplications in newborns has been estimated to be 1 in 8 and 1 in 50, respectively, and it is obvious that not all of them can be assumed to be pathogenic.36 On the other hand, de novo CNVs may be pathogenic. They may provide a risk that becomes manifest because of sequence variants in the other allele or the genetic background. Databases that collect information about CNVs in healthy individuals and individuals with diseases will become indispensable for their risk estimation. This will become even more important when improved array designs allow detection of intragenic deletions and duplications.
Acknowledgments
We thank the patients and their families for participation in this study. We also thank Sandy Loesecke, Corinna Keri, Gabi Lederer, and Doris Sollacher, for technical assistance, and Monika Cohen, for providing DNA samples.
Appendix A
Table A1. .
Clinical Characteristics[Note]
| Patient ID | Sex | Father | Mother | GTG Banding | Subtelomer Screening | EEG | MRI | Metabolism | MR | Delayed Motor Development | Language and/or Speech Retardation | Facial Dysmorphism | Stature | Microcephalus | Miscellaneous |
| 27384 | F | 27739 | 27738 | 2 | + | + | + | + | Mod-sev | + | + | ||||
| 27524 | M | NA | NA | 1 | ND | ||||||||||
| 27525 | M | NA | NA | 1 | ND | + | Short | ||||||||
| 27526 | F | NA | NA | 1 | ND | ||||||||||
| 27581 | F | NA | NA | 1 | + | Mod-sev | + | + | |||||||
| 27733 | M | NA | NA | 1 | Mod-sev | Short | + | Clinodactyly | |||||||
| 27737 | M | NA | 31778 | 1 | + | Mild | + | Hypospadia, hypotonia | |||||||
| 27831 | M | NA | NA | 1 | Mod-sev | + | + | ||||||||
| 27877 | F | 27878 | 27879 | 1 | + | + | + | Mod-sev | + | + | + | + | Wolff-Parkinson-White syndrome | ||
| 28037 | M | 28039 | 28038 | 1 | Mod-sev | + | Brachydactyly | ||||||||
| 28142 | M | 28144 | 28143 | 1 | Mod-sev | Short | Aortic coarctation, hypospadia, omphalocele | ||||||||
| 28181 | M | 28183 | 28182 | 1 | Mod-sev | ||||||||||
| 28188 | F | NA | 28189 | 1 | Mod-sev | + | Hypotelorism | ||||||||
| 28190 | M | NA | 28191 | 1 | + | Mod-sev | + | + | + | Brachycephalus | |||||
| 28192 | M | 28194 | 28193 | 1 | Mod-sev | + | Aggressive behavior | ||||||||
| 28283 | M | 28449 | 28284 | 1 | + | Mod-sev | + | + | |||||||
| 28289 | M | 28290 | 28291 | 1 | + | Mod-sev | + | + | + | Dolichocephaly, hyperopia | |||||
| 28430 | F | 30806 | 30805 | 1 | Mild | + | + | Short | + | Anemia | |||||
| 28448 | F | NA | NA | 1 | ND | Short | + | ||||||||
| 28499 | F | 28500 | 28501 | 1 | ND | ||||||||||
| 28509 | F | 28510 | 28511 | 1 | + | + | Mod-sev | + | Brachymetacarpy, hyperopia | ||||||
| 28526 | M | 28527 | 28528 | 1 | Mod-sev | ||||||||||
| 28529 | M | 28530 | 28531 | 1 | Mod-sev | + | + | Short | |||||||
| 28631 | M | 28633 | 28632 | 1 | + | Mod-sev | + | Short | Brachycephaly, myopia, autoimmune thyreoditis | ||||||
| 28634 | F | NA | 28635 | 1 | + | + | Mild | + | Tall | ||||||
| 28651 | M | NA | NA | 2 | + | Mod-sev | + | + | Short | Nystagmus | |||||
| 28701 | M | 28702 | 28703 | 1 | + | + | Mod-sev | + | Hydrocephalus, cleft palate, tricuspid dysplasia | ||||||
| 28730 | M | 28731 | 28734 | 1 | + | Mod-sev | + | + | + | Muscle hypotonia | |||||
| 28735 | M | 28736 | 28737 | 2 | + | + | Mod-sev | + | + | ||||||
| 29195 | M | 29197 | 29196 | 2 | + | + | + | Mod-sev | + | + | |||||
| 29199 | F | NA | 29198 | 1 | Mod-sev | + | Tall | ||||||||
| 29200 | M | 29202 | 29201 | 1 | Mod-sev | ||||||||||
| 29608 | M | NA | 29610 | 2 | + | Mod-sev | + | + | + | Epilepsy | |||||
| 29609 | M | NA | NA | 2 | + | Mod-sev | + | + | + | ||||||
| 29699 | M | NA | NA | 2 | + | + | + | Mod-sev | + | + | + | Brachycephaly, epilepsy | |||
| 29700 | M | 29701 | 29709 | 1 | Mod-sev | + | |||||||||
| 29833 | F | 29835 | 29834 | 2 | + | Mod-sev | + | + | |||||||
| 29836 | M | 29838 | 29837 | 2 | + | + | Mod-sev | + | + | + | |||||
| 29901 | M | 29903 | 29902 | 2 | + | + | Mod-sev | + | + | Brachycephaly | |||||
| 29922 | M | NA | 29923 | 2 | + | Mild | + | Hypospadia | |||||||
| 29942 | M | 29944 | 29943 | 1 | Mod-sev | + | |||||||||
| 29945 | M | NA | 33360 | 1 | Mod-sev | + | |||||||||
| 29949 | M | NA | NA | 2 | + | + | Mod-sev | + | Short | Brachydactyly, attention-deficit disorder | |||||
| 29950 | M | 29952 | 29951 | 2 | + | + | + | Mod-sev | + | + | + | Brachycephaly, hyperopia | |||
| 29962 | F | NA | NA | 1 | + | + | Mod-sev | + | + | + | |||||
| 29993 | M | 29994 | 29994 | 2 | + | + | + | + | Mod-sev | + | + | Hypotonia | |||
| 29996 | M | NA | NA | 2 | + | Mod-sev | + | + | Brachycephaly | ||||||
| 30221 | F | 30223 | 30222 | 1 | + | + | Mod-sev | + | |||||||
| 30224 | M | 30226 | 30225 | 2 | + | Mod-sev | + | Hypotonia | |||||||
| 30227 | M | 30229 | 30228 | 2 | + | + | + | + | Mod-sev | + | + | Dolichocephaly, autistic behavior | |||
| 30241 | F | 30243 | 30242 | 2 | + | + | Mod-sev | + | + | + | + | ||||
| 30303 | M | 30305 | 30304 | 1 | + | Mod-sev | + | + | + | + | |||||
| 30372 | M | 30374 | 30373 | 2 | + | Mod-sev | + | + | + | + | |||||
| 30375 | M | 30377 | 30376 | 2 | + | ND | + | + | Atrial septal defect II | ||||||
| 30392 | M | 30647 | 30393 | 2 | + | + | + | Mod-sev | + | + | |||||
| 30428 | F | 30430 | 30429 | 1 | + | Mod-sev | + | + | + | + | |||||
| 30431 | M | 30433 | 30432 | 2 | + | Mod-sev | + | + | + | ||||||
| 30434 | M | 30435 | 30436 | 2 | + | Mod-sev | + | ||||||||
| 30437 | M | 30438 | 30550 | 2 | Mod-sev | + | + | ||||||||
| 30508 | M | NA | 30510 | 1 | + | + | + | Mod-sev | + | + | + | ||||
| 30520 | M | 30521 | 30522 | 1 | Mod-sev | ||||||||||
| 30569 | F | 30571 | 30570 | 2 | + | Mod-sev | + | + | + | Epilepsy | |||||
| 30704 | F | 30706 | 30705 | 2 | + | + | + | + | Mod-sev | + | + | ||||
| 30782 | M | 30784 | 30783 | 2 | + | Mod-sev | + | + | Brachycephaly | ||||||
| 30830 | F | 30832 | 30831 | 2 | + | Mod-sev | + | + | + | Brachycephaly | |||||
| 30872 | F | 30874 | 30873 | 1 | Mod-sev | + | + | + | |||||||
| 30875 | M | 30877 | 30876 | 1 | Mod-sev | + | + | Pulmonary stenosis, esophageal atresia, laryngomalacia | |||||||
| 30909 | M | 30911 | 30910 | 1 | + | Mod-sev | + | + | |||||||
| 30944 | F | 30945 | 30946 | 1 | Mod-sev | + | |||||||||
| 30987 | M | 30988 | 30989 | 2 | + | Mod-sev | + | ||||||||
| 31154 | M | NA | NA | 1 | Mod-sev | + | + | ||||||||
| 31922 | M | NA | NA |
Note.— EEG = electroencephalogram; Mod-sev = moderate to severe; MRI = magnetic resonance imaging; NA = not available; ND = not determined; a plus sign (+) = presence of feature.
Table A2. .
Primer Sequences
| Sequence(5′→3′) |
||||
| Use and Primer | Forward | Reverse | Length (bp) |
Annealing Temperature (°C) |
| qPCR: | ||||
| 8920 | TGAAGGCTCTAAATCCCCAG | AGCAACCGCTAAAACCCAG | 126 | 60 |
| 8921 | GCGAGCCCGATTCTCTG | ACCAATCCTCAGGTCCAGC | 126 | 60 |
| 8922 | CTTCACTGGACCGAAACACC | TTTATCCCGATTGCTTCTGC | 135 | 60 |
| 8923 | AGCAGCCTCATGCTCTATGG | TGAAGTGTCCAGTCCAAGGC | 128 | 60 |
| 8924 | ATTTCCTCTATGGAGCGTGG | AATGGCTCCGATACACTTCC | 127 | 60 |
| 8925 | AACTGGGAAGGAGGTATCCG | AGGGAGCTCCAGCCAGC | 133 | 60 |
| 8926 | GGACCACCCTTCGGCTG | GGCTGACGATGTTCGAGG | 124 | 60 |
| 8928 | TGGTGATTACTTTTGACAGTCTTTG | GGAAAGAGTTGTAGCTCCCG | 129 | 60 |
| 8929 | CGAGTCGCTGACCAGTTACC | ATCTGCTTGTCCTGGCTGAC | 126 | 60 |
| 8930 | GACATGTGCAGCCGTGTG | ATGGGCTGTGTTGTCAAGG | 124 | 60 |
| 8932 | TTTTGGCCCAGTTTAGCC | AATATGATGGTGGCTGGCTC | 130 | 60 |
| 8933 | CTCATTCAGCCTCATCTCACC | CCTCCATGAAGGGCTAAAAC | 131 | 60 |
| 8934 | ATGCAAAATAATACCATCCAGAC | GCCACTTGTGGGAAGTGC | 137 | 60 |
| 8935 | CTTTTCATTAAATGTGCATTAACC | TCTGCAAAGCAACTGAACTG | 135 | 60 |
| 8936 | CCTCCTACTATATGAAAATTTTGGTCC | GCTGCTTAAGACTGCAAAGC | 126 | 60 |
| 8937 | TCAACTATGACAGGGTGTCTGC | CCCTTCAGAGGCAAATAATAGG | 121 | 60 |
| 8938 | AGATTTGGGCCCCTCAAC | AACAACGAGGAAGAAATATTGG | 133 | 60 |
| 8939 | GCAGCAGCAATATCCCTTTATG | GCTGGTAGTCAAGTAAGGCTTTG | 128 | 60 |
| 8940 | TGACTTAAATATCAATTGAGGATCAC | TTGAATTAACTGAAACCCAATCTC | 135 | 60 |
| 8941 | TCTGTCTCTGGATTCCTATTTGC | TCAAACACAAGTCTTGGTCTCC | 130 | 60 |
| 8942 | GGAAGATGTATTTGTCACTTTTCTTC | GCACAGTCAACCATTATTTTCTC | 132 | 60 |
| 8944 | AAAAGTGTGTGCAGAGCCAG | GATGAAGCAAGCACATAGCG | 130 | 60 |
| 8945 | ATTTGTCTTTGTCTCAATATGGG | CCCTTAGGTTCTTCACTTCCC | 130 | 60 |
| 8946 | TCACACCTTCTCTGTGGCAG | GCAAAACAAACCCCGAAAAC | 127 | 60 |
| 8948 | AAGGAAGGAGGTCTTCCAGC | CAACTTTGCAGGTACCACAGG | 118 | 60 |
| 9054 | CTTAACCTGATCACGGGGC | AACAGTGGCCTCAACTCCTC | 148 | 60 |
| 9055 | CCTTCTAGGCTCTGAAAATTGG | TGATGAATTCGAGTTTGCTG | 130 | 60 |
| 9056 | GCCCACCCTCATCTACCTG | CGACGAGGGATTGTCCTG | 136 | 60 |
| 9057 | GTCTGCAACACACTGCAACC | TGGTTTCGTGCCTGTAGTAGG | 154 | 60 |
| 9058 | TCTCTCAACTCACACGCCC | GAAACGGTGACCGCCTG | 133 | 60 |
| 9059 | CTGGACATTACACAGGCTCG | CTGCAGAATCTCTGTGGACTG | 133 | 60 |
| 9060 | GAACATCAGCAGTTGCCTTC | TCCCATGGAGATGCACATAG | 152 | 60 |
| 9061 | CTCCCGGAAGTGGGAGG | AGAGGAGAGTTGCTGGAGCC | 147 | 60 |
| 9062 | TAGATCACAGGGTCGGAAGG | CAGGGCAAGGAAGGCAG | 131 | 60 |
| 9063 | CTACCTGCGGGTCCTGG | GCCCTGAAGCAGTCCCTC | 141 | 60 |
| 9064 | TTGACTAACCTACGGCCACG | TCTTAAGGAGAAGGGGAGGG | 153 | 60 |
| 9065 | CCTGATGCTTGACTCTCCTC | GACTTTCCCTCTGCACCAAG | 125 | 60 |
| 9066 | CTCGGATTGCTCAAGGACC | ACTTTACGCTGGCAGGAGG | 136 | 60 |
| 9067 | TCACAGAGCAGCTCCCATC | GGCCACCAGGGAGATACAG | 130 | 60 |
| 9068 | GATTGGCCAGAACCCTGAG | GCGCTGCATGGTCATATTTC | 125 | 60 |
| 9069 | TGTAGCAAATGGTGGCAAAG | GAGACTATTGGCGATGCGAG | 146 | 60 |
| 9070 | TCTCCTTGTTAATCTGAGCTCTTG | GGTTATGATGTCCATCCAGG | 126 | 60 |
| 9073 | GGAAACATCACCAACAGCC | TTCAAACAAGGCAGAATACAGAC | 133 | 60 |
| 9074 | TGGAGATAGATGCCATTTGC | AGGGATCGATGTGCTAGGAG | 133 | 60 |
| 9076 | TCAGTGGAGAAGTGTGTGTGG | AATCCAAGACCCAGAAAGCC | 141 | 60 |
| 9077 | TGGGAGTTCACCTTTCTTGC | ACTTGTTGTAGCTGCCCAGG | 120 | 60 |
| 9078 | TCAACCTGAACCATTACGCC | GGTGACCAGGGTGGTGTAG | 140 | 60 |
| 9648 | AATAGCAGCTCTAAATCCCAGTC | CAGGAAGACGGGAACACAAC | 148 | 60 |
| 9649 | TGTGTTTGTCACAGCCTTGC | CTAGAGATGGACCTCCCACG | 141 | 60 |
| 9650 | GTTGAAGTACGGCCGCTG | CCTGAGAATGACTCCATGCTTC | 149 | 60 |
| 9651 | GGACTCAGTGATCAAACCCC | ATTGTTCTCAGAATGCACCC | 130 | 60 |
| 9652 | CACGATAAATTGTTTCTCTGTCC | GCGTCTCCTTGGTATGGATG | 109 | 60 |
| 9653 | ATTGCAACCAGAAGCTGGAG | TCCAGGTAAAGCAGGTCCTC | 137 | 60 |
| 9655 | GAAAGATCTCTGGGTGTCCG | AATCAGTGATGGCCATGGAG | 160 | 60 |
| 9656 | GTTCACTGCAGAGGAGGTTG | AATCTTGCCTGTTCTTTAGCG | 143 | 60 |
| 9657 | CCCCTTTGGATTAAGTTGC | ACTTCAGGGTCTGGGATGTC | 131 | 60 |
| 9820 | CAAGATGTGTCCAGACATTCCC | CATGGTGGCGTATGAACTTGG | 162 | 60 |
| 9821 | CAACCTTTTAAAACGCTTTGC | TATAGCTGCACGAATCTCCG | 119 | 60 |
| 9822 | GGTTCTTGAGTGATTAGTGTAGG | CCTCAGTTGCGAATATAAAAAGGC | 179 | 60 |
| MBD5 breakpoint determination: | ||||
| 9265 | TGCAAAACTGTGTTTATGATGTTAC | AAGTCTGTGTTAAAAGAAATTTGGTG | 131 | 60 |
| 9266 | GCTATCCCTTTAAAACTCTCACAGC | TGCTCTTTGGGAGCAATAGG | 111 | 60 |
| 9267 | TGACTTTGCTTTCCAATAACCC | CAAAACAATAACTGAAGAACTCATGG | 113 | 60 |
| 9268 | TTTCACACGTGAACCAGGC | GACCTGGGTAGAGCAGCATC | 121 | 60 |
| 9269 | TTGCATATTTTATAGGCATAGATAGC | TCCCTACTCAACACTAAAGAGCC | 134 | 60 |
| 9270 | CAGGTTGTCATCACCACTGC | GAATGACCGCACTGACAGC | 133 | 60 |
| 9271 | AAAAGGTAGGGGTTGATGCC | AATATTTGTCAAAGGTGGCCC | 140 | 60 |
| 9272 | ACAAGGGGATAACCACAGGG | TTGTCAAGAAAGCTGGGAGG | 124 | 60 |
| 9273 | CAGCCAAGCCTTAGTATAGCC | TACCTATGGCACCCTTTCCC | 124 | 60 |
| 9274 | TCCTTGTCTTGCTTTGGTTG | GACTCTACAGGTTATTTGGAGGC | 127 | 60 |
| 9348 | TCAGGGAAATGTAGGATTGC | TCTCTTCGTCTCCCTCCCTC | 133 | 60 |
| 9349 | TCATGTGTCTGATGCCTGTTTAC | TGCTGAGAACAAGGATGGC | 207 | 60 |
| 9350 | AATTTTCTCTTTCTTGACATAAATCTG | GGAAACATGCAGAACTGCTC | 132 | 60 |
| 9351 | TGCTTAAGCCACAAATCTGAAG | TTGTATGTGCACTGGAACTTG | 252 | 60 |
| 9352 | TTTCATACACAGAGGAGGAAGG | ACATGGAGGAGATGCCGTC | 136 | 60 |
| 9353 | CTAACTTTGGGAGGTGCGTC | AGGATTCATTTCCAACAGCG | 181 | 60 |
| 9354 | AGGTTAAGGCTCCCCTCCC | GTGTCTTCAACTTCTCGGGC | 142 | 60 |
| 9355 | TTGCCAGTTGAAACATTCTACC | ACAGAAGATTTGCTCCTCGC | 264 | 60 |
| MBD5 mutation screening: | ||||
| 9275 | TCATCTTATTGCTGATATCTTTGGAG | TTGCAGGTACCACAGGTAATAATAAG | 193 | 60 |
| 9276 | AAAATGCTTTTCCCTAGTGGG | GAAAAGGTAGAAAGGTGGTTTTAATG | 482 | 60 |
| 9277 | TTTTACAGACATATTCTAAACAAAGGC | GGAGAGGAGAATTCCTGAACC | 261 | 60 |
| 9278 | TCCCTCCCACCACAAAAG | GTGGGTCAGTCCTTGGAGAG | 395 | 60 |
| 9279 | GAAATCTCCATTCCGTGGC | AACATTGCTCGTGGTATTTCC | 354 | 60 |
| 9280 | TCTTTCCCCAACCTTGACTAC | GCAATGGGAGATTACTTGGC | 335 | 60 |
| 9281 | TTAATCCAACCAGTTTCCATTC | TTGGGGACCCTATTGTTGAC | 375 | 60 |
| 9282 | ATGATGCCACCTGTAGGACC | GATTTGCTAGCTGTGCTTTGG | 355 | 60 |
| 9283 | TGGGAATGCCTTTAAATCAG | TCCATTTGAGACTGTCTGAGC | 355 | 60 |
| 9284 | AGACGCATTGCGGAAAAG | GTGTTTGCATTGTGGCAGTG | 339 | 60 |
| 9285 | GCCTCAAATACTGCTTTGCC | GAAGAGAATTTCACAATGGGG | 467 | 60 |
| 9286 | TCATGATTAATAACTGGGTTTTGTG | AGTGCTGTTGGATGGAAAATG | 243 | 60 |
| 9287 | ACCCATTGGGAGTGATTTTC | AGGGTGGAGGTTGATCTCAG | 223 | 60 |
| 9288 | TCTCGGGTACAAAGAGAGGC | TTGTTTTCTTCTAAAATGACACAGC | 320 | 60 |
| 9289 | GAGGCCTCAAAATTATTTCCC | AATGGGGCTGATCTGAGTTG | 351 | 60 |
| 9290 | TTACAAAGCAGTTGTCGATGC | TGTGGCAACTTGCTGACTTG | 311 | 60 |
| 9291 | GAGGAATTCAAGAGGGGCTC | CATCTTTCCACAGTGTTCTCTAGTC | 387 | 60 |
| 9292 | CCACCAAGAAACTGTCCAGG | ATCTGAAGGGCTAGGCACAC | 315 | 60 |
| 9293 | GGAGCAGTCTCCAAGTTCC | GAACAAAGGTTAGTATTTGACAATGG | 361 | 60 |
| 9294 | TCTGGGTAATGTGGTTTGGTC | TCAGTGAGATTTCATTGTCCC | 197 | 60 |
| 9295 | TGGAATTGGTACTTTTGTTTTCTG | TTTGACAATACTCAAGAGACTTTACC | 258 | 60 |
| 9296 | TGTGATTTCATCCTCTGTTGTTG | CACTGCGCATTAGTGGAGTAG | 151 | 60 |
| 9658 | TCCCCAACCTTGACTACAAAG | AGAGCACAAGAAGGTGGAGG | 140 | 60 |
| 9659 | TCCACTTGGCATTCTTGACC | CTTGGCAAAGGAACAACAGC | 147 | 60 |
| MBD5 RT-PCR and RACE primer: | ||||
| 9432 | … | GACATTCCAAGCCACACTTGC | … | 64 |
| 9433 | … | CATGTTCCATCAGTAAGCAGG | … | 62 |
| 9434 | … | ACACGACGCTGCCAACCCAC | … | 66 |
| 9468 C1F | AGAGGTACTCCCTTATAGGG | … | … | 60 |
| 9468 C2F | GGACTCGTAAAGACATAGAGC | … | … | 60 |
| 9469 C1F | TACTCCCTTATAGGGACTCG | … | … | 60 |
| 9469 C2F | GGGACTCGTAAAGACATAGAG | … | … | 60 |
| 9470 C1F | GAAATCAAGAAGAGCACACAC | … | … | 60 |
| 9470 C1F | ACACTATTTTCCTTCATCAATCC | … | … | 60 |
| 9725 | ATGTCAGTTTCTACATGTGGG | … | … | 60 |
| 9726 | ATGTCAGTTTCTACATGTGGG | … | … | 60 |
| 9727 | GGGAAGTAGACATTGAAGGC | … | … | 60 |
| 9728 | GCCACCCGTCAGAGAGGGAC | … | … | 60 |
| 9729 | GTCAGAGAGGGACATGCGC | … | … | 60 |
| 10028 | … | GTTGACATCCTCTGTTGCCA | … | 60 |
| X inactivation: | ||||
| 8774 | TCCAGAATCTGTTCCAGAGCGTGC | GCTGTGAAGGTTGCTGTTCCTCAT | 287 | 58 |
Footnotes
Nucleotide sequence data reported herein are available in the DNA Data Bank of Japan (DDBJ), EMBL, and GenBank databases.
Web Resources
Accession numbers and URLs for data presented herein are as follows:
- Database of Genomic Variants, http://projects.tcag.ca/variation/
- DECIPHER, http://www.sanger.ac.uk/PostGenomics/decipher/
- ExonPrimer, http://ihg.gsf.de/ihg/ExonPrimer.html
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for MBD5 breakpoint region [accession number EF504248], FOXP1 breakpoint region [accession number EF504249], and MBD5 mRNA [accession number EF542797])
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for 17p11.2 duplication syndrome, Smith-Magenis syndrome, Diamond-Blackfan anemia, and X-linked ichthyosis)
- Scripts Web site, http://ihg.gsf.de/cnv-scripts (for scripts used for data analysis in this study)
References
- 1.Roeleveld N, Zielhuis GA, Gabreels F (1997) The prevalence of mental retardation: a critical review of recent literature. Dev Med Child Neurol 39:125–132 [DOI] [PubMed] [Google Scholar]
- 2.Leonard H, Wen X (2002) The epidemiology of mental retardation: challenges and opportunities in the new millennium. Ment Retard Dev Disabil Res Rev 8:117–134 10.1002/mrdd.10031 [DOI] [PubMed] [Google Scholar]
- 3.de Vries BB, van den Ouweland AM, Mohkamsing S, Duivenvoorden HJ, Mol E, Gelsema K, van Rijn M, Halley DJ, Sandkuijl LA, Oostra BA, et al, for the Collaborative Fragile X Study Group (1997) Screening and diagnosis for the fragile X syndrome among the mentally retarded: an epidemiological and psychological survey. Am J Hum Genet 61:660–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schinzel A (2001) Catalogue of unbalanced chromosome aberrations in man. Walter de Gruyter, Berlin [Google Scholar]
- 5.Flint J, Wilkie AO, Buckle VJ, Winter RM, Holland AJ, McDermid HE (1995) The detection of subtelomeric chromosomal rearrangements in idiopathic mental retardation. Nat Genet 9:132–140 10.1038/ng0295-132 [DOI] [PubMed] [Google Scholar]
- 6.Koolen DA, Nillesen WM, Versteeg MH, Merkx GF, Knoers NV, Kets M, Vermeer S, van Ravenswaaij CM, de Kovel CG, Brunner HG, et al (2004) Screening for subtelomeric rearrangements in 210 patients with unexplained mental retardation using multiplex ligation dependent probe amplification (MLPA). J Med Genet 41:892–899 10.1136/jmg.2004.023671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Vries BB, Pfundt R, Leisink M, Koolen DA, Vissers LE, Janssen IM, Reijmersdal S, Nillesen WM, Huys EH, Leeuw N, et al (2005) Diagnostic genome profiling in mental retardation. Am J Hum Genet 77:606–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shaw-Smith C, Redon R, Rickman L, Rio M, Willatt L, Fiegler H, Firth H, Sanlaville D, Winter R, Colleaux L, et al (2004) Microarray based comparative genomic hybridisation (array-CGH) detects submicroscopic chromosomal deletions and duplications in patients with learning disability/mental retardation and dysmorphic features. J Med Genet 41:241–248 10.1136/jmg.2003.017731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vissers LE, de Vries BB, Osoegawa K, Janssen IM, Feuth T, Choy CO, Straatman H, van der Vliet W, Huys EH, van Rijk A, et al (2003) Array-based comparative genomic hybridization for the genomewide detection of submicroscopic chromosomal abnormalities. Am J Hum Genet 73:1261–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Menten B, Maas N, Thienpont B, Buysse K, Vandesompele J, Melotte C, de Ravel T, Van Vooren S, Balikova I, Backx L, et al (2006) Emerging patterns of cryptic chromosomal imbalance in patients with idiopathic mental retardation and multiple congenital anomalies: a new series of 140 patients and review of published reports. J Med Genet 43:625–633 10.1136/jmg.2005.039453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Slater HR, Bailey DK, Ren H, Cao M, Bell K, Nasioulas S, Henke R, Choo KH, Kennedy GC (2005) High-resolution identification of chromosomal abnormalities using oligonucleotide arrays containing 116,204 SNPs. Am J Hum Genet 77:709–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Friedman JM, Baross A, Delaney AD, Ally A, Arbour L, Armstrong L, Asano J, Bailey DK, Barber S, Birch P, et al (2006) Oligonucleotide microarray analysis of genomic imbalance in children with mental retardation. Am J Hum Genet 79:500–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharp AJ, Hansen S, Selzer RR, Cheng Z, Regan R, Hurst JA, Stewart H, Price SM, Blair E, Hennekam RC, et al (2006) Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet 38:1038–1042 10.1038/ng1862 [DOI] [PubMed] [Google Scholar]
- 14.Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, et al (2006) Global variation in copy number in the human genome. Nature 444:444–454 10.1038/nature05329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee JA, Inoue K, Cheung SW, Shaw CA, Stankiewicz P, Lupski JR (2006) Role of genomic architecture in PLP1 duplication causing Pelizaeus-Merzbacher disease. Hum Mol Genet 15:2250–2265 10.1093/hmg/ddl150 [DOI] [PubMed] [Google Scholar]
- 16.Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215 10.1093/nar/16.3.1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nannya Y, Sanada M, Nakazaki K, Hosoya N, Wang L, Hangaishi A, Kurokawa M, Chiba S, Bailey DK, Kennedy GC, et al (2005) A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res 65:6071–6079 10.1158/0008-5472.CAN-05-0465 [DOI] [PubMed] [Google Scholar]
- 18.Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW (1992) Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet 51:1229–1239 [PMC free article] [PubMed] [Google Scholar]
- 19.Faivre L, Morichon-Delvallez N, Viot G, Narcy F, Loison S, Mandelbrot L, Aubry MC, Raclin V, Edery P, Munnich A, et al (1998) Prenatal diagnosis of an 8p23.1 deletion in a fetus with a diaphragmatic hernia and review of the literature. Prenat Diagn 18:1055–1060 [DOI] [PubMed] [Google Scholar]
- 20.Devriendt K, Matthijs G, Van Dael R, Gewillig M, Eyskens B, Hjalgrim H, Dolmer B, McGaughran J, Brondum-Nielsen K, Marynen P, et al (1999) Delineation of the critical deletion region for congenital heart defects, on chromosome 8p23.1. Am J Hum Genet 64:1119–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Potocki L, Chen KS, Park SS, Osterholm DE, Withers MA, Kimonis V, Summers AM, Meschino WS, Anyane-Yeboa K, Kashork CD, et al (2000) Molecular mechanism for duplication 17p11.2—the homologous recombination reciprocal of the Smith-Magenis microdeletion. Nat Genet 24:84–87 10.1038/71743 [DOI] [PubMed] [Google Scholar]
- 22.Potocki L, Bi W, Treadwell-Deering D, Carvalho CM, Eifert A, Friedman EM, Glaze D, Krull K, Lee JA, Lewis RA, et al (2007) Characterization of Potocki-Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am J Hum Genet 80:633–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stefansson H, Helgason A, Thorleifsson G, Steinthorsdottir V, Masson G, Barnard J, Baker A, Jonasdottir A, Ingason A, Gudnadottir VG, et al (2005) A common inversion under selection in Europeans. Nat Genet 37:129–137 10.1038/ng1508 [DOI] [PubMed] [Google Scholar]
- 24.Koolen DA, Vissers LE, Pfundt R, de Leeuw N, Knight SJ, Regan R, Kooy RF, Reyniers E, Romano C, Fichera M, et al (2006) A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat Genet 38:999–1001 10.1038/ng1853 [DOI] [PubMed] [Google Scholar]
- 25.Shaw-Smith C, Pittman AM, Willatt L, Martin H, Rickman L, Gribble S, Curley R, Cumming S, Dunn C, Kalaitzopoulos D, et al (2006) Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat Genet 38:1032–1037 10.1038/ng1858 [DOI] [PubMed] [Google Scholar]
- 26.Horn D, Spranger S, Krüger G, Wagenstaller J, Weschke B, Ropers HH, Mundlos S, Ullmann R, Strom TM, Klopocki E (2007) Microdeletions and microduplications affecting the STS gene at Xp22.31 are associated with a distinct phenotypic spectrum. Medizinische Genetik 19:62 [Google Scholar]
- 27.Hernandez-Martin A, Gonzalez-Sarmiento R, De Unamuno P (1999) X-linked ichthyosis: an update. Br J Dermatol 141:617–627 10.1046/j.1365-2133.1999.03098.x [DOI] [PubMed] [Google Scholar]
- 28.Fukami M, Kirsch S, Schiller S, Richter A, Benes V, Franco B, Muroya K, Rao E, Merker S, Niesler B, et al (2000) A member of a gene family on Xp22.3, VCX-A, is deleted in patients with X-linked nonspecific mental retardation. Am J Hum Genet 67:563–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van Esch H, Hollanders K, Badisco L, Melotte C, Van Hummelen P, Vermeesch JR, Devriendt K, Fryns JP, Marynen P, Froyen G (2005) Deletion of VCX-A due to NAHR plays a major role in the occurrence of mental retardation in patients with X-linked ichthyosis. Hum Mol Genet 14:1795–1803 10.1093/hmg/ddi186 [DOI] [PubMed] [Google Scholar]
- 30.Green EK, Priestley MD, Waters J, Maliszewska C, Latif F, Maher ER (2000) Detailed mapping of a congenital heart disease gene in chromosome 3p25. J Med Genet 37:581–587 10.1136/jmg.37.8.581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sock E, Rettig SD, Enderich J, Bosl MR, Tamm ER, Wegner M (2004) Gene targeting reveals a widespread role for the high-mobility-group transcription factor Sox11 in tissue remodeling. Mol Cell Biol 24:6635–6644 10.1128/MCB.24.15.6635-6644.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roloff TC, Ropers HH, Nuber UA (2003) Comparative study of methyl-CpG-binding domain proteins. BMC Genomics 4:1 10.1186/1471-2164-4-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qiu C, Sawada K, Zhang X, Cheng X (2002) The PWWP domain of mammalian DNA methyltransferase Dnmt3b defines a new family of DNA-binding folds. Nat Struct Biol 9:217–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koolen DA, Vissers LE, Nillesen W, Smeets D, van Ravenswaaij CM, Sistermans EA, Veltman JA, de Vries BB (2004) A novel microdeletion, del(2)(q22.3q23.3) in a mentally retarded patient, detected by array-based comparative genomic hybridization. Clin Genet 65:429–432 10.1111/j.0009-9163.2004.00245.x [DOI] [PubMed] [Google Scholar]
- 35.Seidman JG, Seidman C (2002) Transcription factor haploinsufficiency: when half a loaf is not enough. J Clin Invest 109:451–455 10.1172/JCI200215043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Ommen GJ (2005) Frequency of new copy number variation in humans. Nat Genet 37:333–334 10.1038/ng0405-333 [DOI] [PubMed] [Google Scholar]