

Abstract
Poly(ADP-ribose) polymerase (PARP) activation is emerging as a fundamental mechanism in the pathogenesis of diabetes complications including diabetic neuropathy. This study evaluated the role of PARP in diabetic sensory neuropathy. The experiments were performed in control and streptozotocin-induced diabetic rats treated with or without the PARP inhibitor 1,5-isoquinolinediol (ISO; 3 mg · kg−1 · day−1 i.p.) for 2 weeks after 2 weeks without treatment. Diabetic rats developed thermal hyperalgesia (assessed by paw-withdrawal and tail-flick tests), mechanical hyperalgesia (von Frey anesthesiometer/rigid filaments and Randall-Sellito tests), tactile allodynia (flexible von Frey filaments), and increased flinching behavior in phases 1 and 2 of the 2% formalin pain test. They also had clearly manifest increase in nitrotyrosine and poly(ADP-ribose) immunoreactivities in the sciatic nerve and increased superoxide formation (hydroxyethidine method) and nitrotyrosine immunoreactivity in vasa nervorum. ISO treatment alleviated abnormal sensory responses, including thermal and mechanical hyperalgesia and tactile allodynia as well as exaggerated formalin flinching behavior in diabetic rats, without affecting the aforementioned variables in the control group. Poly(ADP-ribose) and, to a lesser extent, nitrotyrosine abundance in sciatic nerve, as well as superoxide and nitrotyrosine formation in vasa nervorum, were markedly reduced by ISO therapy. Apoptosis in dorsal root ganglion neurons (transferase-mediated dUTP nick-end labeling assay) was not detected in any of the groups. In conclusion, PARP activation contributes to early diabetic sensory neuropathy by mechanisms that may include oxidative stress but not neuronal apoptosis.
Growing evidence indicates that oxidative-nitrosative stress is one of the leading factors causing motor and sensory nerve conduction deficits, neurovascular dysfunction, and myelinated fiber atrophy characteristic of peripheral diabetic neuropathy (1–5). Free radicals and peroxynitrite have also been implicated in small sensory fiber neuropathy and, in particular, thermal hyper- and hypoalgesia (3,4,6), mechanical hyperalgesia (3), tactile allodynia (7), and diabetic neuropathic pain (8). Oxidative-nitrosative stress may contribute to diabetic neuropathic pain and abnormal sensory responses via multiple mechanisms, including neurotrophic factor deficit (6,9), mitogen-activated protein kinase and cyclooxygenase-2 activation (10–12), impaired Ca2+ homeostasis and signaling (13), and production of inflammatory cytokines, e.g., tumor necrosis factor-α (14). Free radicals, oxidants, and perhaps some still-unidentified metabolic factors result in activation of the nuclear enzyme poly(ADP-ribose) polymerase (PARP), a recently discovered fundamental mechanism in the pathogenesis of diabetes complications including endothelial dysfunction, cardiomyopathy, and retinopathy (15–17). Our group has demonstrated the important role of this mechanism in motor and sensory nerve conduction deficits, neurovascular dysfunction, and energy failure characteristic for early peripheral diabetic neuropathy (18,19). Here, we provide evidence implicating PARP activation in small sensory fiber neuropathy and, in particular, thermal and mechanical hyperalgesia, tactile allodynia, and exaggerated flinching behavior during formalin pain test in streptozotocin (STZ)-induced diabetic rats.
RESEARCH DESIGN AND METHODS
Reagents
Unless otherwise stated, all chemicals were of reagent-grade quality and were purchased from Sigma (St. Louis, MO). Rabbit polyclonal anti-nitrotyrosine antibody was purchased from Upstate (Lake Placid, NY) and mouse monoclonal anti-poly(ADP-ribose) from Trevigen (Gaithersburg, MD). Secondary Alexa Fluor 488 goat anti-rabbit and Alexa Fluor goat anti-mouse antibodies as well as Prolong Gold antifade reagent were purchased from Invitrogen (Eugene, OR). Other reagents for immunohistochemistry were purchased from Dako Laboratories (Santa Barbara, CA). Hydroethidine was purchased from Molecular Probes (Eugene, OR).
The experiments were performed in accordance with regulations specified by the National Institutes of Health’s Principles of Laboratory Animal Care, 1985, revised version, and the Pennington Biomedical Research Center and University of Michigan protocols for animal studies. Male Wistar rats (Charles River, Wilmington, MA), body weight 250–300 g, were fed a standard rat diet (PMI Nutrition, Brentwood, MO) and had access to water ad libitum. Diabetes was induced by STZ as described (18,19). Blood samples for glucose measurements were taken from the tail vein ∼48 h after the STZ injection and the day before the animals were killed. The rats with blood glucose ≥ 13.8 mmol/l were considered to have diabetes. The experimental groups comprised control and diabetic rats treated with or without the PARP inhibitor 1,5-isoquinolinediol (ISO; 3 mg · kg−1 · day−1 i.p.). The treatments were started 2 weeks after initial 2 weeks without treatment to avoid restoration of normoglycemia or alleviation of hyperglycemia that would occur if a PARP inhibitor administration was started shortly after induction of STZ-induced diabetes (20). The behavioral tests were started 24 h after the last ISO injection and performed in the following order: tactile responses to flexible von Frey filaments, thermal algesia, tail-flick test, paw pressure Randall-Sellito test, mechanical algesia with rigid von Frey filaments and von Frey anesthesiometer, and formalin flinching responses.
Anesthesia, euthanasia, and tissue sampling
The animals were sedated by CO2 (21) and immediately killed by cervical dislocation. Both sciatic nerves were rapidly dissected and fixed in formalin (18) for assessment of nitrotyrosine and poly(ADP-ribose) by immunohistochemistry. Some sciatic nerves were used for isolation of epineurial arterioles (2) and assessment of arteriolar superoxide anion radical and nitrotyrosine. Aortas were also sampled and immediately used for superoxide measurements. Separate groups of control, diabetic, and ISO-treated diabetic animals were anesthetized with inactin (65–85 mg/kg body wt i.p.) and perfused with paraformaldehyde as previously described (22). Thirty minutes after perfusion, the dorsal root ganglia (DRG) were removed, fixed in 4% paraformaldehyde, and used for assessment of apoptosis.
Specific methods: behavioral tests
Tactile responses
Tactile responses were evaluated by quantifying the withdrawal threshold of the hindpaw in response to stimulation with flexible von Frey filaments. Rats were placed in individual plexiglass boxes on a stainless steel mesh floor and were allowed to adjust for at least 20 min. A series of calibrated von Frey filaments (range 4–28 g; IITC Life Science, Woodland Hills, CA) was applied perpendicularly to the plantar surface of a hindpaw with sufficient force to bend the filament for 6 s. Brisk withdrawal or paw flinching was considered as a positive response. In the absence of a response, a filament of next-greater force was applied. In the presence of a response, a filament of next-lower force was applied. The test was repeated four to five times at ∼5-min intervals on each animal, and the mean value is used.
Thermal algesia
To determine the sensitivity to noxious heat, rats were placed within a plexiglass chamber on a transparent glass surface and allowed to acclimate for at least 20 min. A thermal stimulation meter (IITC model 336 TG combination tail-flick and paw algesia meter; IITC Life Science) was used. The device was activated after placing the stimulator directly beneath the plantar surface of the hindpaw. The paw-withdrawal latency in response to the radiant heat (17% intensity, cutoff time 35 s) was recorded. Individual measurements were repeated four to five times, and the mean value was calculated as the thermal threshold.
Tail-flick test
Tail-flick response latencies were determined using the IITC model 336 TG described above, set at 40% heating intensity and with a cutoff at 10 s. At least three readings were taken per animal at a 15-min interval, and the average was calculated.
Paw pressure Randall-Sellito test
Paw pressure thresholds were registered with the paw pressure analgesia meter for the Randall-Selitto test (37215 Analgesy-Meter; UGO-Basile, Comerio VA, Italy). Pressure increasing at a linear rate of 10 g/s, with the cutoff of 250 g to avoid tissue injury, was applied to the center of the hindpaw. When the animal displayed pain by withdrawal of the paw, the applied paw pressure was registered by an analgesia meter and expressed in mass units (grams). Five tests separated by at least 15 min were performed for each animal, and the mean value of these tests was calculated.
Mechanical algesia
Sensitivity to noxious mechanical stimuli was determined by quantifying the withdrawal threshold of the hindpaw in response to mechanical stimulation using a von Frey anesthesiometer (model 2290–4; IITC Life Science) and rigid von Frey filaments. The rats were placed in individual plexiglass boxes on a stainless steel mesh floor and were allowed to acclimate for at least 20 min. A 0.5-mm diameter polypropylene rigid tip was used to apply a force to the plantar surface of the hindpaw. The force causing the withdrawal response was recorded by the anesthesiometer. The anesthesiometer was calibrated before each recording. The test was repeated four to five times at ∼5-min intervals on each animal, and the mean value was calculated.
Formalin flinching test
Rats were manually restrained by wrapping in a towel, and formalin (50 μl of 2% solution) was injected subdermally into the dorsum of the right hindpaw. The rat was then placed in an observation chamber, and flinching behavior was counted in 1-min blocks every 5 min for 1 h. In particular, flinches were counted during the minutes 1–2, 5–6, 10–11, 15–16, 20–21, 25–26, 30–31, 35–36, 40–41, 45–46, 50–51, 55–56, and 60–61 after formalin injection as described (23).
Immunohistochemical studies
All sections were processed by a single investigator and blindly evaluated. Low-power observations of stained sections were made using a Zeiss Axioskop microscope. Color images were captured with a Zeiss Axiocam HRc charge-coupled device camera at 1,195 × 949 resolution. Low-power images were generated with a ×40 acroplan objective using the automatic capturing feature of the Zeiss Axiovision software (version 3.1.2.1).
Nitrotyrosine immunoreactivity in sciatic nerves and epineurial vessels
Nitrotyrosine immunoreactivity in the sciatic nerve was assessed by two approaches, i.e., regular immunohistochemistry, as we have described in detail (24), and immunofluorescent histochemistry. In brief, sections were deparaffinized in xylene, hydrated in decreasing concentrations of ethanol, and washed in water. For immunofluorescent histochemistry, rabbit polyclonal anti-nitrotyrosine antibody was used in a working dilution (1:100). Secondary Alexa Fluor 488 goat anti-rabbit antibody was applied in a working dilution (1:200). Sections were mounted in Prolong Gold antifade reagent. The intensity of fluorescence was graded from 1 to 4 (1, no staining; 2, faint; 3, moderate; and 4, intense), and the immunohistochemistry score was expressed as means ± SE for each experimental group. Nitrotyrosine immunoreactivity in epineurial vessels has been assessed by regular immunohistochemistry as we have described (2,24).
Poly(ADP-ribose) immunoreactivity
Poly(ADP-ribose) immunoreactivity was assessed as described (18,19), with minor modification. In brief, sections were deparaffinized in xylene, hydrated in decreasing concentrations of ethanol, and washed in water. Nonspecific binding was blocked in 10% goat serum containing 1% BSA in Tris-buffered saline (Dako, Carpinteria, CA) for 2 h. Mouse monoclonal anti-poly(ADP-ribose) antibody was diluted (1:100) in 1% BSA in Tris-buffered saline and applied overnight at 4°C in the humidity chamber. Secondary Alexa Fluor 488 goat anti-mouse antibody was diluted (1:200) in Tris-buffered saline and applied for 2 h at room temperature. Sections were mounted in Prolong Gold antifade reagent. At least 10 fields of each section were examined to select one representative image. Representative images were microphotographed, and the number of poly(ADP-ribose)-positive nuclei was calculated for each microphotograph.
Superoxide in epineurial vessels and aorta
Superoxide anion radical abundance in epineurial vessels was assessed by the hydroethidine method as described (2,24). The intensity of superoxide fluorescence was graded from 1 to 4 (1, no fluorescence; 2, weak; 3, moderate; and 4, intense), and the immunohistochemistry score was expressed as mean ± SE for each experimental group. Superoxide anion radical abundance in aorta was measured by lucigenin-enhanced chemiluminescence (2,24).
Apoptosis in DRG neurons
Five-micrometer paraffin DRG sections were deparaffinized according to standard protocols and blocked with 20% goat serum for 20 min. Apoptosis was assessed by the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) method (22), using the ApopTag plus Fluorescein In Situ Apoptosis Detection kit (Chemicon, Temecula, CA). Positive control provided by the Chemicon was processed together with experimental samples. Two hundred neurons were blindly examined for the presence of apoptotic (TUNEL-stained) cells.
Statistical analysis
The results are expressed as means ± SE. Data were subjected to equality of variance F test and then to log transformation, if necessary, before one-way ANOVA. Where overall significance (P < 0.05) was attained, individual between-group comparisons were made using the Student-Newman-Keuls multiple range test. Significance was defined at P < 0.05. When between-group variance differences could not be normalized by log transformation (datasets for body weights and plasma glucose), the data were analyzed by the nonparametric Kruskal-Wallis one-way ANOVA, followed by the Bonferroni/Dunn test for multiple comparisons.
RESULTS
The final body weights were comparably lower in untreated and ISO-treated diabetic rats than in the control group (Table 1). The final blood glucose concentrations were similarly elevated in untreated and ISO-treated diabetic rats compared with the control rats. ISO did not affect either weight gain or blood glucose concentrations in nondiabetic rats.
TABLE 1.
Initial and final body weights and final blood glucose concentrations in control and diabetic rats with and without ISO treatment
| Body weight (g)
|
|||
|---|---|---|---|
| Initial* | Final | Blood glucose (mmol/l) | |
| Control | 317.45 ± 6.09 | 428.33 ± 9.58 | 5.31 ± 0.09 |
| Control + ISO | 318.94 ± 4.86 | 434.44 ± 6.53 | 5.70 ± 0.16 |
| Diabetic | 313.33 ± 6.45 | 311.46 ± 11.11† | 23.84 ± 0.95† |
| Diabetic + ISO | 308.61 ± 5.04 | 307.90 ± 11.83† | 23.83 ± 1.32† |
Data are means ± SE. n = 9–11.
Before induction of STZ-induced diabetes.
Significantly different from controls (P < 0.01).
Diabetic rats with 4-week duration of STZ-induced diabetes had clearly manifested thermal hyperalgesia detected by measuring the times of hindpaw-withdrawal (Fig. 1A) or tail-flick (Fig. 1B) responses to noxious thermal stimuli (radiant heat). In particular, the latency of paw withdrawal in response to radiant heat was reduced by 41% in diabetic rats compared with controls (P < 0.01). ISO partially (to 85% of control value; P < 0.05 vs. controls and <0.001 vs. untreated diabetic group) corrected diabetes-induced decrease in paw-withdrawal latency, without affecting this variable in the control group. In a similar fashion, the tail-flick response latency was reduced by 18% in diabetic rats compared with controls (P < 0.01). ISO corrected this variable in diabetic rats, without affecting it in control rats.
FIG. 1.
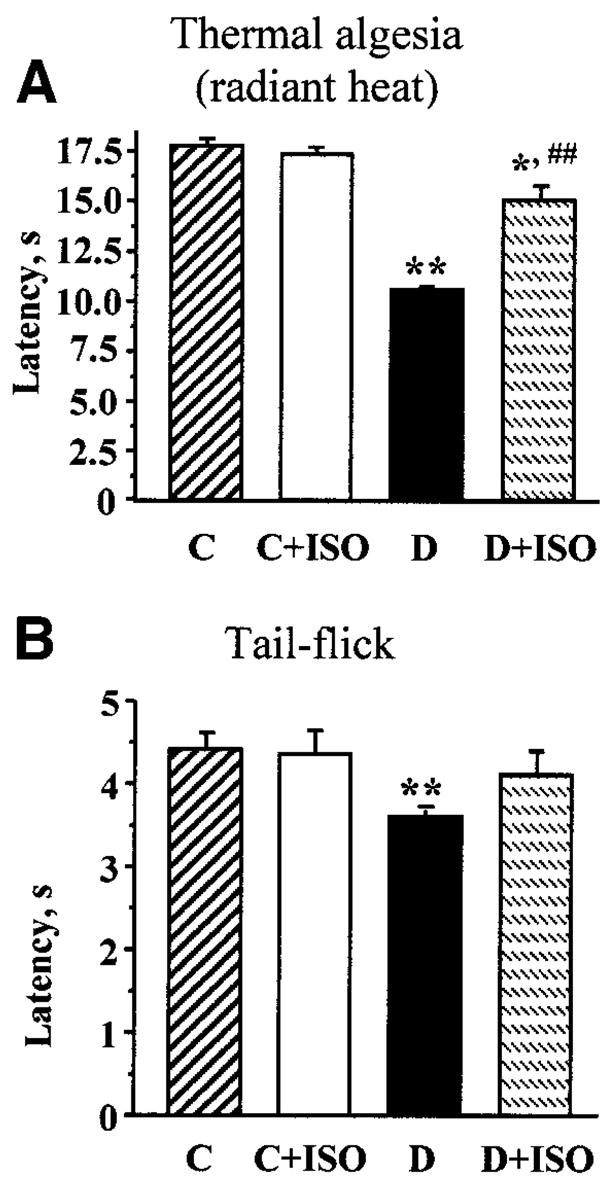
A: Paw-withdrawal latencies in response to thermal noxious stimuli in control (C) and diabetic (D) rats treated with or without ISO. B: Tail-flick response latencies in control and diabetic rats treated with or without ISO. For A and B, means ± SE, n = 9–11 per group. *P < 0.05, **P < 0.01 vs. control group; ##P < 0.01 vs. untreated diabetic group.
Diabetic rats with 4-week duration of STZ-induced diabetes also had mechanical hyperalgesia detected with 1) a von Frey anaesthesiometer by measuring paw withdwaral thresholds in response to noxious stimulation with rigid von Frey filaments (Fig. 2A) and 2) the paw pressure Randall-Sellito test (Fig. 2B). In particular, the paw-withdrawal threshold in response to rigid von Frey filaments was reduced by 49% in diabetic rats compared with controls. ISO partially (to 71% of the control value; P < 0.05 vs. controls and <0.01 vs. untreated diabetic group) corrected diabetes-induced decrease in paw-withdrawal thresholds, without affecting this variable in control rats. In a similar fashion, paw-withdrawal threshold in the Randall-Sellito test was reduced by 38% in diabetic rats compared with controls (P < 0.01). ISO partially (to 82% of the control value; P < 0.01 vs. controls and <0.01 vs. untreated diabetic group) corrected this variable in diabetic rats, without affecting the Randall-Sellito test result in the control group. Another sensory abnormality developing in diabetic rats was tactile allodynia. Tactile withdrawal threshold in response to light touch with flexible von Frey filaments was reduced by 58% in diabetic rats compared with controls (P < 0.01). ISO partially (to 65% of the control value; P < 0.01 vs. controls and <0.01 vs. untreated diabetic group) corrected diabetes-induced decrease in tactile withdrawal thresholds in diabetic rats, without affecting this variable in the control group.
FIG. 2.
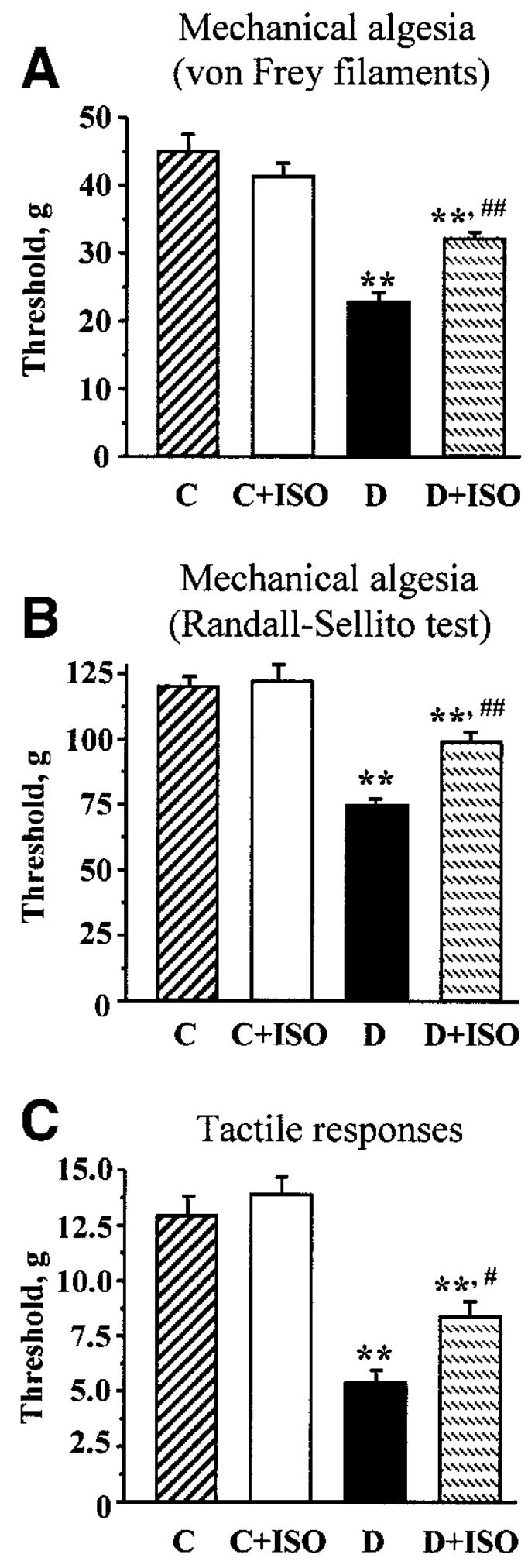
A: Paw-withdrawal thresholds in response to stimulation with rigid von Frey filaments in control (C) and diabetic (D) rats treated with or without ISO. B: Paw-withdrawal thresholds in the paw pressure Randall-Sellito test in control and diabetic rats treated with or without ISO. C: Tactile response thresholds in response to stimulation with flexible von Frey filaments in control and diabetic rats treated with or without ISO. For A–C, means ± SE, n = 9–11 per group. **P < 0.01 vs. control group; #P < 0.05, ##P < 0.01 vs. untreated diabetic group.
Diabetic rats displayed hyperalgesia in the formalin flinching test (Table 2). ISO did not affect formalin-evoked responses in control rats. However, the PARP inhibitor reduced diabetes-associated hyperalgesia in diabetic rats. Poly(ADP-ribose) immunoreactivities were increased in the sciatic nerves of diabetic rats compared with controls, and this increase was essentially corrected by ISO treatment (Fig. 3A). The number of the sciatic nerve poly(ADP-ribose)-positive nuclei was ∼3.1-fold greater in the diabetic group compared with controls (P < 0.01; Fig. 3B). No significant differences in the numbers of sciatic nerve poly(ADP-ribose)-positive nuclei were found between the ISO-treated control or diabetic groups and untreated controls. Nitrotyrosine immunoreactivities were increased in the sciatic nerves of diabetic rats compared with controls, and this increase was slightly, but significantly, reduced by ISO treatment (Fig. 4A–D). Ethidium fluorescence (an index of superoxide production) was increased ∼2.8-fold in epineurial vessels of diabetic rats compared with controls (Fig. 5A and B), and this increase was blunted by ISO treatment. Nitrotyrosine immunoreactivity was increased threefold in epineurial vessels of diabetic rats, and this increase was reduced by ISO treatment (Fig. 5C and D). Superoxide abundance in aorta was increased in diabetic rats (2.95 ± 0.21 relative luminescence units compared with 1.44 ± 0.13 in controls; P < 0.01), and this increase was essentially corrected by ISO treatment (1.80 ± 0.15; P < 0.01 vs. untreated diabetic group). No apoptotic DRG neurons were detected in the control, untreated diabetic, or ISO-treated diabetic rats (Fig. 6).
TABLE 2.
Flinching responses in the 2% formalin test in control and diabetic rats treated with or without ISO
| Formalin-evoked flinches (sum/h) | |
|---|---|
| Control | 163 ± 11 |
| Control + ISO | 173 ± 12 |
| Diabetic | 242 ± 12* |
| Diabetic + ISO | 196 ± 16† |
Data are means ± SE. n = 8–10.
Significantly different from controls (P < 0.01).
Significantly different from untreated diabetic group (P < 0.05).
FIG. 3.
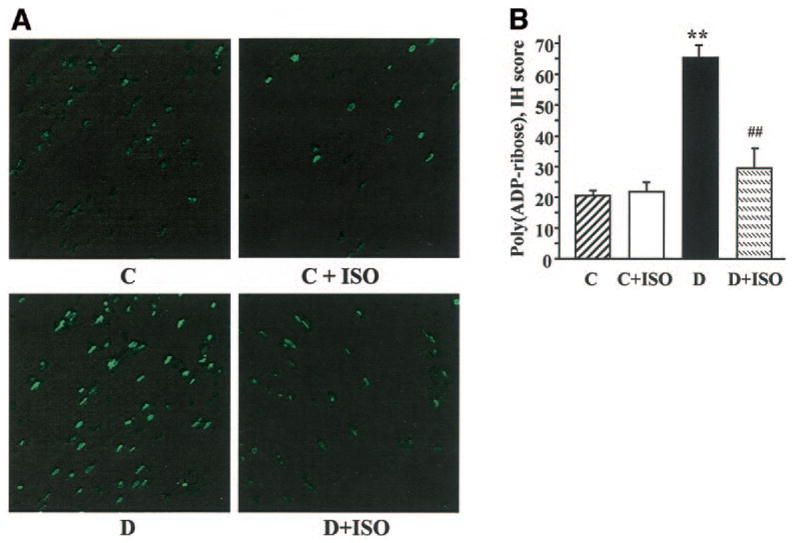
A: Representative microphotographs of immunofluorescent staining of poly(ADP-ribose) in sciatic nerves of control (C) and diabetic (D) rats treated with or without ISO. Magnification ×100. B: Counts of poly(ADP-ribose)-positive nuclei (PAR-positive nuclei) in sciatic nerves of control and diabetic rats treated with or without ISO. Means ± SE, n = 6 per group. *P < 0.01 vs. control group; ##P < 0.01 vs. untreated diabetic group.
FIG. 4.
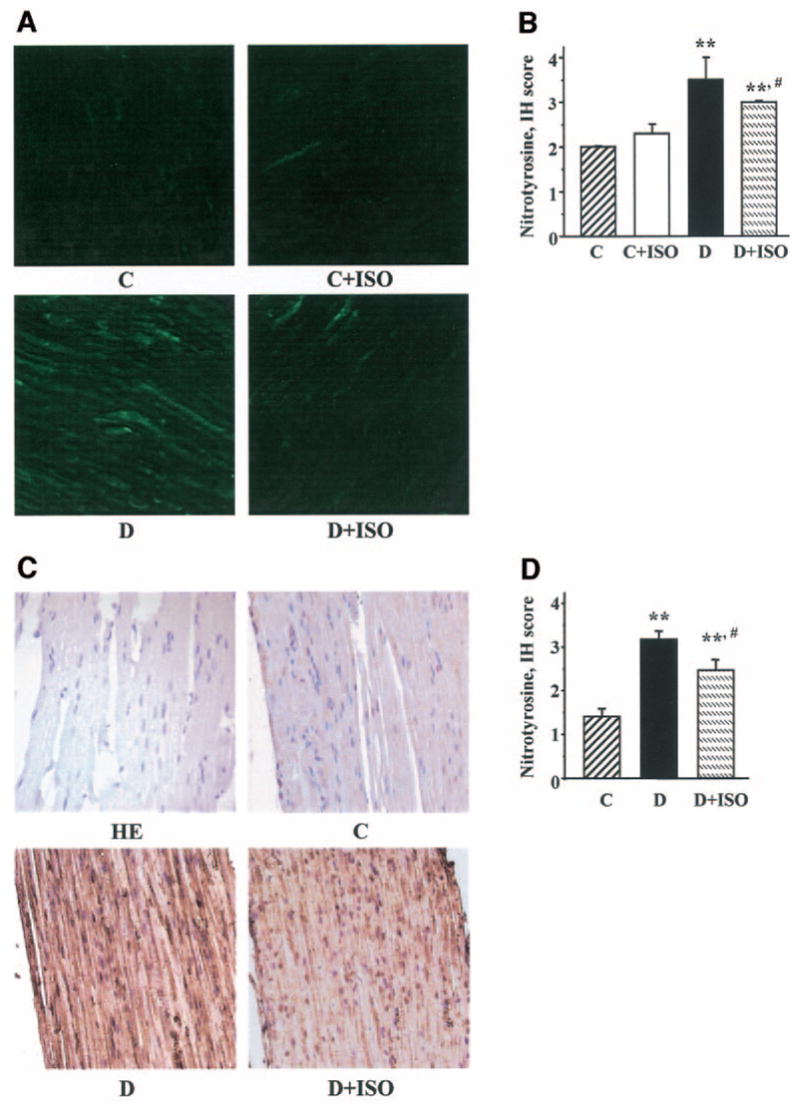
A: Representative microphotographs of immunofluorescent staining of nitrotyrosine in sciatic nerves of control (C) and diabetic (D) rats treated with or without ISO. Magnification ×100. B: Immunohistochemistry scores of sciatic nerve nitrotyrosine immunofluorescent stainings. Means ± SE, n = 9–11 per group. **P < 0.01 vs. control group; #P < 0.05 vs. untreated diabetic group. C: Representative microphotographs of immunohistochemical staining of nitrotyrosine in sciatic nerves of control and diabetic rats treated with or without ISO. Magnification ×40. D: Immunohistochemistry scores of sciatic nerve nitrotyrosine stainings. Means ± SE, n = 6 per group. **P < 0.01 vs. control group; #P < 0.05 vs. untreated diabetic group. HE, sciatic nerves stained with hematoxyline and eosin.
FIG. 5.
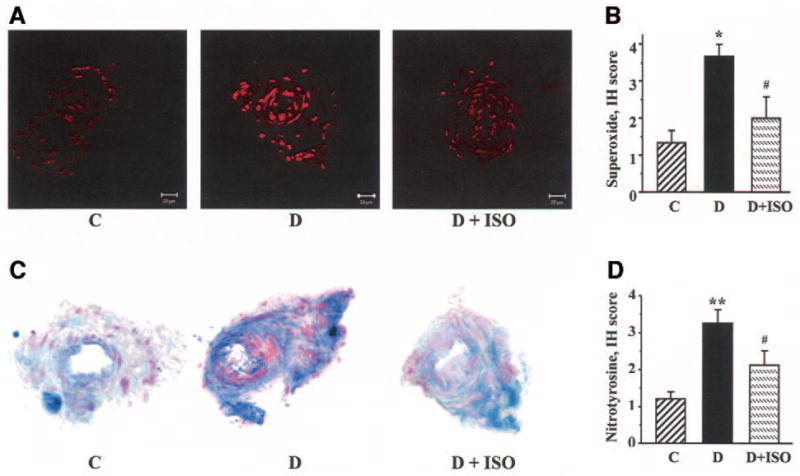
A: Representative microphotographs of superoxide-generated fluorescence in sciatic nerve epineurial vessels of control (C) rats, diabetic (D) rats, and diabetic rats treated with ISO. Magnification ×200. B: Scores of epineurial vessel superoxide-generated immunofluorescence. Means ± SE, n = 3 per group. *P < 0.05 vs. control group; #P < 0.05 vs. untreated diabetic group. C: Representative microphotographs of nitrotyrosine immunohistochemical staining in sciatic nerve epineurial vessels of control rats, diabetic rats, and diabetic rats treated with ISO. Magnification ×200. D: Immunohistochemistry (IH) scores of epineurial vessel nitrotyrosine stainings. Means ± SE, n = 3 per group. **P < 0.01 vs. control group; #P < 0.05 vs. untreated diabetic group.
FIG. 6.
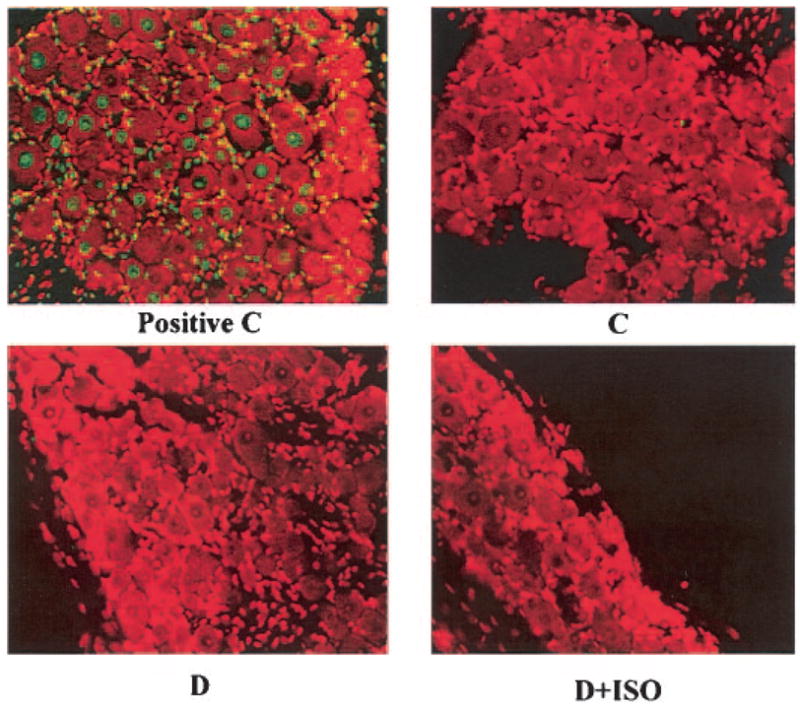
Immunoflurescent assessment of apoptosis (TUNEL staining) in DRG neurons of control (C) and diabetic (D) rats treated with or without ISO. n = 6 per group. Magnification ×200. Positive control was included in the ApopTag plus Fluorescein In Situ Apoptosis Detection kit and processed together with experimental samples.
DISCUSSION
Whereas neuropathic pain and abnormal sensory perceptions occur in a significant proportion of patients with diabetes, thus seriously affecting their quality of life, the mechanisms underlying these disorders remain remarkably understudied. The latter is partially explained by certain limitations of existing animal models (23,25). Diabetic rats and mice have a limited lifespan and rarely show evidence of overt neuropathy such as demyelination, axonal degeneration, fiber loss, or axonal regeneration in their peripheral nerves. This makes diabetic rodents unsuitable for studying the contribution of these phenomena of advanced peripheral diabetic neuropathy to pain or loss of sensory function. In addition, it is not possible to quantify spontaneous pain in animals. Despite these limitations, assessment of behavioral responses to external stimuli in diabetic rodents provides valuable information regarding the mechanisms of abnormal sensation and pain associated with diabetes. Recent behavioral studies have demonstrated that 1) both diabetic rats and mice display altered thermal and mechanical algesia in response to noxious stimuli as well as tactile allodynia, although a type of response (i.e., hyper- or hypoalgesia) often depends on animal species and duration of diabetes (3,4,9,13) and 2) abnormal sensory responses are alleviated by protracted normoglycemia (23) and are amenable to at least some pathogenetic treatments (e.g., aldose reductase inhibitors [9], neurotrophic factors [9], the protein kinase C inhibitor LY333531 [26], rosuvastatin [27], and antioxidants [3,4,7,13]) that also reduce manifestations of large motor and sensory fiber neuropathy (1–5,26–28). Our findings have shown that 1) PARP activation is implicated in hyperalgesia to noxious thermal, mechanical, and chemical stimuli and 2) PARP inhibitor treatment alleviates, but does not completely normalize, tail-flick and paw-withdrawal response latencies, mechanical and tactile withdrawal thresholds, and exaggerated flinching behavior in rats with short-term STZ-induced diabetes.
To assess sensory responses to thermal, mechanical, and chemical noxious stimuli, we used a battery of standard behavioral tests. The tail-flick test, where the time to movement of the tail from a noxious heat source is measured, reflects activity of a simple spinal reflex arc and provides information on peripheral nerve and spinal function in isolation from higher nociceptive processing and cognitive systems (23). The tail-flick response latency data in diabetic rats and mice are quite contradictory (4,23, 25,29,30). In our previous study (4), the tail-flick response latency was increased in 12-week diabetic NOD mice compared with nondiabetic NOD mice. This increase was dose-dependently reduced by a short-term treatment with a peroxynitrite decomposition catalyst (4), which implicates nitrosative stress in abnormal sensory responses to noxious thermal stimuli in experimental type 1 diabetes. Here, the tail-flick response latency appeared decreased in rats with short-term STZ-induced diabetes, consistent with independent measurements of the paw-withdrawal time from noxious thermal stimuli, which, in contrast to the tail-flick test, included supraspinal sensory processing (23). Diabetic rats demonstrated reduced latencies of both responses, which is consistent with the presence of transient thermal hyperalgesia and is in line with our previous studies (13,30) and other reports (3,7,9). Both tail-flick and paw-withdrawal latencies were at least partially corrected by PARP inhibition, consistent with other findings (3,4,7) implicating oxidative-nitrosative stress as well as other mechanisms contributing to free radical and oxidant generation (e.g., increased aldose reductase [9] and protein kinase C [26] activities) to abnormal sensory responses to thermal noxious stimuli in diabetes.
Another diabetes-related phenomenon, mechanical hyperalgesia, was revealed by assessment of paw-withdrawal thresholds in two different tests, i.e., one including stimulation with rigid von Frey filaments with subsequent registration of paw-withdrawal thresholds by von Frey anaesthesiometer and the paw pressure Randall-Sellilo test. Both variables were partially corrected by a PARP inhibitor treatment. These observations are in line with our previous findings (13,31) and other reports (3,7), demonstrating the important contribution of oxidative stress–related mechanisms to increased sensitivity to mechanical noxious stimuli associated with diabetes.
In addition to spontaneous pain, painful diabetic neuropathy in human subjects is sometimes complicated by tactile allodynia, a condition where light touch is perceived as painful (23). Similar phenomenon is observed in diabetic rats where the light touch (<15 g) of von Frey filaments or light stroking of the paw induces a withdrawal response from the stimulus (9,23,25). The mechanisms of tactile allodynia are not studied in detail. Tactile allodynia in diabetic rats is not alleviated by rapid normalization of blood glucose (23) or treatment with aldose reductase inhibitors (9) but can be prevented and reversed by protracted insulin therapy (23) or niterapone, an inhibitor of catechol-O-methyltransferase and antioxidant (7). The current observations suggest that tactile allodynia in rats with short-term STZ-induced diabetes is alleviated by the PARP inhibitor ISO. This finding, as well as alleviation of tactile allodynia in diabetic mice lacking 12/15-lipoxygenase or treated with a 12/15-lipoxygenase inhibitor in our most recent studies (32), are in line with the important role of oxidative-nitrosative stress in abnormal tactile responses associated with diabetes. Tactile allodynia also develops in the model of sciatic nerve ischemia (33), another pathological condition with enhanced oxidative stress (34).
Both mechanical and thermal behavioral tests provide acute sensory stimuli and represent measures of the threshold to nociceptive pain that is transduced by myelinated and unmyelinated fibers, respectively. The formalin test principally differs from both, as the second phase of the flinching response occurs despite minimal input to the spinal cord from primary afferent nociceptors. Thus, the test provides a means for studying mechanisms by which innocuous sensory input can be modulated and amplified in the spinal cord and higher central nervous system to generate a neuropathic pain state, as well as malfunctions of these mechanisms produced by pathological conditions including diabetes. In our study, diabetic rats displayed exaggerated flinching behavior in both the first and second phases of the formalin test. Formalin-induced flinching responses in both phases were blunted by a PARP inhibitor treatment, which supports the role for PARP activation in diabetic neuropathic pain.
Theoretically, PARP activation can lead to neuropathic pain and abnormal sensory responses via several mechanisms, i.e., glutamate excitotoxicity (35), impaired Ca2+ homeostasis and signaling (36), mitogen-activated protein kinase activation (37), and nuclear factor-κB–mediated production of proinflammatory cytokines, e.g., tumor necrosis factor-α and related cyclooxygenase-2 overexpression (38). Such phenomena as altered Ca2+ homeostasis and signaling in DRG neurons and mitogen-activated protein kinase activation and cyclooxygenase-2 overexpression in the spinal cord and Schwann cells of the peripheral nerve have been documented in diabetic rats (11–13). To our knowledge, the presence of inflammatory response in either peripheral nerve or spinal cord of diabetic rodents has not been documented properly, and detailed studies of this phenomenon would be very important for understanding the mechanisms of sensory neuropathy. Note that PARP activation can also participate in the pathogenesis of diabetic sensory neuropathy via exacerbation of oxidative-nitrosative stress (39), a key factor in the development of thermal and mechanical hyperalgesia in STZ-induced diabetic rats (39). In the present study, PARP inhibition with ISO alleviated oxidative-nitrosative stress in sciatic nerve, vasa nervorum, and aorta of diabetic rats, consistent with our previous findings with another PARP inhibitor, 3-aminobenzamide (39). Of interest, the effect of ISO on nitrotyrosine immunoreactivity in the sciatic nerve was quite modest compared with vasa nervorum. The latter is probably a consequence of the pharmacological distribution of ISO rather than differences in oxidative-nitrosative stress mechanisms between vascular and neural compartments because 3-aminobenzamide was equipotent in suppressing nitrotyrosine immunoreactivity in vascular and neural elements of diabetic peripheral nerve (39). Although the role for neurovascular mechanisms in diabetic sensory neuropathy has not properly been sorted out, a correction of thermal hyperalgesia by a protein kinase C inhibitor (26), together with a compelling evidence of protein kinase C activation in the vascular, but not neural (e.g., peripheral nerve and DRG neurons), elements of peripheral nervous system (40–43) strongly suggest that vascular mechanisms may be involved in at least some abnormal sensory responses associated with diabetes. Note that activation of vascular protein kinase C leads to oxidative stress (44,45) via phosphorylation and activation of NAD(P)H oxidase (44), a superoxide-generating enzyme known as the most important contributor to diabetes-associated superoxide generation (46). In addition, a far better alleviation of mechanical algesia by a combined treatment with low doses of ISO and vasodilators (31) than a high-dose ISO monotherapy also may support the role for neurovascular dysfunction in diabetic sensory neuropathy, although, as we have discussed previously (31), vasodilators exert a variety of nonvascular effects. At the same time, our data provide clear indication that development of diabetic sensory neuropathy in rats with short-term STZ-induced diabetes is not associated with neuronal apoptosis. The absence of neuronal apoptosis in STZ-induced diabetic rats in our study is consistent with two other reports (47,48). However, the area remains a subject of debate (22,47–50), and thus it would be of interest to explore the effects of PARP inhibition on functional and structural (reduced skin fiber density) manifestations of sensory loss and to correlate them with the rates of neuronal apoptosis, if any, in rats with longer durations of diabetes. The role for PARP in apoptosis remains controversial (17,51).
In conclusion, PARP activation contributes to early experimental sensory neuropathy by mechanisms that may include oxidative-nitrosative stress but not neuronal apoptosis. A PARP inhibitor treatment alleviates diabetic neuropathic pain and abnormal sensory responses in STZ-induced diabetic rats. The study provides a new evidence for the important role for PARP activation in the pathogenesis of diabetes complications and, in particular, peripheral diabetic neuropathy.
Acknowledgments
The study was supported by a Juvenile Diabetes Research Foundation International Grant, an American Diabetes Association Research Grant, and a National Institutes of Health Grant (DK 071566-01) (all to I.G.O.); a Juvenile Diabetes Research Foundation Center for the Study of Complications of Diabetes Grant (4-200-421 to I.G.O. and M.J.S.); a Veterans Administration Merit Review Grant and American Diabetes Association Research Grant (both to M.A.Y.); and the Intramural Research Program of the National Insitutes of Health/National Institute on Alcohol Abuse and Alcoholism (to P.P.).
The authors thank Dr. Fei Li for help with setting the measurements of thermal and mechanical algesia at Pennington Biomedical Research Center and Tamara Charniauskaya, Omorodola Abatan, and Dennis Larkin for expert technical assistance.
- DRG
dorsal root ganglia
- ISO
1,5-isoquinolinediol
- PARP
poly(ADP-ribose) polymerase
- STZ
streptozotocin
- TUNEL
transferase-mediated dUTP nick-end labeling
References
- 1.Stevens MJ, Obrosova I, Cao X, Van Huysen C, Greene DA. Effects of DL-α-lipoic acid on peripheral nerve conduction, blood flow, energy metabolism, and oxidative stress in experimental diabetic neuropathy. Diabetes. 2000;49:1006–1015. doi: 10.2337/diabetes.49.6.1006. [DOI] [PubMed] [Google Scholar]
- 2.Coppey LJ, Gellett JS, Davidson EP, Dunlap JA, Lund DD, Yorek MA. Effect of antioxidant treatment of streptozotocin-induced diabetic rats on endoneurial blood flow, motor nerve conduction velocity, and vascular reactivity of epineurial arterioles of the sciatic nerve. Diabetes. 2001;50:1927–1937. doi: 10.2337/diabetes.50.8.1927. [DOI] [PubMed] [Google Scholar]
- 3.Cameron NE, Tuck Z, McCabe L, Cotter MA. Effect of the hydroxyl radical scavenger, dimethylthiourea, on peripheral nerve tissue perfusion, conduction velocity and nociception in experimental diabetes. Diabetologia. 2001;44:1161–1169. doi: 10.1007/s001250100626. [DOI] [PubMed] [Google Scholar]
- 4.Obrosova IG, Mabley JG, Zsengeller Z, Charniauskaya T, Abatan OI, Groves JT, Szabo C. Role for nitrosative stress in diabetic neuropathy: evidence from studies with a peroxynitrite decomposition catalyst. FASEB J. 2005;19:401–403. doi: 10.1096/fj.04-1913fje. [DOI] [PubMed] [Google Scholar]
- 5.Sagara M, Satoh J, Wada R, Yagihashi S, Takahashi K, Fukuzawa M, Muto G, Muto Y, Toyota T. Inhibition of development of peripheral neuropathy in streptozotocin-induced diabetic rats with N-acetylcysteine. Diabetologia. 1996;39:263–269. doi: 10.1007/BF00418340. [DOI] [PubMed] [Google Scholar]
- 6.Hounsom L, Corder R, Patel J, Tomlinson DR. Oxidative stress participates in the breakdown of neuronal phenotype in experimental diabetic neuropathy. Diabetologia. 2001;44:424–428. doi: 10.1007/s001250051638. [DOI] [PubMed] [Google Scholar]
- 7.Pertovaara A, Wei H, Kalmari J, Ruotsalainen M. Pain behavior and response properties of spinal dorsal horn neurons following experimental diabetic neuropathy in the rat: modulation by nitecapone, a COMT inhibitor with antioxidant properties. Exp Neurol. 2001;167:425–434. doi: 10.1006/exnr.2000.7574. [DOI] [PubMed] [Google Scholar]
- 8.Ametov AS, Barinov A, Dyck PJ, Hermann R, Kozlova N, Litchy WJ, Low PA, Nehrdich D, Novosadova M, O’Brien PC, Reljanovic M, Samigullin R, Schuette K, Strokov I, Tritschler HJ, Wessel K, Yakhno N, Ziegler D the SYDNEY Trial Study Group. The sensory symptoms of diabetic polyneuropathy are improved with α-lipoic acid: the SYDNEY trial. Diabetes Care. 2003;26:770–776. doi: 10.2337/diacare.26.3.770. [DOI] [PubMed] [Google Scholar]
- 9.Calcutt NA, Freshwater JD, Mizisin AP. Prevention of sensory disorders in diabetic Sprague-Dawley rats by aldose reductase inhibition or treatment with ciliary neurotrophic factor. Diabetologia. 2004;47:718–724. doi: 10.1007/s00125-004-1354-2. [DOI] [PubMed] [Google Scholar]
- 10.Purves T, Middlemas A, Agthong S, Jude EB, Boulton AJ, Fernyhough P, Tomlinson DR. A role for mitogen-activated protein kinases in the etiology of diabetic neuropathy. FASEB J. 2001;15:2508–2514. doi: 10.1096/fj.01-0253hyp. [DOI] [PubMed] [Google Scholar]
- 11.Svensson CI, Marsala M, Westerlund A, Calcutt NA, Campana WM, Freshwater JD, Catalano R, Feng Y, Protter AA, Scott B, Yaksh TL. Activation of p38 mitogen-activated protein kinase in spinal microglia is a critical link in inflammation-induced spinal pain processing. J Neurochem. 2003;86:1534–1544. doi: 10.1046/j.1471-4159.2003.01969.x. [DOI] [PubMed] [Google Scholar]
- 12.Freshwater JD, Svensson CI, Malmberg AB, Calcutt NA. Elevated spinal cyclooxygenase and prostaglandin release during hyperalgesia in diabetic rats. Diabetes. 2002;51:2249–2255. doi: 10.2337/diabetes.51.7.2249. [DOI] [PubMed] [Google Scholar]
- 13.Li F, Obrosova IG, Abatan O, Tian D, Larkin D, Stuenkel EL, Stevens MJ. Taurine replacement attenuates hyperalgesia and abnormal calcium signaling in sensory neurons of streptozotocin-diabetic rats. Am J Physiol Endocrinol Metab. 2005;288:E29–E36. doi: 10.1152/ajpendo.00168.2004. [DOI] [PubMed] [Google Scholar]
- 14.Empl M, Renaud S, Erne B, Fuhr P, Straube A, Schaeren-Wiemers N, Steck AJ. TNF-alpha expression in painful and nonpainful neuropathies. Neurology. 2001;56:1371–1377. doi: 10.1212/wnl.56.10.1371. [DOI] [PubMed] [Google Scholar]
- 15.Garcia Soriano F, Virag L, Jagtap P, Szabo E, Mabley JG, Liaudet L, Marton A, Hoyt DG, Murthy KG, Salzman AL, Southan GJ, Szabo C. Diabetic endothelial dysfunction: the role of poly(ADP-ribose) polymerase activation. Nat Med. 2001;7:108–113. doi: 10.1038/83241. [DOI] [PubMed] [Google Scholar]
- 16.Pacher P, Liaudet L, Soriano FG, Mabley JG, Szabo E, Szabo C. The role of poly(ADP-ribose) polymerase activation in the development of myocardial and endothelial dysfunction in diabetes. Diabetes. 2002;51:514–521. doi: 10.2337/diabetes.51.2.514. [DOI] [PubMed] [Google Scholar]
- 17.Zheng L, Szabo C, Kern TS. Poly(ADP-ribose) polymerase is involved in the development of diabetic retinopathy via regulation of nuclear factor-κB. Diabetes. 2004;53:2960–2967. doi: 10.2337/diabetes.53.11.2960. [DOI] [PubMed] [Google Scholar]
- 18.Obrosova IG, Li F, Abatan OI, Forsell MA, Komjati K, Pacher P, Szabo C, Stevens MJ. Role of poly(ADP-ribose) polymerase activation in diabetic neuropathy. Diabetes. 2004;53:711–720. doi: 10.2337/diabetes.53.3.711. [DOI] [PubMed] [Google Scholar]
- 19.Li F, Szabo C, Pacher P, Southan GJ, Abatan OI, Charniauskaya T, Stevens MJ, Obrosova IG. Evaluation of orally active poly(ADP-ribose) polymerase inhibitor in streptozotocin-diabetic rat model of early peripheral neuropathy. Diabetologia. 2004;47:710–717. doi: 10.1007/s00125-004-1356-0. [DOI] [PubMed] [Google Scholar]
- 20.Virag L, Szabo C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54:375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- 21.Obrosova IG, Fathallah L, Lang HJ, Greene DA. Evaluation of a sorbitol dehydrogenase inhibitor on diabetic peripheral nerve metabolism: a prevention study. Diabetologia. 1999;42:1187–1194. doi: 10.1007/s001250051290. [DOI] [PubMed] [Google Scholar]
- 22.Srinivasan S, Stevens M, Wiley JW. Diabetic peripheral neuropathy: evidence for apoptosis and associated mitochondrial dysfunction. Diabetes. 2000;49:1932–1938. doi: 10.2337/diabetes.49.11.1932. [DOI] [PubMed] [Google Scholar]
- 23.Calcutt NA. Modeling diabetic sensory neuropathy in rats. Methods Mol Med. 2004;99:55–65. doi: 10.1385/1-59259-770-x:225. [DOI] [PubMed] [Google Scholar]
- 24.Obrosova IG, Pacher P, Szabo C, Zsengeller Z, Hirooka H, Stevens MJ, Yorek MA. Aldose reductase inhibition counteracts oxidative-nitrosative stress and poly(ADP-ribose) polymerase activation in tissue sites for diabetes complications. Diabetes. 2005;54:234–242. doi: 10.2337/diabetes.54.1.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Calcutt NA. Experimental models of painful diabetic neuropathy. J Neurol Sci. 2004;220:137–139. doi: 10.1016/j.jns.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 26.Cotter MA, Jack AM, Cameron NE. Effects of the protein kinase C beta inhibitor LY333531 on neural and vascular function in rats with streptozotocin-induced diabetes. Clin Sci (Lond) 2002;103:311–321. doi: 10.1042/cs1030311. [DOI] [PubMed] [Google Scholar]
- 27.Cameron N, Cotter M, Inkster M, Nangle M. Looking to the future: diabetic neuropathy and effects of rosuvastatin on neurovascular function in diabetes models. Diabetes Res Clin Pract. 2003;61 (Suppl 1):S35–S39. doi: 10.1016/s0168-8227(03)00123-2. [DOI] [PubMed] [Google Scholar]
- 28.Obrosova IG. Update on the pathogenesis of diabetic neuropathy. Curr Diab Rep. 2003;3:439–445. doi: 10.1007/s11892-003-0005-1. [DOI] [PubMed] [Google Scholar]
- 29.Kamei J, Zushida K, Morita K, Sasaki M, Tanaka S. Role of vanilloid VR1 receptor in thermal allodynia and hyperalgesia in diabetic mice. Eur J Pharmacol. 2001;422:83–86. doi: 10.1016/s0014-2999(01)01059-7. [DOI] [PubMed] [Google Scholar]
- 30.Anjaneyulu M, Chopra K. Quercetin, a bioflavonoid, attenuates thermal hyperalgesia in a mouse model of diabetic neuropathic pain. Prog Neuro-psychopharmacol Biol Psychiatry. 2003;27:1001–1005. doi: 10.1016/S0278-5846(03)00160-X. [DOI] [PubMed] [Google Scholar]
- 31.Li F, Drel VR, Szabo C, Stevens MJ, Obrosova IG. Low-dose poly(ADP-Ribose) polymerase inhibitor-containing combination therapies reverse early peripheral diabetic neuropathy. Diabetes. 2005;54:1514–1522. doi: 10.2337/diabetes.54.5.1514. [DOI] [PubMed] [Google Scholar]
- 32.Obrosova IG, Marchand J, Nadler JL, Drel VR. 12/15-lipoxygenase and early diabetic neuropathy (Abstract) Diabetologia. 2005;48 (Suppl 1):A356. [Google Scholar]
- 33.Gustafsson H, Flood K, Berge OG, Brodin E, Olgart L, Stiller CO. Gabapentin reverses mechanical allodynia induced by sciatic nerve ischemia and formalin-induced nociception in mice. Exp Neurol. 2003;182:427–434. doi: 10.1016/s0014-4886(03)00097-9. [DOI] [PubMed] [Google Scholar]
- 34.Wang Y, Schmeichel AM, Iida H, Schmelzer JD, Low PA. Ischemia-reperfusion injury causes oxidative stress and apoptosis of Schwann cell in acute and chronic experimental diabetic neuropathy. Antioxid Redox Signal. 2005;7:1513–1520. doi: 10.1089/ars.2005.7.1513. [DOI] [PubMed] [Google Scholar]
- 35.Wang H, Yu SW, Koh DW, Lew J, Coombs C, Bowers W, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Apoptosis-inducing factor substitutes for caspase executioners in NMDA-triggered excitotoxic neuronal death. J Neurosci. 2004;24:10963–10973. doi: 10.1523/JNEUROSCI.3461-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szenczi O, Kemecsei P, Holthuijsen MF, van Riel NA, van der Vusse GJ, Pacher P, Szabo C, Kollai M, Ligeti L, Ivanics T. Poly(ADP-ribose) polymerase regulates myocardial calcium handling in doxorubicin-induced heart failure. Biochem Pharmacol. 2005;69:725–732. doi: 10.1016/j.bcp.2004.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Veres B, Radnai B, Gallyas F, Jr, Varbiro G, Berente Z, Osz E, Sumegi B. Regulation of kinase cascades and transcription factors by a poly(ADP-ribose) polymerase-1 inhibitor, 4-hydroxyquinazoline, in lipopolysaccharide-induced inflammation in mice. J Pharmacol Exp Ther. 2004;310:247–255. doi: 10.1124/jpet.104.065151. [DOI] [PubMed] [Google Scholar]
- 38.Ha HC, Hester LD, Snyder SH. Poly(ADP-ribose) polymerase-1 dependence of stress-induced transcription factors and associated gene expression in glia. Proc Natl Acad Sci U S A. 2002;99:3270–3275. doi: 10.1073/pnas.052712399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Obrosova IG, Drel VR, Pacher P, Ilnytska O, Wang ZQ, Stevens MJ, Yorek MA. Oxidative-nitrosative stress and poly(ADP-ribose) polymerase (PARP) activation in experimental diabetic neuropathy: the relation is revisited. Diabetes. 2005;54:3435–3441. doi: 10.2337/diabetes.54.12.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakamura J, Kato K, Hamada Y, Nakayama M, Chaya S, Nakashima E, Naruse K, Kasuya Y, Mizubayashi R, Miwa K, Yasuda Y, Kamiya H, Ienaga K, Sakakibara F, Koh N, Hotta N. A protein kinase C-β–selective inhibitor ameliorates neural dysfunction in streptozotocin-induced diabetic rats. Diabetes. 1999;48:2090–2095. doi: 10.2337/diabetes.48.10.2090. [DOI] [PubMed] [Google Scholar]
- 41.Cameron NE, Cotter MA, Jack AM, Basso MD, Hohman TC. Protein kinase C effects on nerve function, perfusion, Na(+), K(+)-ATPase activity and glutathione content in diabetic rats. Diabetologia. 1999;42:1120–1130. doi: 10.1007/s001250051280. [DOI] [PubMed] [Google Scholar]
- 42.Yamagishi S, Uehara K, Otsuki S, Yagihashi S. Differential influence of increased polyol pathway on protein kinase C expressions between endoneurial and epineurial tissues in diabetic mice. J Neurochem. 2003;87:497–507. doi: 10.1046/j.1471-4159.2003.02011.x. [DOI] [PubMed] [Google Scholar]
- 43.Uehara K, Yamagishi S, Otsuki S, Chin S, Yagihashi S. Effects of polyol pathway hyperactivity on protein kinase C activity, nociceptive peptide expression, and neuronal structure in dorsal root ganglia in diabetic mice. Diabetes. 2004;53:3239–3247. doi: 10.2337/diabetes.53.12.3239. [DOI] [PubMed] [Google Scholar]
- 44.Inoguchi T, Sonta T, Tsubouchi H, Etoh T, Kakimoto M, Sonoda N, Sato N, Sekiguchi N, Kobayashi K, Sumimoto H, Utsumi H, Nawata H. Protein kinase C-dependent increase in reactive oxygen species (ROS) production in vascular tissues of diabetes: role of vascular NAD(P)H oxidase. J Am Soc Nephrol. 2003;14 (Suppl 3):S227–S232. doi: 10.1097/01.asn.0000077407.90309.65. [DOI] [PubMed] [Google Scholar]
- 45.Abiko T, Abiko A, Clermont AC, Shoelson B, Horio N, Takahashi J, Adamis AP, King GL, Bursell SE. Characterization of retinal leukostasis and hemodynamics in insulin resistance and diabetes: role of oxidants and protein kinase-C activation. Diabetes. 2003;52:829–837. doi: 10.2337/diabetes.52.3.829. [DOI] [PubMed] [Google Scholar]
- 46.Inoguchi T, Nawata H. NAD(P)H oxidase activation: a potential target mechanism for diabetic vascular complications, progressive beta-cell dysfunction and metabolic syndrome. Curr Drug Targets. 2005;6:495–501. doi: 10.2174/1389450054021927. [DOI] [PubMed] [Google Scholar]
- 47.Cheng C, Zochodne DW. Sensory neurons with activated caspase-3 survive long-term experimental diabetes. Diabetes. 2003;52:2363–2371. doi: 10.2337/diabetes.52.9.2363. [DOI] [PubMed] [Google Scholar]
- 48.Burnand RC, Price SA, McElhaney M, Barker D, Tomlinson DR. Expression of axotomy-inducible and apoptosis-related genes in sensory nerves of rats with experimental diabetes. Brain Res Mol Brain Res. 2004;132:235–240. doi: 10.1016/j.molbrainres.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 49.Schmeichel AM, Schmelzer JD, Low PA. Oxidative injury and apoptosis of dorsal root ganglion neurons in chronic experimental diabetic neuropathy. Diabetes. 2003;52:165–171. doi: 10.2337/diabetes.52.1.165. [DOI] [PubMed] [Google Scholar]
- 50.Kamiya H, Zhangm W, Sima AA. Apoptotic stress is counterbalanced by survival elements preventing programmed cell death of dorsal root ganglions in subacute type 1 diabetic BB/Wor rats. Diabetes. 2005;54:3288–3295. doi: 10.2337/diabetes.54.11.3288. [DOI] [PubMed] [Google Scholar]
- 51.Jagtap P, Szabo C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov. 2005;4:421–440. doi: 10.1038/nrd1718. [DOI] [PubMed] [Google Scholar]