

Abstract
Our previous genomewide linkage scan of 428 nuclear families (GeneQuest) identified a significant genetic susceptibility locus for premature myocardial infarction (MI) on chromosome 1p34-36. We analyzed candidate genes in the locus with a population-based association study involving probands with premature coronary artery disease (CAD) and/or MI from the GeneQuest families (381 cases) and 560 controls without stenosis detectable by coronary angiography. A nonconservative substitution, R952Q, in LRP8 was significantly associated with susceptibility to premature CAD and/or MI by use of both population-based and family-based designs. Three additional white populations were used for follow-up replication studies: another independent cohort of CAD- and/or MI-affected families (GeneQuest II: 441 individuals from 22 pedigrees), an Italian cohort with familial MI (248 cases) and 308 Italian controls, and a separate Cleveland GeneBank cohort with sporadic MI (1,231 cases) and 560 controls. The association was significantly replicated in two independent populations with a family history of CAD and/or MI, the GeneQuest II family-based replication cohort and the Italian cohort, but not in the population with sporadic disease. The R952Q variant of LRP8 increased activation of p38 mitogen-activated protein kinase by oxidized low-density lipoprotein. This extensive study, involving multiple independent populations, provides the first evidence that genetic variants in LRP8 may contribute to the development of premature and familial CAD and MI.
Atherosclerotic coronary artery disease (CAD) and myocardial infarction (MI) are complex traits that account for the leading cause of death in the Western world, heart disease, projected by 2020 to be the number one cause of death and disability worldwide.1 Multiple previous studies have documented the heritability of CAD and, in particular, its most acute manifestation, MI.2,3 Genomewide linkage analysis is an unbiased approach that may lead to the identification of previously unknown genetic loci or genes for CAD and MI. Genomewide linkage scans with hundreds of sibling pairs have identified several major genetic susceptibility loci for CAD or MI.2–4 The MI susceptibility gene on chromosome 13q12 has been identified as ALOX5AP (MIM 603700), encoding 5-lipoxygenase–activating protein.5,6 For the chromosome 3q CAD locus, two candidate genes, GATA2 (MIM 137295) and Kalirin (MIM 604605), were associated with CAD.7,8 The genes at other loci have not yet been reported.
This study focuses on a susceptibility locus for MI on chromosome 1p34-36, reported by us.9 We studied 428 multiplex families (GeneQuest) with at least two sibs affected with premature CAD in each family and with one nonaffected sib in a majority of the families. Genomewide linkage analysis with the GeneQuest cohort identified a genetic susceptibility locus for premature MI on chromosome 1p34-36 in the American white population.9 Here, we report the results from the analysis of potential candidates for MI at the 1p34-36 locus. The LRP8 gene (MIM 602600) showed significant association with CAD and MI in three independent populations affected with familial CAD and MI: the GeneQuest and GeneQuest II populations, which are two family-based American cohorts with early-onset disease, and an Italian cohort. The LRP8 protein is also known as the “apolipoprotein E receptor 2.” It is a lipoprotein receptor from the low-density lipoprotein (LDL) receptor family.10 The structure of LRP8 closely resembles the LDL receptor and the very-low-density lipoprotein receptor (VLDLR). The LRP8 gene is highly expressed in the brain and testes, but it is also expressed in the heart, endothelial cells, vascular smooth muscle cells, and platelets.11 Binding of LDL to LRP8 activates the phosphorylation of LRP8, which further activates the p38 mitogen-activated protein kinase (MAPK) stress-response signaling pathway, leading to platelet aggregation and, potentially, thrombosis and MI.12 Functional studies revealed that the nonsynonymous SNP R952Q of LRP8, which was associated with CAD and MI, significantly increased the activation (phosphorylation) of p38 MAPK by LRP8 after treatment with oxidized LDL. Our results suggest that LRP8 is a novel susceptibility gene for familial and premature CAD and MI.
Material and Methods
Study Populations
The GeneQuest population consists of 1,613 individuals from 428 multiplex families with premature CAD and MI.9 Each family has at least two affected sibs. The details, including diagnostic criteria for CAD and MI, have been described elsewhere.9 For the case-control association study, the probands from the GeneQuest families were grouped together as the case group. Only 381 white patients with available DNA samples were selected, among which 183 were patients with MI. Patients of other ethnic origins were excluded, to avoid the confounding effects of population admixture. The 560 controls were selected from >9,800 individuals who underwent coronary angiography in Cardiac Catheterization Laboratories at Cleveland Clinic (Cleveland GeneBank), and only white individuals without atherosclerotic lesions detectable by angiography were included. A total of 1,231 patients with MI were selected from GeneBank and were used as a replication cohort.
The second familial and premature CAD- and/or MI-affected population, GeneQuest II, was ascertained using criteria9 identical to those that defined the original GeneQuest population at the Center for Cardiovascular Genetics of the Cleveland Clinic. A total of 22 white families with 441 family members were enrolled. The average size of the GeneQuest II families is 20 ± 14 members, and the number of affected individuals is 140.
The Italian cohort was enrolled in Verona, Italy, and details about this study population were described elsewhere.13 The cohort consisted of 416 unrelated individuals with MI (248 with family history), and 308 controls with no detectable stenosis by coronary angiography.
This study was approved by local institutional review boards on human subjects, and informed consent was obtained from the participants. Whole blood was drawn from each participant, and genomic DNA was isolated from the blood by use of standard protocols.
Genotyping of SNPs
High-throughput SNP genotyping was performed using the 5′ nuclease allelic discrimination assay (TaqMan Assay) on an ABI PRISM 7900HT Sequence Detection System. The assay includes the forward target-specific PCR primer, the reverse primer, and the TaqMan MGB probes labeled with two special dyes: FAM and VIC. The probes were purchased through TaqMan Assays-on-Demand or Assays-by-Design from Applied Biosystems (ABI). Genotyping was performed in a total 5-μl PCR volume containing 2.5 μl of TaqMan Universal PCR Master Mix, 0.25 μl of 20× TaqMan MGB Assay Mix, and 25 ng of genomic DNA. Automatic allele calling was performed by ABI PRISM 7900HT data collection and analysis software, version 2.1.
Direct DNA sequence analysis of SNPs in 24 DNA samples was used to ensure the quality of SNP genotyping. Direct DNA sequence analysis was performed using an ABI PRISM 3100 Genetic Analyzer. A DNA fragment containing the SNP was PCR amplified in a 25-μl volume containing 2.5 μl of 10× PCR buffer (1.5 mM MgCl2, 2.5 μl of 0.2 mM deoxynucleotide triphosphates, 0.5 μM of each PCR primer, 1 U of Taq polymerase, and 50 ng of genomic DNA). PCR products were separated from agarose gels, were isolated and purified using the QIAquick PCR Purification Kit (QIAGEN), and were sequenced with both forward and/or reverse primers. The sequencing reaction was performed using the BigDye Terminator v1.1 Cycle Sequencing Kit (ABI).
Activation Assay of p38 MAPK
The green fluorescent protein (GFP)–tagged human LRP8 gene was PCR amplified with primers containing inframe HindIII and BamHI restriction sites. The PCR fragment was digested with HindIII and BamHI and was subcloned into the pEGFP C1 vector cut with the same enzymes. Mutant LRP8 containing the SNP R952Q was generated by PCR-based site-directed mutagenesis, as described elsewhere.14 Both the wild-type and mutant LRP8 expression constructs were verified by direct DNA sequence analysis of the entire inserts.
Meg-01 cells (American Type Culture Collection) were grown in the RPMI-1640 medium containing 10% fetal bovine serum and were maintained at a density of 3×105 cells/ml. In general, cells were used for experiments after 5 d of supplementation. Transfection of Meg-01 cells was performed with 1 μg plasmid DNA/well with a Nucleofector device and corresponding kits (Amaxa) in 6-well plates. At 48 h after transfection, cells were divided equally into five wells and were incubated with oxidized LDL15 for 0, 20, 40, 60, and 120 min. Cells were lysed, and an equal amount of total cellular proteins per sample was separated with 12% SDS-PAGE gels and was electrotransferred onto polyvinylidene fluoride membranes. The blots were blocked in 5% nonfat milk powder in phosphate-buffered saline Tween-20 (PBST) for 1 h, were washed briefly in Tris-buffered saline Tween-20, and then were incubated with a primary monoclonal antibody directed against phosphor-p38 MAPK (D-8, catalog number SC-7973 [Santa Cruz Biotechnology]) or a polyclonal antibody against total p38 MAPK (C-20, catalog number SC-535 [Santa Cruz Biotechnology]) in 5% nonfat milk and PBST overnight. Membranes were then extensively washed in PBST (eight times for 5 min) and were incubated with a 1:2,000 dilution of horseradish peroxidase–conjugated anti-mouse IgM (Sigma) for 1 h at room temperature. Membranes were again washed (three times for 5 min each) in FBST, and ECL western-blotting detection reagents (Amersham Pharmacia Biotech) were used to visualize the protein signal. A prestained low-molecular-mass protein ladder (Bio-Rad) was run in adjacent lanes. The images from western blots were scanned and quantified. Relative signal density of phosphorylated p38 MAPK versus total p38 MAPK was calculated.
Platelet Aggregation Assays
Platelet aggregation assays were performed within 3 h after blood collection. Aggregation was measured using an impedance method on a Chrono-log Whole Blood Impedance Aggregometer. A sample of 500 μl of blood was mixed with 500 μl of saline, and the change in the impedance of the sample was measured in the presence of adenosine diphosphate (ADP) during a 6-min test run and was recorded on a computer with the Chrono-log AGGRO/LINK software. Impedance aggregation in ohms was used to index the rate and the degree of platelet aggregation.
To avoid the confounding effects of antiplatelet medications, we conducted statistical analysis of the data from the platelet aggregation assay only for 56 well-characterized patients with MI who were verified as having no history of taking antiplatelet drugs (e.g., aspirin). To reveal the relationship between platelet aggregation and LRP8 SNPs, an autosomal dominant model (i.e., meanAA=meanAa≠meanaa) was evaluated in the framework of a general linear model (SAS, version 9.00).
Statistical Analysis
All SNPs were tested for Hardy-Weinberg equilibrium among controls by use of the Haploview software package, version 3.0. All SNPs were in Hardy-Weinberg equilibrium (P>.05).
Association of SNPs with the disease was assessed using Pearson’s 2×2 contingency table χ2 test or Fisher’s exact test (SAS, version 9.00). Odds ratios (ORs) and 95% CIs were estimated using the χ2 test (SAS, version 9.00). Multivariate analysis was performed by incorporating age and sex as covariates by use of multivariate logistic regression or in combination with additional covariates, including total cholesterol and triglyceride levels. Smoking was not included in the analysis because of lack of detailed phenotypic data. Genotyping data were analyzed additionally for association with CAD or MI by use of the Z-score method.16 Empirical P values were calculated using 10,000 Monte Carlo simulations, by the CLUMP program17 (Software Written by Dave Curtis Web site).
A sib–transmission/disequilibrium test (TDT) analysis was performed using the TDT/S-TDT program 1.1.18–20 The sib-TDT was performed for CAD only because the sample size for MI was small for a sib-TDT analysis.
To remove the effects of the LRP8 genetic variant encoding R952Q from the original 1p34-36 linkage signal, we used multiple regression models with the LRP8 SNP identical by descent (IBD) considered as a covariate, to demonstrate its influence on the original linkage profile. In detail, the new Haseman-Elston regression uses the following form of a general linear model:
where y is the quadratic form of the phenotypes of the siblings (here, the mean-corrected trait cross-product); α is the intercept; the subscripts m, s, and k represent the original microsatellite marker, the LRP SNP, and the covariate sex, respectively; d is the dominant genetic variance due to the marker or the SNP; a is the additive genetic variance due to the marker or the SNP; ck is a nuisance parameter accounting for the effect of mean-corrected cross-product of sex (f(zk)); and ɛ is the residual error. 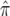 and
and 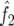 are the probabilities of mean IBD sharing and exactly 2 alleles IBD sharing, respectively. Note that S.A.G.E. 5.3 reports only 8 decimal places, instead of 12 decimal places as in the previous version used to analyze the original GeneQuest pedigrees.21
are the probabilities of mean IBD sharing and exactly 2 alleles IBD sharing, respectively. Note that S.A.G.E. 5.3 reports only 8 decimal places, instead of 12 decimal places as in the previous version used to analyze the original GeneQuest pedigrees.21
Results
Risk of CAD and MI Conferred by LRP8 SNP R952Q in the GeneQuest Population
Initially, we employed a population-based case-control association study design to characterize the candidate genes for MI at the 1p34-36 locus. The 381 CAD cases were the white probands, one per family, from the GeneQuest families with familial premature CAD and MI.9 Among them, 183 had well-characterized MI and served as the MI cases. We excluded individuals of other ethnic origins, to minimize the confounding effects of population admixture. The 560 controls were independent white individuals who were enrolled at Cleveland Clinic and showed no detectable stenosis by coronary angiography. The clinical features of cases and controls are shown in table 1.
Table 1. .
Clinical Characteristics of Study Populations and Unaffected Controls[Note]
| Finding in Population |
||||||
| Characteristic | GeneQuest CAD (n=381) |
GeneQuest MI (n=183) |
GeneBank MI (n=1,231) |
Control (n=560) |
Italian MI (n=248) |
Italian Control (n=308) |
| No. of males; females | 248; 133a | 121; 62a | 1,010; 221a | 269; 291 | 199; 49a | 214; 94 |
| Age (years) | 40.2 ± 4.9a | 39.6 ± 5.1a | 60.6 ± 12.1a | 53.5 ± 12.1 | 60.9 ± 9.6b | 58.1 ± 12.6 |
| Ethnicity | White | White | White | White | Italian | Italian |
| Family history of disease | Yes | Yes | Sporadic | NA | Yes | NA |
| Smoking (%) | 79.0a | 86.3a | 75.4a | 47.2 | 70.0a | 45.1 |
| BMId | 29.6 ± 5.6 | 28.9 ± 6.3 | 29.7 ± 8.2 | 29.2 ± 7.1 | 26.4 ± 3.3c | 25.3 ± 3.3 |
| Hypertension (%) | 47.9 | 43.7 | 71.9a | 43.4 | 65.3a | 33.1 |
| Diabetes (%) | 16.5a | 12.0 | 30.6a | 7.9 | 23.4a | 13.5 |
| Total cholesterol (mg/dL) | 219.2 ± 57.0a | 205.2 ± 58.2a | 172.9 ± 44.2a | 188.2 ± 43.2 | 221.9 ± 45.9a | 213.4 ± 45.5 |
| HDL cholesterol (mg/dL) | 39.0 ± 11.2a | 37.2 ± 13.5a | 41.1 ± 11.3a | 45.1 ± 14.6 | NA | NA |
| LDL cholesterol (mg/dL) | 135.1 ± 43.2a | 122.7 ± 50.5a | 96.9 ± 35.1a | 116.6 ± 35.9 | 151.5 ± 38.4a | 138.7 ± 36.8 |
| Triglycerides (mg/dL) | 239.4 ± 24.6a | 222.0 ± 20.5a | 180.5 ± 26.2a | 135.0 ± 82.5 | 178.0 ± 79.9a | 136.5 ± 52.0 |
Note.— Data are shown as mean ± SD, unless otherwise indicated. Age = age at onset for cases and age at examination for controls; HDL = high-density lipoprotein; NA = not applicable.
P<.001, compared with controls.
P<.01, compared with controls.
P<.05, compared with controls.
BMI measured as body weight in kilograms divided by the square of height in meters.
We employed a type of systematic approach to analyze candidate genes across the entire 1p34-36 region (4.0–59.1 Mb) on the basis of a gene’s position and function. For the genetic interval under the linkage peak (13.2–34.1 Mb), one gene every 1–3 Mb was selected. For the genetic intervals outside the linkage peak (4.0–13.2 and 34.1–59.1 Mb), one gene every 3–6 Mb was selected. Genes with physiological functions relevant to those involved in the development of CAD or MI were prioritized. Twelve candidate genes were identified for analysis in this manner (table 2). One SNP was studied for each gene, on the basis of its availability in the ABI Assays-on-Demand, minor-allele frequency (MAF) >30% (on the basis of the assumption that common disease may be associated with evolutionarily old, common variants), exonic position, and nonsynonymous nature, if possible. Of the 12 genes tested, only a single SNP, a nonsynonymous variant located within the LRP8 gene (rs5174 [R952Q]; dbSNP database), showed significant association with CAD and MI (P=.003 for CAD and P=.004 for MI; P=.036 for CAD and P=.048 for MI after adjustment by approximate Bonferroni correction22) (table 2).
Table 2. .
Analysis of Candidate Genes at the 1p34-36 MI Locus for Association with CAD and MI[Note]
|
P |
||||
| Gene | Positiona (Mb) |
SNP | CADb | MIc |
| MFAP2 | 17.1 | rs2235932 | .552 | .632 |
| PLA2G2A | 20.1 | rs4744 | .202 | .476 |
| HMGCL | 23.9 | rs719400 | .375 | .338 |
| LDLRAP1 | 25.6 | rs11563 | .686 | .246 |
| NR0B2 | 26.9 | rs7504 | .131 | .288 |
| SLC9A1 | 27.2 | rs11247613 | .683 | .852 |
| AK2 | 33.1 | rs998664 | .197 | .300 |
| GJB5 | 34.9 | rs2275229 | .730 | .700 |
| CSF3R | 36.6 | rs3917924 | .630 | .500 |
| CDC20 | 43.5 | rs839763 | .815 | .744 |
| FAAH | 46.6 | rs324420 | .916 | .736 |
| LRP8 | 53.5 | rs5174 | .003 | .004 |
Note.— The entire 1p34-36 locus spans a region from 4.0 Mb (CATC015) to 59.1 Mb (GATA26G09P), with the plateau of genomewide significance from 13.2 Mb (GATA27E01) to 34.1 Mb (ATA79C10) (information on the markers can be found at Marshfield Genotyping Service).
Positions as shown in the UCSC Genome Browser.
P value for association with CAD.
P value for association with MI.
Permutation testing also showed a significant empirical P value for the association between SNP R952Q and CAD (empirical P=.003) or MI (empirical P=.004) (table 3). Multivariate analysis was performed to examine the possible confounding effects of age, sex, and other factors, and we found that SNP R952Q can be considered an independent risk factor for CAD (P=.009 after adjustment for age and sex; P=.006 after adjustment for age, sex, total cholesterol levels, triglyceride levels, hypertension, and diabetes) and MI (adjusted P values of .010 and .003, respectively) (table 3). We also used the Z-score method16 to analyze the SNP data; by this method, the R952Q variant was significantly associated with CAD and MI (table 4). These results suggest that LRP8 SNP R952Q is associated with premature CAD and MI in an American white population.
Table 3. .
Association of SNP R952Q of LRP8 with Family-Based CAD and MI
| Allelea |
P |
||||||||
| Populations | Risk | Minor | No. (%) of Cases | No. (%) of Controls | OR | HWb | Observedc | Adjustedd | Empiricale |
| GeneQuest CAD and control | A | A | 381 (44.9) | 560 (37.8) | 1.35 | .28 | .003 | .009 | .003 |
| GeneQuest MI and control | A | A | 183 (46.2) | 560 (37.8) | 1.42 | .28 | .004 | .010 | .004 |
| Italian MI and control | A | A | 248 (40.6) | 308 (33.6) | 1.35 | .34 | .016 | .020 | .019 |
| Combinedf | A | A | 651 (42.9) | 868 (36.3) | 1.31 | .36 | .0004 | .0003 | .0006 |
Allele A at the nucleotide level corresponds to variant 952Q at the protein level.
P value for Hardy-Weinberg disequilibrium analysis.
Uncorrected P value.
P value obtained after adjustment for sex and age. Multivariate analysis was also performed for plasma total cholesterol levels, triglyceride levels, hypertension, and diabetes in addition to age and sex for GeneQuest CAD (P=.006), GeneQuest MI (P=.003), Italian MI (P=.041), and combined (P=.0005) populations.
Permutation P value calculated using 10,000 Monte Carlo simulations.
Combined population comprised 381 CAD cases from GeneQuest population, 22 MI probands from GeneQuest II population, and 248 Italian cases from familial MI population.
Table 4. .
Comparison of Two Different Statistics in Analyzing the Association of LRP8 SNP R952Q with CAD and MI
| Statistic | GeneQuest CAD | GeneQuest MI | Italian MI |
| χ2: | 9.05 | 8.16 | 6.95 |
| P | .003 | .004 | .008 |
| Z score: | 3.00 | 2.82 | 2.66 |
| P | .003 | .005 | .008 |
To determine whether the SNP in LRP8 that was associated with CAD and MI is relevant to the previous 1p34-36 MI linkage, we investigated the covariate effects of the LRP8 SNP on the original chromosome 1 linkage profile. SNP R952Q appeared to explain a significant amount of the original linkage signals (particularly the additive component) (fig. 1).
Figure 1. .

Relationship between LRP8 SNP R952Q and the chromosome 1p34-36 premature MI susceptibility locus. The X-axis shows the position of markers, and the Y-axis is −log(P).
Variant Associated with Risk of CAD and MI in the LRP8 Section of Haplotype Block LD5
The LRP8 gene contains 19 exons spanning ∼60 kb. SNP R952Q, showing positive association in the above-described study, is located in exon 19. Analysis of the HapMap data showed that LRP8 is composed of five linkage disequilibrium (LD) blocks (LD1–LD5, from the 5′ end to the 3′ end of the gene) among individuals of European American ancestry. SNP R952Q is in LD5 at the 3′ terminus of the LRP8 gene. To test the association of other LD blocks of LRP8 with CAD and MI, one SNP was selected from each block on the basis of the criteria described above (rs3820198 in LD1, rs1288480 in LD2, rs867884 in LD3, and rs12039021 in LD4) and was used for fine-scale association mapping in LRP8. No association was detected between these SNPs and CAD or MI (P=.16–.94) (table 5). These data suggest that only LD5, the most 3′ block of LRP8, is associated with risk of CAD and MI.
Table 5. .
Analysis of Association between SNPs in Blocks LD1–LD4 of the LRP8 Gene and CAD and MI
| MAF(%) |
||||
| Association and SNP |
Cases | Controls | OR (95% CI) | P |
| With CADa: | ||||
| rs3820198 | 34.7 | 36.5 | .92 (.75–1.13) | .439 |
| rs1288480 | 34.3 | 33.6 | 1.03 (.84–1.27) | .761 |
| rs867884 | 40.3 | 37.9 | 1.09 (.90–1.35) | .327 |
| rs12039021 | 41.8 | 41.6 | 1.01 (.83–1.23) | .938 |
| With MIb: | ||||
| rs3820198 | 34.2 | 36.5 | .90 (.69–1.18) | .454 |
| rs1288480 | 36.4 | 33.6 | 1.13 (.86–1.48) | .370 |
| rs867884 | 42.4 | 38.0 | 1.20 (.93–1.56) | .162 |
| rs12039021 | 43.1 | 41.6 | 1.06 (.82–1.38) | .631 |
GeneQuest CAD population (381 cases) and 560 controls.
GeneQuest MI population (183 cases) and 560 controls.
Further analysis of SNP genotyping data from the HapMap data revealed that LD5 of LRP8 extends beyond the 3′ end of the LRP8 gene and spans additional genes, MAGOH and FLJ20580. MAGOH, encoding a component of the multiprotein exon-junction complex, resides at 14.6 kb from the 3′ end of LRP8. Tag SNPs capturing MAGOH (rs6673692 and rs10788949) were selected and tested, but we did not find any association with CAD (P=.61 and .95, respectively) or MI (P=.13 and .58, respectively) (table 6). Additionally, we tested two SNPs in FLJ20580 (rs1056425 and rs1134688), which is the gene distal to MAGOH (30 kb from LRP8), but, again, we did not find any association with CAD (P=.74 and .93, respectively) or MI (P=.57 and .96, respectively). These data suggest that MAGOH and FLJ20580 are not associated with CAD or MI and that functional variants conferring risk of CAD and MI reside within LRP8.
Table 6. .
Analysis of Association between Two Additional Genes in LD5 of LRP8 and CAD and MI
|
P |
|||
| Gene and Positiona |
SNP | CADb | MIc |
| MAGOH: | |||
| 53,469,727 | rs6673692 | .608 | .131 |
| 53,468,639 | rs10788949 | .945 | .583 |
| FLJ20580: | |||
| 53,454,287 | rs1134688 | .926 | .958 |
| 43,018,309 | rs1056425 | .743 | .565 |
Positions in bases as shown in the UCSC Genome Browser.
P value for association with CAD.
P value for association with MI.
Similar to the HapMap data, in the GeneQuest control population, LRP8 SNP R952Q is in a continuous haplotype block (LD5) with rs6673692 and rs10788949 in the MAGOH gene and with rs1056425 and rs1134688 in the FLJ20580 gene (fig. 2). However, in the CAD- and MI-affected populations, LD5 was disrupted, and SNP R952Q was cut off from the block (if LD significance is equal to D′>.8). These results suggest that disruption of an LD block in a disease population may be considered as supportive evidence of its association with the disease.
Figure 2. .

Pairwise LD analysis. LD between SNPs in LRP8, including R952Q (rs5174), and SNPs in two nearby genes (rs6673692 and rs10788949 in MAGOH and rs1134688 and rs1056425 in FLJ20580) was derived from genotyping data from the GeneQuest control (top) and MI (bottom) populations. LD for the CAD population was nearly identical to the MI population (data not shown). The pairwise correlation between SNPs was measured as D′ and is shown (×100) in each diamond. The red-to-white color gradient indicates the magnitude of pairwise LD, ranging from higher to lower values of LD.
Family-Based TDT
Sib-TDT was used to determine whether the risk allele of a SNP was transmitted preferentially to affected offspring. If a specific allele is transmitted more frequently to affected offspring, the allele is both linked and associated with CAD.20 Sib-TDT is also an effective strategy to minimize the confounding effects of population admixture. LRP8 SNP R952Q was genotyped in the full GeneQuest cohort (table 7), including probands and other family members. Sib-TDT of the genotyping data revealed that R952Q was significantly associated with CAD (P=.005) (table 8). Together, these data suggest that SNP R952Q is associated with CAD by use of both population- and family-based study designs with the GeneQuest cohort.
Table 7. .
Demographic Features of the GeneQuest and GeneQuest II Family-Based Study Populations
| Feature | GeneQuest | GeneQuest II |
| No. of individuals | 1,769 | 441 |
| No. of males; females | 1,018; 751 | 216; 225 |
| Ethnicity | White | White |
| Pedigree structure: | ||
| No. of pedigrees | 378 | 22 |
| No. of sibships | 384 | 118 |
| Pedigree size (mean ± SD) | 4.7±1.3 | 20.1±13.8 |
| Sibship size (mean ± SD) | 2.6±1.2 | 2.6±1.9 |
| No. affected with CAD/MI | 757 | 140 |
| No. of relative pairs: | ||
| Sibling/sibling | 1,303 | 442 |
| Sister/sister | 258 | 145 |
| Brother/brother | 476 | 105 |
| Brother/sister | 569 | 192 |
Table 8. .
Sib-TDT Analysis in the GeneQuest and GeneQuest II Populations
| Population | Risk Allelea | Minor Allelesb | Z Score | P |
| GeneQuest | A | 170 | 2.63 | .005 |
| GeneQuest II | A | 72 | 2.34 | .009 |
Allele A at the nucleotide level corresponds to variant 952Q at the protein level.
The number of minor alleles among affected sibs across all sibships.
Association of LRP8 SNP R952Q with CAD in the GeneQuest II Families
Recently, we enrolled 441 individuals from 22 large white families (GeneQuest II replication population, average pedigree size of 20) (table 7), to test further the association of LRP8 with CAD. Selection criteria for GeneQuest II were identical to those for the original GeneQuest cohort.9 Briefly, each proband must have presented with “premature” CAD, which was defined as any previous or current evidence of significant atherosclerotic CAD occurring in males aged ⩽45 years or females aged ⩽50 years. For recruitment, each proband was required to have at least one other living sibling who met the same criteria. SNP R952Q was genotyped in the GeneQuest II cohort. Sib-TDT of the genotyping data showed that SNP R952Q was significantly associated with CAD (P=.009) (table 8). These results from the GeneQuest II families provide the first replication of association between SNP R952Q and CAD in American white families with premature CAD.
Validation of Association of LRP8 SNP R952Q with MI in an Italian Cohort
To replicate further the association of LRP8 with MI, we studied a separate white population from Italy with familial MI. Of a total of 416 MI cases, 248 (60%) were individuals with a family history of MI and were selected for further study. We genotyped SNP R952Q in the Italian cohort (248 MI cases and 308 controls) (table 1). The SNP showed significant association with MI (table 3) and was further confirmed by permutation testing, yielding a significant empirical P value. Significant association was also identified when all 416 Italian MI cases were analyzed together (P=.008). These results were also significant after adjustment for age, sex, triglyceride levels, total cholesterol levels, hypertension, and diabetes (table 3). These results provide the second replication of our finding of association between LRP8 SNP R952Q and CAD and MI.
Assessment of Association of LRP8 SNP R952Q with CAD in a Combined White Cohort
To provide a comprehensive assessment of the association of LRP8 SNP R952Q with CAD, we performed an analysis of all familial white CAD populations combined (651 cases and 868 controls), as suggested by Skol et al.16 The significant P values for association of CAD with the LRP8 SNP R952Q were improved by >10-fold (P=.0003–.006). Multivariate analysis with age and sex as covariates showed a significant P value of .0003. The P value remained significant after the factors plasma total cholesterol levels, triglyceride levels, hypertension, and diabetes, in addition to age and sex, were included as covariates (P=.0005) (table 3).
SNP R952Q and the Function of LRP8
LRP8 SNP R952Q is a nonsynonymous substitution of a positively charged arginine residue by a negatively charged glutamine residue. To clarify whether this SNP can affect the function of the LRP8 protein, we transfected mammalian expression constructs with either wild-type LRP8 or mutant LRP8 with the variant encoding R952Q into Meg-01 cells. Cells were treated with oxidized LDL for various times, and activation of p38 MAPK was assayed. Six replicate experiments showed that the peak phosphorylation level of p38 MAPK by mutant LRP8 R952Q was greater than that by the wild type (P=.009), and the high level of phosphorylation of p38 MAPK was maintained for a much longer time (P=.001 at 120 min) (fig. 3). These results suggest that the R952Q variant of LRP8 is a functional SNP that results in increased phosphorylation (activation) of p38 MAPK.
Figure 3. .

Effects of the LRP8 R952Q SNP on phosphorylation of p38 MAPK induced by oxidized LDL (ox-LDL). Meg-01 cells were transfected with pEGFP-hLRP8 wild type (WT) or pEGFP-hLRP8 mutant (R952Q) by use of Nucleofector. After 48 h, cells were incubated with ox-LDL (2 μg/ml) for different times and were lysed. An equal amount of total cellular proteins (30 μg) was analyzed for phosphorylated (p-p38) and total p38 MAPK by western-blot analysis. A representative image of the blot is shown (A). The images were scanned and quantified. The experiments were replicated six times, and the graph (B) represents the data from six independent experiments. Data are shown as mean ± SD densitometric units.
We further studied the correlation of LRP8 SNP R952Q with activity of platelet aggregation in 56 patients with MI who did not take any antiplatelet medications. As shown in figure 4, the R952Q variant was associated with a small but significant increase (14%) of platelet activity at two concentrations of the ADP agonist (P=.02 and .03).
Figure 4. .

Effects of LRP8 SNP R952Q on platelet aggregation
Lack of Association of LRP8 SNP R952Q with MI in Sporadic Disease
Since we identified a consistent association of LRP8 SNP R952Q with CAD and MI in three populations with familial and premature disease, we wanted to test whether the association held in patients with sporadic and late-onset disease. To this end, we studied one population with primarily sporadic CAD and MI, the Cleveland GeneBank cohort (table 1). It is interesting to note that one other major difference in this population, compared with the previous populations, is the markedly reduced total and LDL cholesterol and triglyceride levels (table 1). We genotyped the GeneBank cohort, consisting of 1,231 patients mostly with late-onset, sporadic MI, and compared allelic frequencies with those for the 560 unaffected controls. No significant association was identified (table 9). These results suggest that the functional LRP8 SNP R952Q may be associated with familial, premature CAD and MI but not with the sporadic form of the disease.
Table 9. .
Lack of Association of LRP8 SNP R952Q with Sporadic CAD and MI in the GeneBank Population (1,231 Cases) and 560 Controls
| MAF(%) |
P |
|||||
| SNP | Allelea | Cases | Controls | OR (95% CI) | Observedb | Adjustedc |
| R952Q | A | 39.1 | 37.8 | 1.06 (.82–1.09) | .439 | .253 |
Allele A at the nucleotide level corresponds to variant 952Q at the protein level.
Uncorrected P value.
P value obtained after adjustment for sex and age.
Discussion
Here, we report the identification of association of a previously unknown susceptibility gene, LRP8, with familial and premature CAD and MI. Several lines of evidence strongly support this conclusion. (1) By use of a population-based approach, SNP R952Q, residing in the last LD block (LD5) at the 3′ terminus of LRP8, showed a significantly increased risk of premature, familial CAD and MI in an American white population (GeneQuest). (2) LRP8 SNPs in the other four LD blocks (LD1–LD4) showed negative association with CAD and MI. (3) SNPs in other candidate genes within the 1p34-36 linkage interval were not associated with CAD and MI, including MFAP2, PLA2G2A, HMGCL, LDLRAP1, NR0B2, SLC9A1, AK2, GJB5, CSF3R, CDC20, FAAH, MAGOH, and FLJ20580. (4) Family-based (TDT) analysis showed that the risk allele of SNP R952Q was transmitted preferentially to affected individuals in GeneQuest families and provided further support that the LRP8 variant conferred risk of CAD. (5) Family-based (TDT) analysis showed that SNP R952Q conferred risk of CAD in another, independent population with familial CAD, the GeneQuest II American white families with premature CAD. (6) Another replication was made in an Italian white population with familial MI by use of a population-based approach. (7) Further analysis with all familial combined populations with CAD and/or MI showed highly significant association between LRP8 SNP R952Q and CAD and MI (empirical P=.0006). (8) Functional studies demonstrated that SNP R952Q has a pronounced effect on the function of LRP8. Together, these results suggest that LRP8 SNP R952Q conferred risk of familial and premature CAD and MI.
Platelets are a critical component of the atherosclerotic process.23 Platelets are known to secrete and express substances for coagulation and inflammation that may play roles in atherosclerosis.23 The activation of platelets is a key risk factor for atherothrombosis. Interestingly, we found that the LRP8 SNP R952Q, associated with CAD and MI, was also associated with a modest but significant increase in platelet aggregation activity in 56 patients with MI who had not received antiplatelet therapy. Thus, one mechanism by which the LRP8 variant increases risk of CAD and MI may be through sensitization of platelets.
LRP8 has been shown to be a receptor for LDL and acts by homodimerization or heterodimerization with other receptors (VLDLR and β2-glycoprotein I).12,24,25 LDL can bind to LRP8, and the interaction induces tyrosine phosphorylation of LRP8 and activation of p38 MAPK in platelets.12 LRP8 SNP R952Q increased the activation of p38 MAPK (fig. 3). Activation of p38 MAPK may influence the sensitization of platelets by LDL, resulting in the formation of arachidonate metabolites and the release of inflammatory molecules, which may increase the risk of atherosclerosis or atherothrombosis. The p38 MAPK is a stress-activated protein kinase that may play a role in the development of atherosclerosis. The p38 MAPK could affect leukocyte emigration and lead to increased leukocyte accumulation in ischemic-reperfused tissue.26 The important role of p38 MAPK in inflammation has been well established, and overexpression of p38 MAPK has been shown to induce myocardial fibrosis and inflammation.27,28 Kumar et al.29 showed that the activation of p38 MAPK was responsible for endothelial cell apoptosis induced by γ-irradiation. It has also been shown that p38 MAPK becomes activated during ischemia, and this activation leads to myocyte death and myocardial injury.30 SB203580, an inhibitor of p38 MAPK catalytic site, inhibited ischemia-induced phosphorylation of p38 MAPK and reduced myocardial infarction volume in mice.30 These reported results are consistent with our finding that the risk allele of SNP R952Q increased activation of p38 MAPK and susceptibility to CAD and MI. Furthermore, the potential roles of the LRP8 SNP in the pathogenic processes of CAD and MI may go beyond the p38 MAPK activation and platelet aggregation and may involve endothelial cells, vascular smooth-muscle cells, and cardiac cells where LRP8 is expressed. In addition, LRP8 is a receptor for apolipoprotein E (apoE) and can bind and internalize apoE-containing lipid vesicles. Mice deficient in apoE develop atherosclerotic lesions, and depressed expression of apoE has been associated with atherosclerosis in humans.31 Future studies will define more precisely the mechanism by which LRP8 variants play a role in the pathogenesis of CAD and MI.
To the best of our knowledge, this study is the first to employ the family-based SNP association design to identify a susceptibility gene for CAD and MI. Functional LRP8 SNP R952Q demonstrated significant association with CAD and MI in two independent populations: GeneQuest (P=.005) and GeneQuest II (P=.009) (tables 7 and 8). Interestingly, the SNP R952Q was not associated with sporadic CAD and MI. This result likely reflects the sporadic nature of MI in patients (mostly with no family history of disease) in the Cleveland GeneBank cohort, in contrast to CAD and MI in patients from the GeneQuest and GeneQuest II families and the Italian cohort, with their unique enrollment characteristics, such as early onset of the disease (premature MI) and/or a family history of disease. These results further stress the importance of using homogeneous populations, such as the GeneQuest, GeneQuest II, and Italian cohorts, to identify the most-reliable associations with MI with use of a case-control design. Furthermore, unlike the Italian cohort, which was enrolled on the basis of MI, the patients in GeneBank were participants in a large 10,000-patient cardiovascular DNA repository. Accordingly, the criteria for enrollment of a population may also be important. Overall, these results suggest that LRP8 is associated with early-onset and familial CAD and MI but not with late-onset, sporadic CAD and MI. These results also suggest that familial and premature CAD and MI may be distinctively different genetically from the late-onset, sporadic form of the disease.
It is important to point out that, as in other studies of common complex diseases or traits, we cannot exclude the possibilities that (1) other SNPs in LRP8 may be in LD with SNP R952Q and also associated with CAD and MI or that (2) LRP8 SNP R952Q may be in LD with variants in additional genes in the 1p34-36 linkage interval that also confer risk of CAD and MI. Such a case was reported for the 3q CAD linkage, in which two candidate genes, GATA2 and Kalirin, were both associated with CAD.7,8,32 Nevertheless, our studies identified LRP8 SNP R952Q as a genetic marker for association with premature, familial CAD and MI. Future studies employing a systematic approach to analyze each candidate gene under the 1p34-36 linkage interval may identify other susceptibility genes for CAD and MI.
In conclusion, we have demonstrated that the LRP8 SNP R952Q is significantly associated with familial and premature CAD and MI but not with the sporadic and late-onset form of the disease. Our results implicate a new, LRP8-mediated molecular pathway for susceptibility to familial CAD and MI.
Acknowledgments
This work was supported by National Institutes of Health grants P50 HL77107 (to E.J.T., Q.K.W., E.F.P., and S.L.H.), R01 HL073817 (to Q.K.W.), R01 GM28356 (to R.C.E.), and R01 LM008991 (to J.L.); a Doris Duke Innovation Award in Clinical Research (to Q.K.W. and E.J.T.); Italian Ministry of University and Research MIUR grant 2005065152 (to D.G.); and American Heart Association Established Investigator award 0440157N (to Q.K.W.). We thank D. Schmitt for technical assistance and help, Shenghan Chen for isolation of genomic DNA for the GeneQuest II population, Dr. A. Helgadottir for critical reading of the manuscript, and Dr. S. Misra and members of the Wang Laboratory for discussion and advice.
Web Resources
Accession numbers and URLs for data presented herein are as follows:
- dbSNP, http://www.ncbi.nlm.nih.gov/SNP/
- Haploview, http://www.broad.mit.edu/mpg/haploview/download.php/ (for Haploview software version 3.0 package)
- HapMap, http://www.hapmap.org/
- Marshfield Genotyping Service, http://research.marshfieldclinic.org/genetics/GeneticResearch/screeningsets.asp
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for ALOX5AP, GATA2, Kalirin, and LRP8)
- S.A.G.E., http://darwin.cwru.edu/sage/
- Software Written by Dave Curtis, http://www.mds.qmw.ac.uk/statgen/dcurtis/software.html (for CLUMP program)
- TDT/S-TDT, http://genomics.med.upenn.edu/spielman/TDT.htm
- UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Thom T, Haase N, Rosamond W, Howard VJ, Rumsfeld J, Manolio T, Zheng ZJ, Flegal K, O’Donnell C, Kittner S, et al (2006) Heart disease and stroke statistics—2006 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 113:e85–e151 10.1161/CIRCULATIONAHA.105.171600 [DOI] [PubMed] [Google Scholar]
- 2.Wang Q (2005) Molecular genetics of coronary artery disease. Curr Opin Cardiol 20:182–188 10.1097/01.hco.0000160373.77190.f1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Q (2005) Advances in the genetic basis of coronary artery disease. Curr Atheroscler Rep 7:235–241 10.1007/s11883-005-0012-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Topol EJ, Smith JC, Plow EF, Wang QK (2006) Genetic susceptibility to myocardial infarction and coronary artery disease. Hum Mol Genet Suppl 2 15:R117–R123 10.1093/hmg/ddl183 [DOI] [PubMed] [Google Scholar]
- 5.Helgadottir A, Manolescu A, Thorleifsson G, Gretarsdottir S, Jonsdottir H, Thorsteinsdottir U, Samani NJ, Gudmundsson G, Grant SF, Thorgeirsson G, et al (2004) The gene encoding 5-lipoxygenase activating protein confers risk of myocardial infarction and stroke. Nat Genet 36:233–239 10.1038/ng1311 [DOI] [PubMed] [Google Scholar]
- 6.Helgadottir A, Gretarsdottir S, St Clair D, Manolescu A, Cheung J, Thorleifsson G, Pasdar A, Grant SF, Whalley LJ, Hakonarson H, et al (2005) Association between the gene encoding 5-lipoxygenase-activating protein and stroke replicated in a Scottish population. Am J Hum Genet 76:505–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Connelly JJ, Wang T, Cox JE, Haynes C, Wang L, Shah SH, Crosslin DR, Hale AB, Nelson S, Crossman DC, et al (2006) GATA2 is associated with familial early-onset coronary artery disease. PLoS Genet 2:e139 10.1371/journal.pgen.0020139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang L, Hauser ER, Shah SH, Pericak-Vance MA, Haynes C, Crosslin D, Harris M, Nelson S, Hale AB, Granger CB, et al (2007) Peakwide mapping on chromosome 3q13 identifies the kalirin gene as a novel candidate gene for coronary artery disease. Am J Hum Genet 80:650–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Q, Rao S, Shen GQ, Li L, Moliterno DJ, Newby LK, Rogers WJ, Cannata R, Zirzow E, Elston RC, et al (2004) Premature myocardial infarction novel susceptibility locus on chromosome 1p34-36 identified by genomewide linkage analysis. Am J Hum Genet 74:262–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riddell DR, Vinogradov DV, Stannard AK, Chadwick N, Owen JS (1999) Identification and characterization of LRP8 (apoER2) in human blood platelets. J Lipid Res 40:1925–1930 [PubMed] [Google Scholar]
- 11.Korschineck I, Ziegler S, Breuss J, Lang I, Lorenz M, Kaun C, Ambros PF, Binder BR (2001) Identification of a novel exon in apolipoprotein E receptor 2 leading to alternatively spliced mRNAs found in cells of the vascular wall but not in neuronal tissue. J Biol Chem 276:13192–13197 10.1074/jbc.M011795200 [DOI] [PubMed] [Google Scholar]
- 12.Korporaal SJ, Relou IA, van Eck M, Strasser V, Bezemer M, Gorter G, van Berkel TJ, Nimpf J, Akkerman JW, Lenting PJ (2004) Binding of low density lipoprotein to platelet apolipoprotein E receptor 2′ results in phosphorylation of p38MAPK. J Biol Chem 279:52526–52534 10.1074/jbc.M407407200 [DOI] [PubMed] [Google Scholar]
- 13.Girelli D, Russo C, Ferraresi P, Olivieri O, Pinotti M, Friso S, Manzato F, Mazzucco A, Bernardi F, Corrocher R (2000) Polymorphisms in the factor VII gene and the risk of myocardial infarction in patients with coronary artery disease. N Engl J Med 343:774–780 10.1056/NEJM200009143431104 [DOI] [PubMed] [Google Scholar]
- 14.Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, Potenza D, Moya A, Borggrefe M, Breithardt G, et al (1998) Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 392:293–296 10.1038/32675 [DOI] [PubMed] [Google Scholar]
- 15.Podrez EA, Poliakov E, Shen Z, Zhang R, Deng Y, Sun M, Finton PJ, Shan L, Febbraio M, Hajjar DP, et al (2002) A novel family of atherogenic oxidized phospholipids promotes macrophage foam cell formation via the scavenger receptor CD36 and is enriched in atherosclerotic lesions. J Biol Chem 277:38517–38523 10.1074/jbc.M205924200 [DOI] [PubMed] [Google Scholar]
- 16.Skol AD, Scott LJ, Abecasis GR, Boehnke M (2006) Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet 38:209–213 10.1038/ng1706 [DOI] [PubMed] [Google Scholar]
- 17.Sham PC, Curtis D (1995) Monte Carlo tests for associations between disease and alleles at highly polymorphic loci. Ann Hum Genet 59:97–105 [DOI] [PubMed] [Google Scholar]
- 18.Spielman RS, McGinnis RE, Ewens WJ (1993) Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet 52:506–516 [PMC free article] [PubMed] [Google Scholar]
- 19.Spielman RS, Ewens WJ (1996) The TDT and other family-based tests for linkage disequilibrium and association. Am J Hum Genet 59:983–989 [PMC free article] [PubMed] [Google Scholar]
- 20.Spielman RS, Ewens WJ (1998) A sibship test for linkage in the presence of association: the sib transmission/disequilibrium test. Am J Hum Genet 62:450–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.S.A.G.E. (2006) Statistical analysis for genetic epidemiology, release 5.3. Department of Epidemiology and Biostatistics, Case Western Reserve University, Cleveland [Google Scholar]
- 22.Bland JM, Altman DG (1995) Multiple significance tests: the Bonferroni method. BMJ 310:170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vorchheimer DA, Becker R (2006) Platelets in atherothrombosis. Mayo Clin Proc 81:59–68 [DOI] [PubMed] [Google Scholar]
- 24.Lutters BC, Derksen RH, Tekelenburg WL, Lenting PJ, Arnout J, de Groot PG (2003) Dimers of β2-glycoprotein I increase platelet deposition to collagen via interaction with phospholipids and the apolipoprotein E receptor 2′. J Biol Chem 278:33831–33838 10.1074/jbc.M212655200 [DOI] [PubMed] [Google Scholar]
- 25.Strasser V, Fasching D, Hauser C, Mayer H, Bock HH, Hiesberger T, Herz J, Weeber EJ, Sweatt JD, Pramatarova A, et al (2004) Receptor clustering is involved in Reelin signaling. Mol Cell Biol 24:1378–1386 10.1128/MCB.24.3.1378-1386.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johns DG, Ao Z, Willette RN, Macphee CH, Douglas SA (2005) Role of p38 MAP kinase in postcapillary venule leukocyte adhesion induced by ischemia/reperfusion injury. Pharmacol Res 51:463–471 [DOI] [PubMed] [Google Scholar]
- 27.Tenhunen O, Soini Y, Ilves M, Rysa J, Tuukkanen J, Serpi R, Pennanen H, Ruskoaho H, Leskinen H (2006) p38 Kinase rescues failing myocardium after myocardial infarction: evidence for angiogenic and anti-apoptotic mechanisms. FASEB J 20:1907–1909 10.1096/fj.05-5618fje [DOI] [PubMed] [Google Scholar]
- 28.Tenhunen O, Rysa J, Ilves M, Soini Y, Ruskoaho H, Leskinen H (2006) Identification of cell cycle regulatory and inflammatory genes as predominant targets of p38 mitogen-activated protein kinase in the heart. Circ Res 99:485–493 10.1161/01.RES.0000238387.85144.92 [DOI] [PubMed] [Google Scholar]
- 29.Kumar P, Miller AI, Polverini PJ (2004) p38 MAPK mediates γ-irradiation-induced endothelial cell apoptosis, and vascular endothelial growth factor protects endothelial cells through the phosphoinositide 3-kinase-Akt-Bcl-2 pathway. J Biol Chem 279:43352–43360 10.1074/jbc.M405777200 [DOI] [PubMed] [Google Scholar]
- 30.Tanno M, Bassi R, Gorog DA, Saurin AT, Jiang J, Heads RJ, Martin JL, Davis RJ, Flavell RA, Marber MS (2003) Diverse mechanisms of myocardial p38 mitogen-activated protein kinase activation: evidence for MKK-independent activation by a TAB1-associated mechanism contributing to injury during myocardial ischemia. Circ Res 93:254–261 10.1161/01.RES.0000083490.43943.85 [DOI] [PubMed] [Google Scholar]
- 31.Greenow K, Pearce NJ, Ramji DP (2005) The key role of apolipoprotein E in atherosclerosis. J Mol Med 83:329–342 10.1007/s00109-004-0631-3 [DOI] [PubMed] [Google Scholar]
- 32.Hauser ER, Crossman DC, Granger CB, Haines JL, Jones CJ, Mooser V, McAdam B, Winkelmann BR, Wiseman AH, Muhlestein JB, et al (2004) A genomewide scan for early-onset coronary artery disease in 438 families: the GENECARD study. Am J Hum Genet 75:436–447 [DOI] [PMC free article] [PubMed] [Google Scholar]