

Abstract
Epidemiological studies demonstrated that even in the absence of other risk factors (e.g. diabetes, hypertension, hypercholesterolemia), advanced age itself significantly increases cardiovascular morbidity. Although aging is inevitable, cardiovascular gerontologists recognize that a better understanding of the aging process in the not-so-distant future will lead to pharmacological interventions that considerably delay the functional decline of the cardiovascular system. Since the original publishing of the free radical theory of aging, an increased production of reactive oxygen species has been implicated both in the aging process and the development of age-related cardiovascular diseases. This review focuses on the role of oxidative and nitrosative stress in cardiovascular dysfunction in aging, downstream mechanisms including activation of NF-κB, and the role of poly(ADP-ribose)polymerase (PARP) and longevity genes that are linked to regulation of cellular redox status and oxidative stress resistance (p66shc, sirtuins, FOXO transcription factors).
Keywords: Endothelium, heart, coronary circulation, senescence, inflammation, gene expression, redox status, peroxynitrite
INTRODUCTION
The population in the Western world is aging. With the baby boomers approaching retirement by the end of this decade, the number of senior citizens (65 years old or older) will be close to 40 million people in the United States. In spite of unprecedented development in early diagnostic techniques and intervention treatments, cardiovascular disease will remain a significant health risk for the elderly. Epidemiological studies suggest that even in the absence of other risk factors (e.g. diabetes, hypertension, hypercholesterolemia), advanced age itself significantly increases cardiovascular morbidity. A better understanding of the pathophysiological mechanisms underlying the complex phenomenon of cardiovascular aging will likely lead to novel pharmacological treatments that prevent or delay the development of ischemic heart disease and stroke.
Cardiovascular aging is characterized by a gradual deterioration of endothelial function and myocardial performance [1], which begins to accelerate after mid-life. To illustrate this point, we have expressed age-related changes in endothelial dysfunction, represented as changes in endothelium-dependent responses (vascular relaxation, dilation or NO release) of vessels from various vascular beds (Fig. 1). There is increasing evidence that aging-induced changes in the proteome (Fig. 2) underlie this age-related functional decline and that cellular redox regulation plays a major role in these phenotypic and functional alterations. The present review focuses on some of the mechanisms by which advanced age may promote vascular oxidative and nitrosative stress and the possible downstream mechanisms by which reactive oxygen and nitrogen species may impair cardiovascular function in aging.
Fig 1.

(A). Aging-induced endothelial dysfunction, represented as changes in endothelium-dependent responses (vascular relaxation, dilation or NO release) of vessels from various vascular beds (aorta, coronary, carotid, femoral or mesentery artery), induced by sub-maximal dose of a vasodilator (acetylcholine and the calcium ionophore A23187). This figure has been compiled from both published [3, 6, 7, 62, 74–81] and unpublished data. Age is shown as percentage of maximal lifespan of each species. (B). Aging-induced decline in cardiac performance in F344 rats (representative pressure-volume loops, pressure signals and dP/dt (C) obtained with the Millar P-V conductance catheter system).
Fig 2.
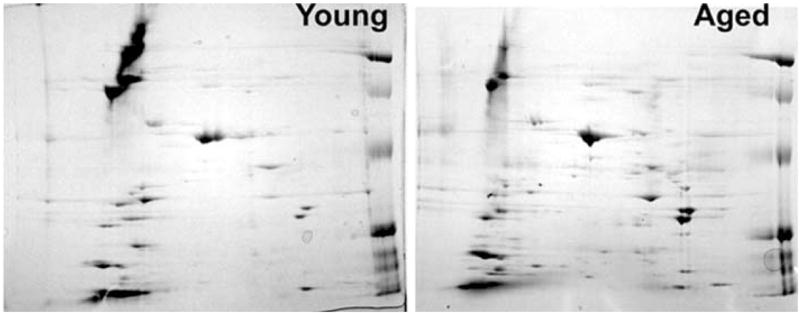
A set of representative 2DE separation of proteins from the carotid artery of 3 month old (young) and 26 month old (aged) F344 rats illustrating age-related changes in the vascular proteome. Arteries were homogenized and the proteins were extracted as described [3]. Samples were focused on 7 cm pH 3-10 NL IEF strips and separated on 12% second-dimension gels. Future studies should characterize both aging-induced gene expression changes at the protein level and post-translational protein modifications related to an increased oxidative-nitrosative stress present in the aged vasculature.
1. ROLE OF INCREASED OXIDATIVE AND NITRO-SATIVE STRESS IN CARDIOVASCULAR AGING
Since Harmon proposed the original free radical theory of aging [2], considerable evidence has been published that the aging in various tissues is associated with an increased oxidative stress. Recently, we and others showed that in small coronary arteries [3] and mesenteric arteries [4] of aged rats, there is an increased O2− production. Similarly, increased ROS production has also been reported in the aorta and carotid arteries of aged rats [5, 6] and mice [7]. Aging-induced vascular oxidative stress seems to be associated with a pro-oxidant shift in vascular phenotype, including an increased expression of iNOS [1, 7] and increased activity of NAD(P)H oxidases [3, 6, 8] and/or other oxidase mechanisms [9] and a down-regulation of antioxidants, such as ecSOD [4].
One of the consequences of increased oxidative stress in aging is a functional inactivation of NO by high concentrations of O2.−, resulting in an enhanced ONOO− formation [3, 4, 7, 8]. It is generally accepted that tonic release of NO from the endothelium exerts vasculoprotective and cardioprotective effects, such as maintenance of normal coronary blood flow, inhibition of platelet aggregation and inflammatory cell adhesion to endothelial cells and disruption of pro-inflammatory cytokine-induced signaling pathways. The severe impairment of NO bioavailability in aging, also aggravated by an age-related decline in eNOS expression [3, 10–13] and/or a decreased intracellular L-arginine availability [14], limits cardiac blood supply and alters myocardial O2 consumption and cardiac contractility [8]. Recent studies also suggest that decreased endothelial NO production in aging enhances apoptosis of endothelial cells [13, 15].
In addition, increased production of reactive oxygen species (ROS), such as O2.−, .OH, H2O2, and reactive nitrogen species, such as ONOO−, are thought to be implicated in the inflammatory process by acting as second messengers. NF-κB is a redox-sensitive transcription factor, expressed by both endothelial and smooth muscle cells. Activation of NF-κB is thought to induce the transcription of a large range of genes implicated in inflammation, including cytokines (e.g. TNFα, IL-6 and IL-1β), chemokines and adhesion molecules [16–18], and it is generally believed that chronic activation of NF-κB predisposes arteries to atherosclerosis [19]. Numerous studies demonstrated that increased levels of ROS may activate NF-κB in endothelial and smooth muscle cells and many other cell types, leading to the up-regulation of TNFα and other cytokines. Importantly, there are studies suggesting that NF-κB binding increases in aging [20]. There is also evidence for an pro-inflammatory shift in vascular [3, 15, 21] and cardiac [22] cytokine expression profile (including an up-regulation of TNFα and IL-6). Thus, further studies are definitely needed to establish the link between oxidative stress, NF-κB activation and vascular inflammation in aging.
Many of the adverse consequences of oxidative stress are not directly due to O2. − itself, but are mediated via production of highly reactive oxidant peroxynitrite, the reaction product of NO and superoxide [23, 24]. There are several studies extant showing a substantially enhanced cardiovascular ONOO− formation in aging [3, 4, 7, 8]. The possible downstream targets of peroxynitrite-induced cytotoxicity are multiple [23, 24]. The reaction of peroxynitrite with enzymes, macromolecules and lipids, has been shown to influence cellular functions. For example, tyrosine nitration may lead to dysfunction of nitrated proteins, as has been shown or suggested in the case of Mn-superoxide dismutase, cytoskeletal actin, neuronal tyrosine hydroxylase, cytochrome P450 and prostacyclin synthase. For example, peroxynitrite-mediated nitration of myofibrillar creatine kinase was suggested to impair myocardial contractility [25]. Importantly, a recent study analyzing protein nitration in cardiac tissue from old rats using proteomics techniques identified, among others, several enzymes of the glycolytic machinery (α-enolase-1, α-aldolase and GAPDH) as targets for protein nitration [26]. Mitochondrial proteins appear to be especially sensitive to aging-related nitration. The nitrated mitochondrial proteins identified by Kanski et al. include aconitase, creatine kinase, voltage-dependent anion channel, ATP synthase and other proteins involved in electron transfer [26]. In addition, peroxynitrite-modified cellular proteins are subject to accelerated degradation via the proteosome. Peroxynitrite may also inhibit a variety of ion pumps including calcium pumps, calcium-activated potassium channels and Na+/K+ ATP-ase [23, 24]. The reaction of lipids with peroxynitrite leads to peroxidation (malondialdehyde and conjugated diene formation) and formation of nitrito-, nitro-, nitrosoperoxo- and/or nitrated lipid oxidation adducts [23, 24]. Peroxynitrite also potently oxidizes various biomolecules including tetrahydrobiopterin (BH4) to quinonoid 5,6-dihydrobiopterin. A large proportion of the quinonoid isomer readily loses its side chain to form 7,8-dihydropterin, which is not a co-factor for NO synthase. Low cellular levels of BH4 can promote a cycle of its own destruction mediated by NO synthase-dependent formation of peroxynitrite. This mechanism may contribute to vascular endothelial dysfunction induced by oxidative stress in aging. Peroxynitrite generation also decreases the nitric oxide available for G-protein stimulation and vasodilation, thus contributing to endothelial dysfunction. Peroxynitrite may also inhibit superoxide dismutase, glutaredoxin and other antioxidant molecules and systems, which leads to positive feedback cycles of intra-cellular oxidant generation and oxidative injury [23, 24]. Recent work suggests that oxidative-nitrosative stress supports MMP activation [27, 28], which seems to be present in aged vessels [29–31]. Peroxynitrite in large concentration may induce DNA single strand breaks, which may contribute to the endothelial damage in aging [32]. Oxidative DNA damage may lead to the activation of the nuclear enzyme PARP, which can trigger a cellular suicide pathway [23]. High concentrations of peroxynitrite trigger rapid derangements in energy metabolism and PARP activation leading to necrosis while lower concentrations of peroxynitrite induce cytochrome c release from the mitochondria and trigger apoptosis [23]. It appears that peroxynitrite decomposition catalysts improve cardiac function and overall outcome in animal models of heart failure. For instance, metalloporphyrin peroxynitrite decomposition catalysts (FP15 and FeTPPS) reduced myocardial necrosis in a rat model of acute myocardial failure [33], and improved mechanical dysfunction in rat hearts exposed to pro-inflammatory cytokines [34]. Future studies should test the potential cardioprotective effects of these drugs in aging.
2. LONGEVITY GENES, OXIDATIVE STRESS RESISTANCE AND CARDIOVASCULAR AGING
Since the last decade, there is growing evidence that the aging process is genetically determined and there is reasonable hope that the function of genes that control lifespan can be eventually therapeutically modulated. We can expect that the number of papers on the relationship between free radical production, oxidative stress resistance, the rate of development of age-related cardiovascular dysfunction and longevity will explode in the coming years.
An interesting avenue of research is to compare these mechanisms in species with markedly different maximum lifespan potential (MLSP). For example, the extended longevity of bats, despite their high metabolic rate, may provide insight to mechanisms of cardiovascular aging. A recent landmark study revealed that mitochondria from the heart of the little brown bat, Myotis lucifugus (MLSP=34 years), produced half to one-third the amount of hydrogen peroxide per unit of oxygen consumed compared to mitochondria from the short-tailed shrew, Blarina brevicauda (MLSP=2 years), and the white-footed mouse, Peromyscus leucopus (MLSP=8 years) [35, 36]. Similar results have been obtained in birds as well, which like bats have high metabolic rates and extended longevity. Further studies are evidently needed for cross-species comparison of a wide range of genes that are known to participate in cardiovascular aging and to correlate age-related changes in endothelial function and cardiac performance with oxidative-nitrosative stress and oxidative stress resistance in long- and short-lived animal models.
It is worth to point out that caloric restriction (which increases maximum life span in virtually every species) was shown to decrease ROS production and pro-inflammatory gene expression (e.g. TNFα) in various organs (including the heart) of aged animals [37, 38]. Further studies are needed to determine whether this is the case in aged blood vessels, especially in coronary arteries, and to test the hypothesis that caloric restriction modulates vascular function and phenotype by exerting antioxidant effects [39–41]. Importantly, caloric restriction was shown to decrease cellular DNA damage in the aorta of aged mice [32]. To better understand the effects of caloric restriction, recently, several mutant mice were created that possess defects in the growth hormone/insulin-like growth factor 1 neurohormonal pathway. These animals exhibit extended life span with a phenotype that resembles those of mice subjected to caloric restriction. Studies are underway to characterize the rate of cardiovascular aging in these mice.
Interestingly, the long-lived p66shc−/− mice were shown to be protected against age-related endothelial dysfunction [7]. Investigation of the underlying mechanisms revealed that deletion of the p66shc gene (that encodes an adaptor protein participating in regulation of the intracellular redox state) lowers aortic O2 − and ONOO− production, thereby reducing NO breakdown and increasing its bioavailability [7]. Of note, the expression of iNOS increased significantly in the old wild type mice, whereas no age-dependent changes were found in the p66shc−/− mice [7], suggesting that upregulation of iNOS is involved in ONOO− formation and hence increased oxidative damage of aging vascular tissue.
Another exciting area of research that may yield valuable insight in the cardiovascular aging process centers on the members of the Foxo subfamily of forkhead transcription factors, including Foxo3a (FKHRL1), Foxo1 (FKHR), and Foxo4 (AFX) [42–44]. Foxo transcription factors are homologs of DAF-16, which is known to regulate longevity as well as resistance to oxidative stress in the nematode Caenorhabditis elegans as a part of the insulin/insulin-like growth factor-1/phosphatidylinositol-3 kinase/Akt signaling pathway [45–47]. Expression of forkhead transcription factors has been demonstrated in murine heart [48], human endothelial cells [49] and we have recently identified Foxo subfamily members expressed in rat coronary arteries. In C. elegans, DAF-16 appears to regulate oxidative stress responses by transactivating a number of antioxidant enzymes and stress-related gene products [50, 51]. In mammalian, cells overexpression of Foxo3a also results in an increase in both H2O2 scavenging and oxidative stress resistance [52]. Among others, putative Foxo binding sites [48] have been identified on the human catalase promoter [52]. Comparison of the catalase promoter from different species revealed that Foxo binding sites are present in a region that shows some degree of evolutionary conservation (Fig. 2). There are also studies suggesting a link between Foxo3a and p66shc [52]. Interestingly, the level of Foxo transcription factors appears to change in skeletal muscle with aging [44] and in cultured coronary vessels with an increased number of passages (Fig. 3). Yet, there is no information available on how vascular and cardiac FOXO activity correlates with aging-induced oxidative and nitrosative stress.
Fig 3.

(A). Increased number of passages are associated with a decreased mRNA expression of forkhead transcription factors Foxo1 and Foxo4 in rat coronary arterial endothelial cells (CAEC). Real-time PCR measurements were performed as reported [3, 15]. mVISTA plots showing the percent of conservation between the mouse and human at the 5' flanking region (3000 bp) of the catalase (B) and Mn-SOD genes (C). The arrows point to the location of conserved putative Foxo binding sites.
Longevity regulatory genes also include the NAD-dependent histone deacetylase silent information regulator 2 (Sir2), which acts to extend lifespan in C. elegans upstream of a Foxo transcription factor, in the insulin-like signaling pathway. Recent findings established Foxo1 as a direct and functional target for Sir2 homologs in mammalian cells and suggested that it plays a cardioprotective role [53]. Further studies are needed to determine whether pharmacological activation of sirtuins (e.g. by polyphenol compounds) can delay or reverse age-related cardiovascular dysfunction.
3. ROLE OF POLY(ADP-RIBOSE) POLYMERASE ACTIVATION IN CARDIOVASCULAR AGING
The enzyme poly (ADP-ribose) polymerase (PARP-1) is a nuclear protein belonging to the DNA damage surveillance network [54]. PARP-1 responds to DNA damage by transferring 50 to 200 molecules of ADP-ribose to various nuclear proteins, including transcription factors and histones. Poly(ADP-ribose) polymers and PARP-1 have been known since 1964, and although their essential functions have not yet been fully elucidated, it appears that poly(ADP-ribosyl)ation activity of PARP-1 is important for maintaining genomic integrity [54, 55]. The catalytic activity of PARP-1 is markedly stimulated upon binding to DNA strand interruptions, and the resulting polymer is thought to participate in signaling the presence of damage to DNA repair complexes. PARP-1 activity is responsible for approximately 90% of the total poly(ADP-ribose) formation in a living cell. In recent years, other PARPs have been discovered based on sequence homology, such as PARP-2, which is activated by DNA strand breaks like PARP-1. Moderate PARP activation facilitates the efficient repair of DNA damage arising from reactive oxygen and nitrogen species such as H2O2 and ONOO−. However, under conditions of tissue ischemia, this action can lead to significant decreases in NAD+, massive ATP depletion and cell death. The latter aspect of PARP-1 activity has been implicated in the pathogenesis of cardiovascular dysfunction associated with various clinical conditions such as shock, ischemia-reperfusion and diabetes. Inhibition of PARP-1 has been shown to be effective on one hand, in the treatment of cancer in combination with alkylating agents by suppressing DNA repair and thus driving tumor cells into apoptosis, and on the other hand, as a promising drug target for the treatment of pathological conditions involving oxidative and nitrosative stress.
Accumulation of macromolecular damage, including damage to DNA and genomic instability, is considered a driving force for the aging process and age-related diseases. It has been repeatedly shown that a positive correlation exists between poly(ADP-ribosyl)ation capacity and the longevity of various mammalian species [56, 57]. Also, recent studies indicate that a functional defect of the poly(ADP-ribosyl)ation pathway is present in the premature aging disease Werner syndrome that is characterized by genomic instability and hypersensitivity to DNA-damaging agents [58, 59]. Although it was reported that PARP-1 −/− mice did not develop an increased number of spontaneous neoplasias [59, 60], PARP-1 deficiency resulted in a slight decrease in survival and the general health of mice [60], suggesting a role of PARP-1 on mouse viability during aging. PARP-2 knockout mice do not display any obvious spontaneous phenotype, but PARP-1/PARP-2 double-knockout mice exhibit embryonic lethality, pointing to an important function of PARPs in developmental processes. Telomeres can also be influenced by poly(ADP-ribosyl)ation and there is data that tankyrases (TNKS-1 and TNKS-2, also termed PARP-5) and PARP-1 and PARP-2 are localized telomeric DNA and are involved in telomere maintenance [61]. Although telomeres are thought to be important regulators of cellular viability, the role of these mechanisms in vascular aging is yet to be determined [60].
Although there is a paucity of data regarding the age-related cardiovascular morbidity of PARP-1 deficient animals, there are reports that chronic administration (2 months) of the phenantridinone derivative PARP inhibitor PJ34 improved cardiac function (decreased the left ventricular end diastolic pressure [LVEDP]) and augmented endothelial function in the aorta of aged (22 month old) Wistar rats [62] (Fig. 4C). Also, treatment with the PARP inhibitor INO-1001 (for 2 months) was shown to improve both the diastolic and systolic cardiac function in 24 month old F344 rats [63, 64]. Inhibition of PARP-1 also decreased LVEDP in middle aged (40 week old) spontaneously hypertensive rats [65]. There are also reports extant on the beneficial cardiovascular effects of PARP inhibitors in animal disease models that are characterized by an accelerated cardiovascular aging (e.g. diabetes [66]). The vascular expression of PARP-1 does not seem to change substantially in aging, and more data is evidently needed on the post-translational regulation of PARP activity in aging. Also, homeostasis of poly(ADP-ribosylation) is a dynamic process, but age-related changes in the activity of poly(ADP-ribose) glycohydrolase (PARG), the major enzyme that removes oligo(ADP-ribose) fragments via endoglycosidic cleavage [67], have not yet been investigated. Interestingly, there is an increased presence of the 89-kDa cleaved fragment of PARP-1 in aged coronary arteries (Fig. 4A), which is consistent with the enhanced endothelial apoptosis in these vessels [15]. An increased apoptosis-related PARP-1 cleavage has also been documented in the brain of aged rats [68]. It is generally thought that PARP-1 is one of the first proteins to be cleaved in the execution phase of apoptosis in order to inhibit DNA repair by abrogating DNA damage signaling through separating the catalytic C terminus of PARP-1 from its DNA binding domain (Fig. 4B).
Fig 4.

(A). Representative Western blot showing PARP-1 content in coronary arteries of young (3 month old) and aged (27 month old) F344 rats. Note that an increased amount of 89-kDa cleaved fragment of PARP-1 was present in the aged vessels, which likely corresponds to an enhanced endothelial apoptosis in these arteries. It is important to point out that PARP-1 expression does not necessarily correlate with PARP activity. (B). 3D structure of PARP-1 with the evolutionary conserved catalytic subunit (red) (based on the results of automated comparative analysis at Entrez’s Molecular Modeling Database, which is available at: http://www.ncbi.nlm.nih.gov/Entrez/structure.html). Asterisk shows the putative binding site of PARP inhibitors. Purple: regulatory subunit. C: The PARP inhibitor PJ34 improves endothelial function in the aorta of aged rats (redrawn from data presented in ref [62]).
The mechanisms by which PARP-1 inhibitors may exert beneficial anti-aging cardiovascular effects are not yet fully understood and should be unveiled in up-coming studies. Importantly, potent pharmacological PARP inhibitors have recently entered the stage of clinical investigations, among others, for cardioprotective indications [69]. PARP-1 may contribute to a number of pro-inflammatory mechanisms (e.g. NF-κB activation [70, 71], expression of pro-inflammatory cytokines [72]). Also a recent study showed that TNFα-induced NF-κB -dependent transcriptional activation of various chemokines and adhesion molecules is partially inhibited in PARP-1(−/−) endothelial cells [73]. Future studies are likely to reveal additional vascular effects of the pharmacological disruption of PARP-1-dependent pathways, including an attenuation of vascular inflammation, which may have anti-aging effects.
4. CONCLUSION
In conclusion, aging is associated with oxidative/nitrosative stress and inflammatory changes in the phenotype of blood vessels and the heart (Fig. 5). Whether conventional treatments (e.g. statins, ACE-inhibitors) with anti-oxidant properties are able to reverse or delay the aging-induced considerable functional decline of the cardiovascular system remains a subject of current debate. Overall, we can expect that recent advances in our understanding of the role of reactive oxygen and nitrogen species and redox-sensitive cellular mechanisms underlying cardiovascular aging will, in the not so distant future, yield novel therapeutic approaches that will be exploited for the benefit of elderly patients.
Fig 5.
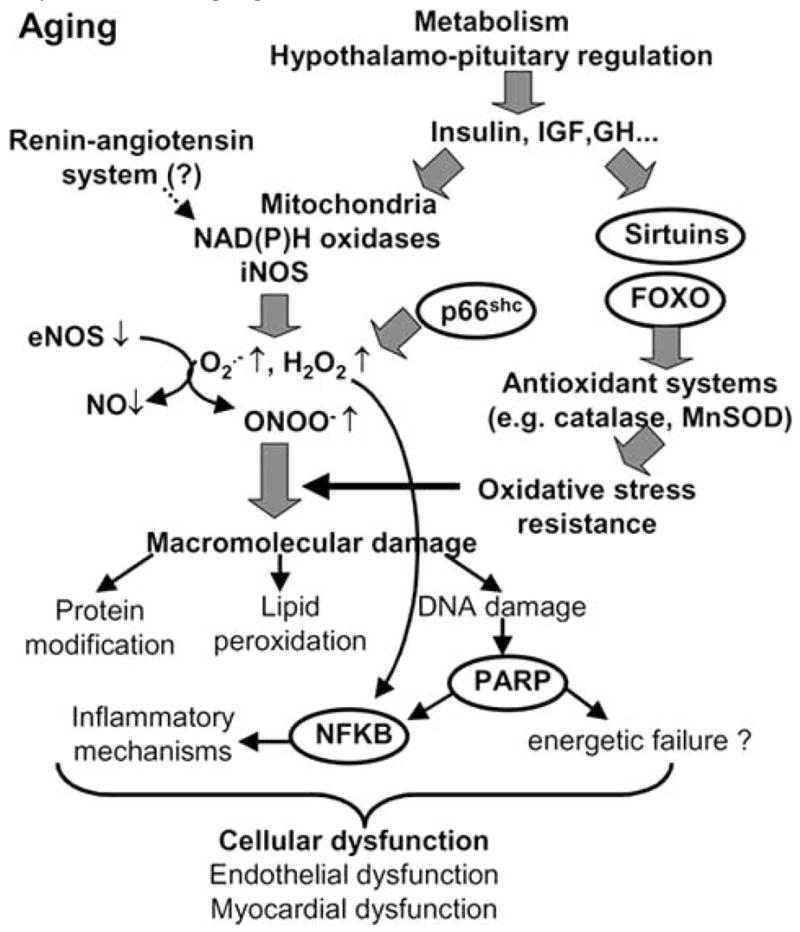
Proposed scheme for the role of oxidative and nitrosative stress, longevity genes and poly(ADP-ribose) polymerase in cardiovascular dysfunction associated with aging.
Acknowledgments
This work was supported by grants from the American Heart Association 0430108N and 0435140N, American Health Assistance Foundation H2004-024, American Federation for Aging Research and NIH PO-1-HL-43023.
References
- 1.Yang B, Larson DF, Watson RR. Modulation of iNOS activity in age-related cardiac dysfunction. Life Sci. 2004;75(6):655–67. doi: 10.1016/j.lfs.2003.09.076. [DOI] [PubMed] [Google Scholar]
- 2.Harman D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 3.Csiszar A, Ungvari Z, Edwards JG, Kaminski PM, Wolin MS, Koller A, et al. Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ Res. 2002;90(11):1159–66. doi: 10.1161/01.res.0000020401.61826.ea. [DOI] [PubMed] [Google Scholar]
- 4.Sun D, Huang A, Yan EH, Wu Z, Yan C, Kaminski PM, et al. Reduced release of nitric oxide to shear stress in mesenteric arteries of aged rats. Am J Physiol Heart Circ Physiol. 2004;286(6):H2249–56. doi: 10.1152/ajpheart.00854.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hamilton CA, Brosnan MJ, McIntyre M, Graham D, Dominiczak AF. Superoxide excess in hypertension and aging: a common cause of endothelial dysfunction. Hypertension. 2001;37(2):529–34. doi: 10.1161/01.hyp.37.2.529. [DOI] [PubMed] [Google Scholar]
- 6.van der Loo B, Labugger R, Skepper JN, Bachschmid M, Kilo J, Powell JM, et al. Enhanced peroxynitrite formation is associated with vascular aging. J Exp Med. 2000;192(12):1731–44. doi: 10.1084/jem.192.12.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Francia P, delli Gatti C, Bachschmid M, Martin-Padura I, Savoia C, Migliaccio E, et al. Deletion of p66shc gene protects against age-related endothelial dysfunction. Circulation. 2004;110(18):2889–95. doi: 10.1161/01.CIR.0000147731.24444.4D. [DOI] [PubMed] [Google Scholar]
- 8.Adler A, Messina E, Sherman B, Wang Z, Huang H, Linke A, et al. NAD(P)H oxidase-generated superoxide anion accounts for reduced control of myocardial O2 consumption by NO in old Fischer 344 rats. Am J Physiol Heart Circ Physiol. 2003;285(3):H1015–22. doi: 10.1152/ajpheart.01047.2002. [DOI] [PubMed] [Google Scholar]
- 9.Bachschmid M, van der Loo B, Schuler K, Labugger R, Thurau S, Eto M, et al. Oxidative stress-associated vascular aging is independent of the protein kinase C/NAD(P)H oxidase pathway. Arch Gerontol Geriatr. 2004;38(2):181–90. doi: 10.1016/j.archger.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 10.Tanabe T, Maeda S, Miyauchi T, Iemitsu M, Takanashi M, Irukayama-Tomobe Y, et al. Exercise training improves ageing-induced decrease in eNOS expression of the aorta. Acta Physiol Scand. 2003;178(1):3–10. doi: 10.1046/j.1365-201X.2003.01100.x. [DOI] [PubMed] [Google Scholar]
- 11.Woodman CR, Price EM, Laughlin MH. Aging induces muscle-specific impairment of endothelium-dependent dilation in skeletal muscle feed arteries. J Appl Physiol. 2002;93(5):1685–90. doi: 10.1152/japplphysiol.00461.2002. [DOI] [PubMed] [Google Scholar]
- 12.Matsushita H, Chang E, Glassford AJ, Cooke JP, Chiu CP, Tsao PS. eNOS activity is reduced in senescent human endothelial cells: Preservation by hTERT immortalization. Circ Res. 2001;89(9):793–8. doi: 10.1161/hh2101.098443. [DOI] [PubMed] [Google Scholar]
- 13.Hoffmann J, Haendeler J, Aicher A, Rossig L, Vasa M, Zeiher AM, et al. Aging enhances the sensitivity of endothelial cells toward apoptotic stimuli: important role of nitric oxide. Circ Res. 2001;89(8):709–15. doi: 10.1161/hh2001.097796. [DOI] [PubMed] [Google Scholar]
- 14.Berkowitz DE, White R, Li D, Minhas KM, Cernetich A, Kim S, et al. Arginase reciprocally regulates nitric oxide synthase activity and contributes to endothelial dysfunction in aging blood vessels. Circulation. 2003;108(16):2000–6. doi: 10.1161/01.CIR.0000092948.04444.C7. [DOI] [PubMed] [Google Scholar]
- 15.Csiszar A, Ungvari Z, Koller A, Edwards JG, Kaley G. Proinflammatory phenotype of coronary arteries promotes endothelial apoptosis in aging. Physiol Genomics. 2004;17(1):21–30. doi: 10.1152/physiolgenomics.00136.2003. [DOI] [PubMed] [Google Scholar]
- 16.Tedgui A, Mallat Z. Anti-inflammatory mechanisms in the vascular wall. Circ Res. 2001;88(9):877–87. doi: 10.1161/hh0901.090440. [DOI] [PubMed] [Google Scholar]
- 17.Libermann TA, Baltimore D. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol Cell Biol. 1990;10(5):2327–34. doi: 10.1128/mcb.10.5.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang YH, Lin JX, Vilcek J. Interleukin-6 induction by tumor necrosis factor and interleukin-1 in human fibroblasts involves activation of a nuclear factor binding to a kappa B-like sequence. Mol Cell Biol. 1990;10(7):3818–23. doi: 10.1128/mcb.10.7.3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci USA. 2000;97(16):9052–7. doi: 10.1073/pnas.97.16.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Helenius M, Hanninen M, Lehtinen SK, Salminen A. Aging-induced up-regulation of nuclear binding activities of oxidative stress responsive NF-kB transcription factor in mouse cardiac muscle. J Mol Cell Cardiol. 1996;28(3):487–98. doi: 10.1006/jmcc.1996.0045. [DOI] [PubMed] [Google Scholar]
- 21.Csiszar A, Ungvari Z, Koller A, Edwards JG, Kaley G. Aging-induced proinflammatory shift in cytokine expression profile in rat coronary arteries. Faseb J. 2003;17(9):1183–5. doi: 10.1096/fj.02-1049fje. [DOI] [PubMed] [Google Scholar]
- 22.Lee CK, Allison DB, Brand J, Weindruch R, Prolla TA. Transcriptional profiles associated with aging and middle age-onset caloric restriction in mouse hearts. Proc Natl Acad Sci USA. 2002;99(23):14988–93. doi: 10.1073/pnas.232308999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szabo C. Multiple pathways of peroxynitrite cytotoxicity. Toxicol Lett. 2003;140–141:105–12. doi: 10.1016/s0378-4274(02)00507-6. [DOI] [PubMed] [Google Scholar]
- 24.Turko IV, Murad F. Protein nitration in cardiovascular diseases. Pharmacol Rev. 2002;54(4):619–34. doi: 10.1124/pr.54.4.619. [DOI] [PubMed] [Google Scholar]
- 25.Mihm MJ, Coyle CM, Schanbacher BL, Weinstein DM, Bauer JA. Peroxynitrite induced nitration and inactivation of myofibrillar creatine kinase in experimental heart failure. Cardiovasc Res. 2001;49(4):798–807. doi: 10.1016/s0008-6363(00)00307-2. [DOI] [PubMed] [Google Scholar]
- 26.Kanski J, Behring A, Pelling J, Schoneich C. Proteomic Identification of 3-Nitrotyrosine-Containing Rat Cardiac Proteins: Effect of Biological Aging. Am J Physiol Heart Circ Physiol. 2005;288(1):H371–81. doi: 10.1152/ajpheart.01030.2003. [DOI] [PubMed] [Google Scholar]
- 27.Okamoto T, Akaike T, Sawa T, Miyamoto Y, van der Vliet A, Maeda H. Activation of matrix metalloproteinases by peroxynitrite-induced protein S-glutathiolation via disulfide S-oxide formation. J Biol Chem. 2001;276(31):29596–602. doi: 10.1074/jbc.M102417200. [DOI] [PubMed] [Google Scholar]
- 28.Lalu MM, Wang W, Schulz R. Peroxynitrite in myocardial ischemia-reperfusion injury. Heart Fail Rev. 2002;7(4):359–69. doi: 10.1023/a:1020766502316. [DOI] [PubMed] [Google Scholar]
- 29.Li Z, Froehlich J, Galis ZS, Lakatta EG. Increased expression of matrix metalloproteinase-2 in the thickened intima of aged rats. Hypertension. 1999;33(1):116–23. doi: 10.1161/01.hyp.33.1.116. [DOI] [PubMed] [Google Scholar]
- 30.Wang M, Takagi G, Asai K, Resuello RG, Natividad FF, Vatner DE, et al. Aging increases aortic MMP-2 activity and angiotensin II in nonhuman primates. Hypertension. 2003;41(6):1308–16. doi: 10.1161/01.HYP.0000073843.56046.45. [DOI] [PubMed] [Google Scholar]
- 31.Wang M, Lakatta EG. Altered regulation of matrix metalloprotein-ase-2 in aortic remodeling during aging. Hypertension. 2002;39(4):865–73. doi: 10.1161/01.hyp.0000014506.13322.66. [DOI] [PubMed] [Google Scholar]
- 32.Guo ZM, Yang H, Hamilton ML, VanRemmen H, Richardson A. Effects of age and food restriction on oxidative DNA damage and antioxidant enzyme activities in the mouse aorta. Mech Ageing Dev. 2001;122(15):1771–86. doi: 10.1016/s0047-6374(01)00298-6. [DOI] [PubMed] [Google Scholar]
- 33.Pacher P, Liaudet L, Bai P, Mabley JG, Kaminski PM, Virag L, et al. Potent metalloporphyrin peroxynitrite decomposition catalyst protects against the development of doxorubicin-induced cardiac dysfunction. Circulation. 2003;107(6):896–904. doi: 10.1161/01.cir.0000048192.52098.dd. [DOI] [PubMed] [Google Scholar]
- 34.Ferdinandy P, Danial H, Ambrus I, Rothery RA, Schulz R. Per-oxynitrite is a major contributor to cytokine-induced myocardial contractile failure. Circ Res. 2000;87(3):241–7. doi: 10.1161/01.res.87.3.241. [DOI] [PubMed] [Google Scholar]
- 35.Brunet-Rossinni AK. Reduced free-radical production and extreme longevity in the little brown bat (Myotis lucifugus) versus two non-flying mammals. Mech Ageing Dev. 2004;125(1):11–20. doi: 10.1016/j.mad.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 36.Brunet-Rossinni AK, Austad SN. Ageing studies on bats: a review. Biogerontology. 2004;5(4):211–22. doi: 10.1023/B:BGEN.0000038022.65024.d8. [DOI] [PubMed] [Google Scholar]
- 37.Spaulding CC, Walford RL, Effros RB. Calorie restriction inhibits the age-related dysregulation of the cytokines TNF-alpha and IL-6 in C3B10RF1 mice. Mech Ageing Dev. 1997;93(1–3):87–94. doi: 10.1016/s0047-6374(96)01824-6. [DOI] [PubMed] [Google Scholar]
- 38.Merry BJ. Oxidative stress and mitochondrial function with aging--the effects of calorie restriction. Aging Cell. 2004;3(1):7–12. doi: 10.1046/j.1474-9728.2003.00074.x. [DOI] [PubMed] [Google Scholar]
- 39.Chung HY, Kim HJ, Kim JW, Yu BP. The inflammation hypothesis of aging: molecular modulation by calorie restriction. Ann N Y Acad Sci. 2001;928:327–35. [PubMed] [Google Scholar]
- 40.Kim HJ, Jung KJ, Yu BP, Cho CG, Choi JS, Chung HY. Modulation of redox-sensitive transcription factors by calorie restriction during aging. Mech Ageing Dev. 2002;123(12):1589–95. doi: 10.1016/s0047-6374(02)00094-5. [DOI] [PubMed] [Google Scholar]
- 41.Chung HY, Kim HJ, Kim KW, Choi JS, Yu BP. Molecular inflammation hypothesis of aging based on the anti-aging mechanism of calorie restriction. Microsc Res Tech. 2002;59(4):264–72. doi: 10.1002/jemt.10203. [DOI] [PubMed] [Google Scholar]
- 42.Potente M, Fisslthaler B, Busse R, Fleming I. 11,12-Epoxyeicosatrienoic acid-induced inhibition of FOXO factors promotes endothelial proliferation by down-regulating p27Kip1. J Biol Chem. 2003;278(32):29619–25. doi: 10.1074/jbc.M305385200. [DOI] [PubMed] [Google Scholar]
- 43.Castrillon DH, Miao L, Kollipara R, Horner JW, DePinho RA. Suppression of ovarian follicle activation in mice by the transcription factor Foxo3a. Science. 2003;301(5630):215–8. doi: 10.1126/science.1086336. [DOI] [PubMed] [Google Scholar]
- 44.Furuyama T, Yamashita H, Kitayama K, Higami Y, Shimokawa I, Mori N. Effects of aging and caloric restriction on the gene expression of Foxo1, 3, and 4 (FKHR, FKHRL1, and AFX) in the rat skeletal muscles. Microsc Res Tech. 2002;59(4):331–4. doi: 10.1002/jemt.10213. [DOI] [PubMed] [Google Scholar]
- 45.Furukawa-Hibi Y, Yoshida-Araki K, Ohta T, Ikeda K, Motoyama N. FOXO forkhead transcription factors induce G(2)-M checkpoint in response to oxidative stress. J Biol Chem. 2002;277(30):26729–32. doi: 10.1074/jbc.C200256200. [DOI] [PubMed] [Google Scholar]
- 46.Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, et al. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature. 2003;424(6946):277–83. doi: 10.1038/nature01789. [DOI] [PubMed] [Google Scholar]
- 47.Modur V, Nagarajan R, Evers BM, Milbrandt J. FOXO proteins regulate tumor necrosis factor-related apoptosis inducing ligand expression. Implications for PTEN mutation in prostate cancer. J Biol Chem. 2002;277(49):47928–37. doi: 10.1074/jbc.M207509200. [DOI] [PubMed] [Google Scholar]
- 48.Furuyama T, Nakazawa T, Nakano I, Mori N. Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochem J. 2000;349(Pt 2):629–34. doi: 10.1042/0264-6021:3490629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abid MR, Guo S, Minami T, Spokes KC, Ueki K, Skurk C, et al. Vascular endothelial growth factor activates PI3K/Akt/forkhead signaling in endothelial cells. Arterioscler Thromb Vasc Biol. 2004;24(2):294–300. doi: 10.1161/01.ATV.0000110502.10593.06. [DOI] [PubMed] [Google Scholar]
- 50.Honda Y, Honda S. The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. Faseb J. 1999;13(11):1385–93. [PubMed] [Google Scholar]
- 51.Barsyte D, Lovejoy DA, Lithgow GJ. Longevity and heavy metal resistance in daf-2 and age-1 long-lived mutants of Caenorhabditis elegans. FASEB J. 2001;15(3):627–34. doi: 10.1096/fj.99-0966com. [DOI] [PubMed] [Google Scholar]
- 52.Nemoto S, Finkel T. Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science. 2002;295 (5564):2450–2. doi: 10.1126/science.1069004. [DOI] [PubMed] [Google Scholar]
- 53.Alcendor RR, Kirshenbaum LA, Imai S, Vatner SF, Sadoshima J. Silent information regulator 2alpha, a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes. Circ Res. 2004;95(10):971–80. doi: 10.1161/01.RES.0000147557.75257.ff. [DOI] [PubMed] [Google Scholar]
- 54.Burkle A, Beneke S, Muiras ML. Poly(ADP-ribosyl)ation and aging. Exp Gerontol. 2004;39(11–12):1599–1601. doi: 10.1016/j.exger.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 55.Tong WM, Cortes U, Wang ZQ. Poly(ADP-ribose) polymerase: a guardian angel protecting the genome and suppressing tumorigenesis. Biochim Biophys Acta. 2001;1552(1):27–37. doi: 10.1016/s0304-419x(01)00035-x. [DOI] [PubMed] [Google Scholar]
- 56.Burkle A. PARP-1: a regulator of genomic stability linked with mammalian longevity. Chembiochem. 2001;2(10):725–8. doi: 10.1002/1439-7633(20011001)2:10<725::AID-CBIC725>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 57.Grube K, Burkle A. Poly(ADP-ribose) polymerase activity in mononuclear leukocytes of 13 mammalian species correlates with species-specific life span. Proc Natl Acad Sci USA. 1992;89(24):11759–63. doi: 10.1073/pnas.89.24.11759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.von Kobbe C, Harrigan JA, May A, Opresko PL, Dawut L, Cheng WH, et al. Central role for the Werner syndrome protein/poly(ADP-ribose) polymerase 1 complex in the poly(ADP-ribosyl)ation pathway after DNA damage. Mol Cell Biol. 2003;23(23):8601–13. doi: 10.1128/MCB.23.23.8601-8613.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lebel M, Lavoie J, Gaudreault I, Bronsard M, Drouin R. Genetic cooperation between the Werner syndrome protein and poly(ADP-ribose) polymerase-1 in preventing chromatid breaks, complex chromosomal rearrangements, and cancer in mice. Am J Pathol. 2003;162(5):1559–69. doi: 10.1016/S0002-9440(10)64290-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Espejel S, Klatt P, Murcia JM, Martin-Caballero J, Flores JM, Taccioli G, et al. Impact of telomerase ablation on organismal viability, aging, and tumorigenesis in mice lacking the DNA repair proteins PARP-1, Ku86, or DNA-PKcs. J Cell Biol. 2004;167(4):627–38. doi: 10.1083/jcb.200407178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dantzer F, Giraud-Panis MJ, Jaco I, Ame JC, Schultz I, Blasco M, et al. Functional interaction between poly(ADP-Ribose) polymerase 2 (PARP-2) and TRF2: PARP activity negatively regulates TRF2. Mol Cell Biol. 2004;24(4):1595–607. doi: 10.1128/MCB.24.4.1595-1607.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pacher P, Mabley JG, Soriano FG, Liaudet L, Komjati K, Szabo C. Endothelial dysfunction in aging animals: the role of poly(ADP-ribose) polymerase activation. Br J Pharmacol. 2002;135(6):1347–50. doi: 10.1038/sj.bjp.0704627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pacher P, Vaslin A, Benko R, Mabley JG, Liaudet L, Hasko G, et al. A new, potent poly(ADP-ribose) polymerase inhibitor improves cardiac and vascular dysfunction associated with advanced aging. J Pharmacol Exp Ther. 2004;311(2):485–91. doi: 10.1124/jpet.104.069658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pacher P, Mabley JG, Liaudet L, Evgenov OV, Marton A, Hasko G, et al. Left ventricular pressure-volume relationship in a rat model of advanced aging-associated heart failure. Am J Physiol Heart Circ Physiol. 2004;287(5):H2132–7. doi: 10.1152/ajpheart.00405.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pacher P, Mabley JG, Soriano FG, Liaudet L, Szabo C. Activation of poly(ADP-ribose) polymerase contributes to the endothelial dysfunction associated with hypertension and aging. Int J Mol Med. 2002;9(6):659–64. [PubMed] [Google Scholar]
- 66.Pacher P, Liaudet L, Soriano FG, Mabley JG, Szabo E, Szabo C. The role of poly(ADP-ribose) polymerase activation in the development of myocardial and endothelial dysfunction in diabetes. Diabetes. 2002;51(2):514–21. doi: 10.2337/diabetes.51.2.514. [DOI] [PubMed] [Google Scholar]
- 67.Cortes U, Tong WM, Coyle DL, Meyer-Ficca ML, Meyer RG, Petrilli V, et al. Depletion of the 110-kilodalton isoform of poly(ADP-ribose) glycohydrolase increases sensitivity to genotoxic and endotoxic stress in mice. Mol Cell Biol. 2004;24(16):7163–78. doi: 10.1128/MCB.24.16.7163-7178.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hiona A, Leeuwenburgh C. Effects of age and caloric restriction on brain neuronal cell death/survival. Ann NY Acad Sci. 2004;1019:96–105. doi: 10.1196/annals.1297.018. [DOI] [PubMed] [Google Scholar]
- 69.Southan GJ, Szabo C. Poly(ADP-ribose) polymerase inhibitors. Curr Med Chem. 2003;10(4):321–40. doi: 10.2174/0929867033368376. [DOI] [PubMed] [Google Scholar]
- 70.Andreone TL, O'Connor M, Denenberg A, Hake PW, Zingarelli B. Poly(ADP-ribose) polymerase-1 regulates activation of activator protein-1 in murine fibroblasts. J Immunol. 2003;170(4):2113–20. doi: 10.4049/jimmunol.170.4.2113. [DOI] [PubMed] [Google Scholar]
- 71.Zingarelli B, Hake PW, O'Connor M, Denenberg A, Kong S, Aro-now BJ. Absence of poly(ADP-ribose)polymerase-1 alters nuclear factor-kappa B activation and gene expression of apoptosis regulators after reperfusion injury. Mol Med. 2003;9(5–8):143–53. doi: 10.2119/2003-00011.zingarelli. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ha HC, Hester LD, Snyder SH. Poly(ADP-ribose) polymerase-1 dependence of stress-induced transcription factors and associated gene expression in glia. Proc Natl Acad Sci USA. 2002;99(5):3270–5. doi: 10.1073/pnas.052712399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Carrillo A, Monreal Y, Ramirez P, Marin L, Parrilla P, Oliver FJ, et al. Transcription regulation of TNF-alpha-early response genes by poly(ADP-ribose) polymerase-1 in murine heart endothelial cells. Nucleic Acids Res. 2004;32(2):757–66. doi: 10.1093/nar/gkh239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Asai K, Kudej RK, Shen YT, Yang GP, Takagi G, Kudej AB, et al. Peripheral vascular endothelial dysfunction and apoptosis in old monkeys. Arterioscler Thromb Vasc Biol. 2000;20(6):1493–9. doi: 10.1161/01.atv.20.6.1493. [DOI] [PubMed] [Google Scholar]
- 75.Stewart KG, Zhang Y, Davidge ST. Aging increases PGHS-2-dependent vasoconstriction in rat mesenteric arteries. Hypertension. 2000;35(6):1242–7. doi: 10.1161/01.hyp.35.6.1242. [DOI] [PubMed] [Google Scholar]
- 76.Taddei S, Galetta F, Virdis A, Ghiadoni L, Salvetti G, Franzoni F, et al. Physical activity prevents age-related impairment in nitric oxide availability in elderly athletes. Circulation. 2000;101(25):2896–901. doi: 10.1161/01.cir.101.25.2896. [DOI] [PubMed] [Google Scholar]
- 77.Taddei S, Virdis A, Mattei P, Ghiadoni L, Gennari A, Fasolo CB, et al. Aging and endothelial function in normotensive subjects and patients with essential hypertension. Circulation. 1995;91(7):1981–7. doi: 10.1161/01.cir.91.7.1981. [DOI] [PubMed] [Google Scholar]
- 78.Taddei S, Virdis A, Ghiadoni L, Salvetti G, Bernini G, Magagna A, et al. Age-related reduction of NO availability and oxidative stress in humans. Hypertension. 2001;38(2):274–9. doi: 10.1161/01.hyp.38.2.274. [DOI] [PubMed] [Google Scholar]
- 79.Tschudi MR, Barton M, Bersinger NA, Moreau P, Cosentino F, Noll G, et al. Effect of age on kinetics of nitric oxide release in rat aorta and pulmonary artery. J Clin Invest. 1996;98(4):899–905. doi: 10.1172/JCI118872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cernadas MR, Sanchez de Miguel L, Garcia-Duran M, Gonzalez-Fernandez F, Millas I, Monton M, et al. Expression of constitutive and inducible nitric oxide synthases in the vascular wall of young and aging rats. Circ Res. 1998;83(3):279–86. doi: 10.1161/01.res.83.3.279. [DOI] [PubMed] [Google Scholar]
- 81.Yan ZQ, Sirsjo A, Bochaton-Piallat ML, Gabbiani G, Hansson GK. Augmented expression of inducible NO synthase in vascular smooth muscle cells during aging is associated with enhanced NF-kappaB activation. Arterioscler Thromb Vasc Biol. 1999;19(12):2854–62. doi: 10.1161/01.atv.19.12.2854. [DOI] [PubMed] [Google Scholar]