

Abstract
Aberrant centrosome numbers are detected in virtually all human cancers where they can contribute to chromosomal instability by promoting mitotic spindle abnormalities. Despite their widespread occurrence, the molecular mechanisms that underlie centrosome amplification are only beginning to emerge. Here, we present evidence for a novel regulatory circuit involved in centrosome overduplication that centers on RNA polymerase II (pol II). We found that human papillomavirus type 16 E7 (HPV-16 E7)- and hydroxyurea (HU)-induced centriole overduplication are abrogated by α-amanitin, a potent and specific RNA pol II inhibitor. In contrast, normal centriole duplication proceeded undisturbed in α-amanitin-treated cells. Centriole overduplication was significantly reduced by siRNA-mediated knock-down of CREB-binding protein (CBP), a transcriptional co-activator. We identified cyclin A2 as a key transcriptional target of RNA pol II during HU-induced centriole overduplication. Collectively, our results show that ongoing RNA pol II transcription is required for centriole overduplication whereas it may be dispensable for normal centriole duplication. Given that many chemotherapeutic agents function through inhibition of transcription, our results may help to develop strategies to target centrosome-mediated chromosomal instability for cancer therapy and prevention.
Introduction
Centrosomes are small cytoplasmic organelles that function as major microtubule organizing centers during interphase and mitosis in most animal and human cells (Bornens, 2002). A normal centrosome consists of a pair of centrioles, short microtubule cylinders, embedded in pericentriolar material (Urbani & Stearns, 1999). The single centrosome duplicates precisely once prior to mitosis in order to form the two spindle poles (Hinchcliffe & Sluder, 2001). Centrosome duplication is synchronized with the cell division cycle and starts in late G1 phase with the splitting of the two centrioles. During S phase, a newly synthesized centriole forms adjacent to each of the older centrioles in order to generate two new centrosomes. The molecular mechanisms involved in the regulation of this process as well as the structural basis of centriole reproduction, however, remain poorly characterized.
Numerical and structural centrosome anomalies are widespread in human cancers, where they can promote chromosomal instability through an increased risk for cell division errors (Doxsey, 2002; Duensing, 2005; Fukasawa, 2005; Nigg, 2002; Salisbury et al., 1999). In principal, aberrant centrosome numbers can develop through two distinct mechanisms: centrosome accumulation and centrosome overduplication (Duensing & Munger, 2001; Nigg, 2002). It is conceivable that an accumulation of centrosomes due to impaired cytokinesis accounts for a large number of centrosome aberrations in malignant tumors (Bunz et al., 1998). However, it has recently been shown that a bona fide centriole overduplication can be induced in cells arrested in early S phase using the ribonucleotide reductase inhibitor hydroxyurea (HU) or following overexpression of the human papillomavirus type 16 (HPV-16) E7 oncoprotein (Duensing et al., 2000; Guarguaglini et al., 2005). HPV-16 E7 is a major transforming oncoprotein of high-risk HPVs and disrupts the G1/S cell cycle checkpoint on multiple levels including binding and degradation of the retinoblastoma tumor suppressor protein (pRB) and inactivation of the cyclin-dependent-kinase (CDK) inhibitors p21Cip1 (Munger & Howley, 2002). Cells treated with HU or overexpressing HPV-16 E7 show a phenotype in which multiple immature daughter centrioles are frequently found adjacent to a single mature centriole (Guarguaglini et al., 2005).
Both, ectopic expression of HPV-16 E7 or treatment with HU cause an abnormally prolonged S phase of the cell division cycle (Balczon et al., 1995; Martin et al., 1998). S phase is governed by E2F-mediated gene transcription and previous studies have shown that E2F activity is involved in the induction of aberrant centrosome duplication by both HPV-16 E7 or HU (Duensing et al., 2000; Meraldi et al., 1999). However, the role of the transcriptional machinery itself in centrosome duplication and differences in the requirement for ongoing transcription between normal and aberrant centriole duplication have not been determined in detail in human cells.
We report here that both HPV-16 E7- and HU-induced centriole overduplication require ongoing RNA pol II-mediated gene transcription whereas normal centriole duplication was not sensitive to an inhibition of RNA pol II activity. Ongoing transcription/translation of cyclin A2 was indentified as a critical step in centriole overduplication induced by a prolonged S phase, which is in line with previous studies showing a requirement for CDK2 activity for centriole overduplication (Duensing et al., 2006). Various chemotherapeutic agents function through an inhibition of transcriptional activity and their effects on centriole duplication errors as therapeutic principle or basis for their use as cancer prevention agents deserves further investigation.
Results
α-Amanitin inhibits HPV-16 E7- and HU-induced centriole overduplication
We analyzed whether α-amanitin, a potent and specific inhibitor of RNA pol II derived from the green death cap mushroom Amanita phalloides (Gong et al., 2004), interferes with the effects of HPV-16 E7 and HU on centriole duplication. The mode of action of α-amanitin is not fully understood but there is evidence that it interacts with the largest RNA pol II subunit, Rpb1, and thereby inhibits transcription initiation and elongation (Bushnell et al., 2002). α-Amanitin blocks nucleotide incorporation into nascent mRNA transcripts in the nucleus whereas nucleolar RNA transcription carried out by other polymerases than RNA pol II remains unaffected (Fig. 1A).
Figure 1. RNA pol II-mediated gene transcription is required for HPV-16 E7- and HU-induced centriole overduplication.
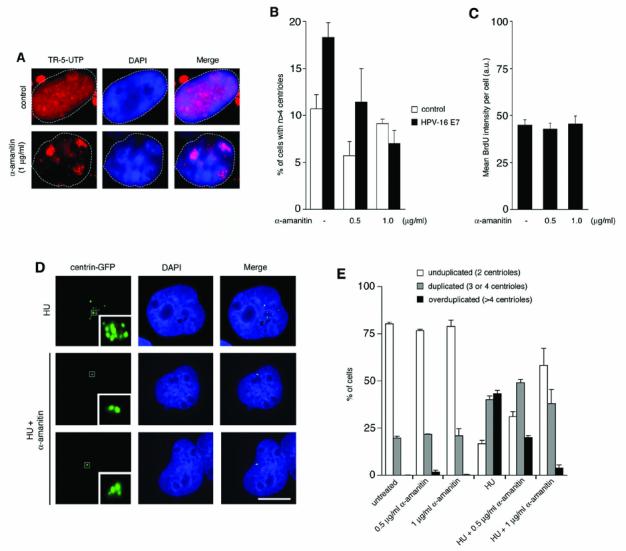
A. Abrogation of RNA pol II activity by 1 μg/ml α-amanitin as determined by Texas Red-5-UTP (TR-5-UTP) incorporation. Note the diffuse nuclear staining in a control cell (top panels) and residual nucleolar staining due to RNA polymerases other than pol II in a α-amanitin-treated cell (bottom panels).
B. Quantification of the percentage of cells displaying abnormal centriole numbers (more than four per cell) in HPV-16 E7 or empty vector (neo)-transfected U2OS/centrin-GFP cells either untreated or treated with the indicated concentrations of α-amanitin for 24 h. Only transfected cells as visualized by mitochondrial DsRED expression were assessed for centriole numbers. Each bar represents mean and standard error of four independent experiments with at least 100 cells analyzed per experiment.
C. Quantification of the mean BrdU signal intensity of U2OS cells either untreated or treated with the indicated concentrations of α-amanitin for 24 h. Arbitrary units (a.u.) are presented. Each bar represents mean and standard error of three counts of at least 50 cells from a representative experiment.
D. Fluorescence microscopy analysis of centriole duplication in U2OS/centrin-GFP cells treated for 72 h with 1 mM HU alone or in combination with 1 μg/ml α-amanitin. Overduplicated centrioles in a HU-arrested cell (top panels) are shown in comparison to a cell with unduplicated centrioles (middle panels) and a cell with normally duplicated centrioles (bottom panels). Nuclei stained with DAPI. Scale bar represents 10 μm.
E. Quantification of cells with unduplicated centrioles (two per cell, open bars), duplicated centrioles (three or four per cell, grey bars) or overduplicated centrioles (>4 per cell, black bars) in U2OS/centrin-GFP populations that were either untreated or treated with 1 mM HU alone or in combination with α-amanitin at the indicated concentrations for 72 h. At least four independent experiments were performed with similar results. Each bar represents mean and standard error of triple quantifications of at least 100 cells of a representative experiment.
Asynchronously growing U2OS cells stably expressing centrin-GFP to visualize individual centrioles (Duensing et al., 2001) were transiently transfected with HPV-16 E7 or empty vector control and subsequently treated with α-amanitin for 24 h (Fig. 1B). A dose-dependent decrease of cells with centriole overduplication was detected in α-amanitin-treated cells transfected with HPV-16 E7 from 18.3% in mock-treated controls to 11.4% in cells treated with 0.5 μg/ml α-amanitin (p>0.05) and 7% in cells treated with 1 μg/ml α-amanitin (p≤0.001; Fig. 1B). This inhibition was not due to a non-specific cell cycle effect since the α-amanitin concentrations used did not affect S phase progression as evidenced by BrdU incorporation (Fig. 1C). Our results indicate that HPV-16 E7 requires RNA pol II activity to induce abnormal centriole duplication and that this process can be effectively inhibited by α-amanitin.
To further dissect the role of RNA pol II in centriole duplication, U2OS/centrin-GFP cells were treated with 1 mM HU either alone or in combination with 0.5 μg/ml or 1 μg/ml α-amanitin for 72 h (Fig. 1D). No major changes of the normal centriole duplication status were detected between untreated cells and asynchronously growing cells treated with α-amanitin alone (Fig. 1E). Treatment with HU alone led to a significant increase of cells with overduplicated centrioles (more than four per cell) from 0% to 43.1% (p≤0.0001). Simultaneous treatment with HU and α-amanitin led to a significant reduction of cells with centriole overduplication from 43.1% in cells treated with HU alone to 20% in cells treated with HU and 0.5 μg/ml α-amanitin (2.2-fold; p≤0.0005) and 3.9% in cells treated with HU and 1 μg/ml α-amanitin (11.1-fold; p≤0.0005), respectively (Fig. 1E). Since HU-treated cells are cell cycle arrested in S phase and the α-amanitin concentrations used did not cause a cell cycle arrest (Fig. 1C), this decrease of numerical centriole abnormalities is unrelated to non-specific cell cycle effects of α-amanitin. The finding that not all cells treated with HU showed overduplication of centrioles is in line with previous reports (Balczon et al., 1995) and most likely based on the fact that a relaxation of an intrinsic block to reduplication (Wong & Stearns, 2003) occurs only in a proportion of cells (D'Assoro et al., 2004; Matsumoto et al., 1999; Meraldi et al., 1999).
A significant increase of cells with normally duplicated centrioles (3 or 4 per cell) was detected in cells treated with HU alone (40.1%) when compared to untreated controls (19.8%; 2.0-fold; p≤0.001) indicating that normal centriole duplication without overduplication continues in a fraction of HU-arrested cells (Fig. 1E). Simultaneous treatment of cells with HU and 0.5 μg/ml α-amanitin or 1 μg/ml α-amanitin did not lead to a decrease of cells displaying normally duplicated centrioles (Fig. 1E). These findings, together with the fact that α-amanitin alone does not reduce the number of cells with normally duplicated centrioles in asynchronously growing populations, suggest that RNA pol II transcriptional activity is required for HU-induced and HPV-16 E7-induced centriole overduplication but may be dispensable for normal centriole duplication.
Depletion of a transcriptional co-activator of RNA pol II inhibits centriole overduplication
We next asked whether indirect inhibition of RNA pol II transcriptional activity affects centriole duplication. Small interfering RNA (siRNA; Elbashir et al., 2001) was used to transiently knock-down protein expression of CREB-binding protein (CBP) in U2OS/centrin-GFP cells (Fig. 2A). CBP is a transcriptional co-activator and key regulator of RNA pol II-mediated gene transcription (Parvin & Young, 1998). CBP functions by connecting sequence-specific transcription factors to the basal transcription machinery (Janknecht & Hunter, 1996) thereby activating gene transcription.
Figure 2. Depletion of CBP inhibits HU-induced centriole overduplication.
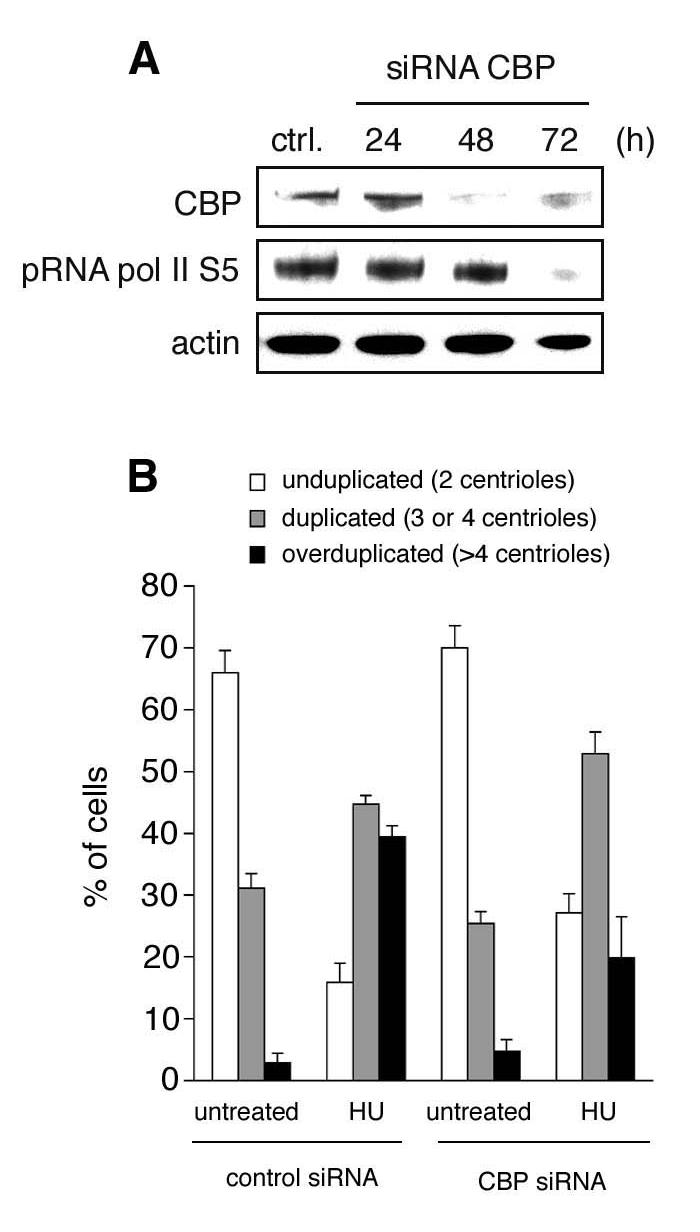
A. Immunoblot analysis of U2OS transfected with small interfering RNA (siRNA) duplexes to knock-down CBP expression. Note the decrease of the serine 5 phosphorylated form of RNA pol II. Immunoblot for actin is shown to demonstrate loading of equal amounts of protein.
B. Quantification of cells with unduplicated centrioles (two per cell, open bars), duplicated centrioles (three or four per cell, grey bars) or overduplicated centrioles (>4 per cell, black bars) in U2OS/centrin-GFP populations transfected either with control siRNA duplexes or siRNA duplexes targeting CBP and left untreated or treated with 1 mM HU for 72 h. Only transfected cells as visualized by mitochondrial DsRED expression were assessed for centriole numbers. At least four independent experiments were performed with similar results. Each bar represents mean and standard error of triple quantifications of at least 100 cells of a representative experiment.
RNA pol II becomes phosphorylated and dephosphorylated at the C-terminal domain (CTD) of Rpb1 during the transcription cycle. The CTD consists of tandem heptad repeats with the consensus sequence Y1S2P3T4S5P6S7. Phosphorylation at serine 5 is associated with transcription initiation whereas serine 2 phosphorylation is predominantly found during transcription elongation (Hahn, 2004). According to the function of CBP as transcriptional co-activator, we detected a reduction of serine 5 phosphorylation of RNA pol II following CBP knock-down (Fig. 2A).
Knock-down of CBP protein expression did not cause major changes of the centriole duplication status in asynchronously growing cells in comparison to cell transfected with control siRNAs (Fig. 2B). However, knock-down of CBP was found to inhibit the HU-induced increase of cells with centriole overduplication that was detected in control siRNA-transfected cells (39.4% versus 19.9%; p≤0.05; Fig. 2B). At the same time, a significant proportion of CBP siRNA-transfected and HU-treated cells showed normally duplicated centrioles (52.9%) in comparison to untreated cells (25.4; p≤0.005; Fig. 2B). These results indicate that knock-down of CBP inhibits centriole overduplication whereas normal centriole duplication is maintained.
Centriole overduplication requires ongoing protein synthesis
Prolonged inhibition of mRNA synthesis inevitably causes reduction of protein production (Braude, 1979). We therefore analyzed next the role of protein synthesis in aberrant and normal centrosome duplication using cycloheximide (CHX), which acts at the large ribosomal subunit and inhibits translation (Fig. 3). We found that simultaneous treatment of U2OS/centrin-GFP cells with 1 mM HU and 5 μg/ml CHX for 72 h efficiently reduced the proportion of cells with centriole overduplication from 45% to 4.7% (9.6-fold; p≤0.0005; Fig. 3A). In contrast to a previous report (Phillips & Rattner, 1976), we found an increase of cells with duplicated centrioles in asynchronously growing populations that were treated with CHX alone. Such increase was not seen in cells treated with α-amanitin alone or transfected with CBP siRNAs and may be related to the fact that CHX can induce a G2 cell cycle arrest leading to an accumulation of cells with duplicated centrioles. Since the reduction of protein synthesis may also affect the expression of centrin-GFP, we repeated these experiments in parental U2OS followed by immunofluorescence analysis for centrin (Uzawa et al., 1995). A similar reduction of HU-induced centriole overduplication was detected after simultaneous treatment of cells with HU and CHX (Fig. 3B). Our results show that centriole overduplication induced by HU does not only require ongoing RNA pol II transcription but also active protein synthesis.
Figure 3. Protein synthesis is required for aberrant centriole duplication.
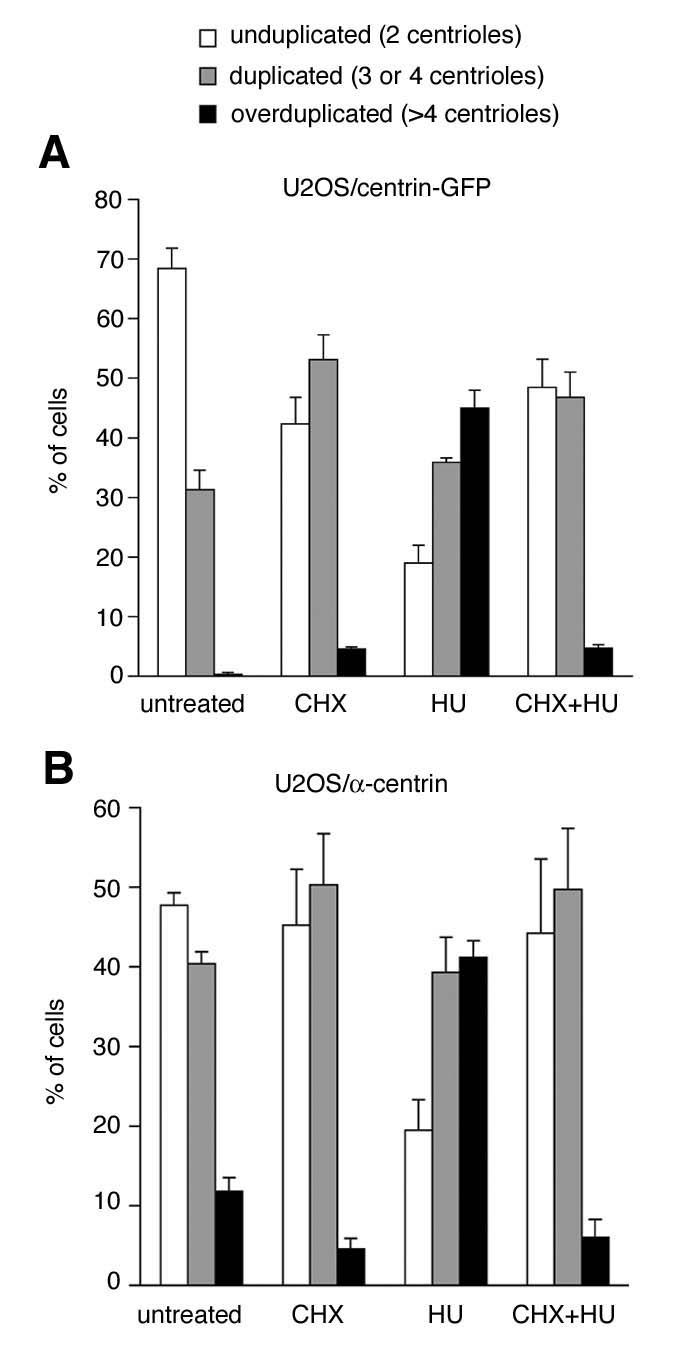
A. Quantification of cells with unduplicated centrioles (two per cell, open bars), duplicated centrioles (three or four per cell, grey bars) or overduplicated centrioles (>4 per cell, black bars) in U2OS/centrin-GFP populations that were either untreated or treated with 5 μg/ml cycloheximide (CHX), 1 mM HU or 5 μg/ml CHX in combination with 1 mM HU. All treatments were carried out over a 72 h time interval. At least four independent experiments were performed with similar results. Each bar represents mean and standard error of triple quantifications of at least 100 cells of a representative experiment.
B. Identical experiment as in A.; instead of U2OS/centrin-GFP cells, parental U2OS cells were used and analyzed by immunofluorescence microscopy for centrin to determine centriole duplication status
Transcription and translation of cyclin A2 is a critical step in HU-induced centriole overduplication
We therefore sought next to identify translational/transcriptional targets by which α-amanitin abrogates centriole overduplication. We have previously shown that CDK2 activity is critical for centriole overduplication induced by the HPV-16 E7 oncoprotein (Duensing et al., 2006; Duensing et al., 2004). CDK2 is activated by cyclin E in late G1 and by cyclin A during S phase (Malumbres & Barbacid, 2001). We therefore tested whether the α-amanitin-mediated inhibition of centriole overduplication was due to a decrease in cyclin A, cyclin E or CDK2 expression. Immunoblot analysis of HU-treated U2OS/centrin-GFP cells revealed a significant reduction of cyclin A expression after 72 h of co-treatment with α-amanitin (Fig. 4A). By contrast, protein expression levels of cyclin E and CDK2 were unaffected. This reduction of cyclin A expression was caused by impaired gene transcription as shown by the absence of cyclin A2 mRNA in cells simultaneously treated with HU and α-amanitin for 72 h (Fig. 4B). To directly test the role of cyclin A2 in HU-induced centriole overduplication, we performed siRNA experiments to deplete cells of cyclin A2 protein (Fig. 4C). HU-treated cells transfected with control siRNA duplexes showed an increase of cells with centriole overduplication from 16.3% in untreated populations to 40.8% after HU treatment for 72 h (p≤0.005) as expected. This increase of cells with overduplicated centrioles was abrogated in cells transfected with cyclin A2 siRNA duplexes when untreated cell populations (24.9%) were compared to populations treated with HU for 72 h (28.5%, p>0.05; Fig. 4D).
Figure 4. Cyclin A2 transcription is required for HU-induced centriole overduplication.
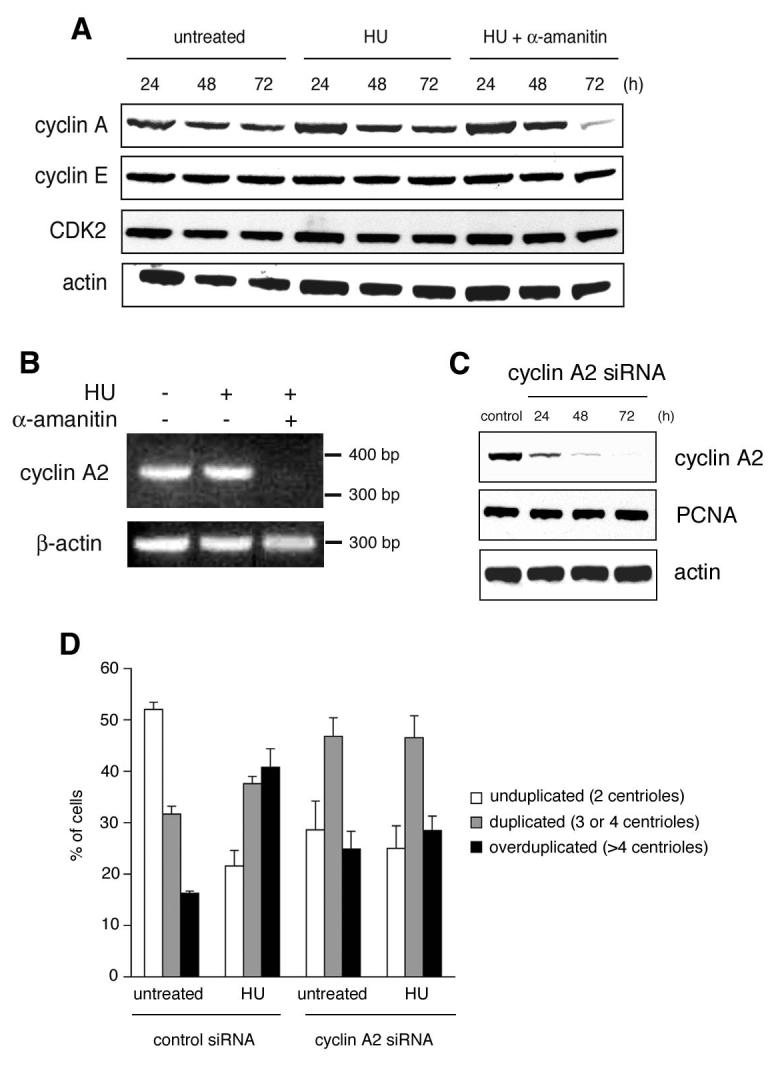
A. Immunoblot analysis of U2OS/centrin-GFP cells for cyclin A, cyclin E and CDK2 after treatment with 1 mM HU alone or in combination with 1 μg/ml α-amanitin or left untreated. Whole cell extracts were prepared at the indicated time points. Immunoblot for actin is shown as loading control.
B. RT-PCR analysis of cyclin A2 mRNA expression in U2OS/centrin-GFP cells either untreated or treated with 1 mM HU alone or in combination with 1 μg/ml α-amanitin.
C. Small interfering RNA (siRNA)-mediated knock-down of cyclin A2 protein expression in U2OS cells. Immunoblots for cyclin A2, PCNA and actin are shown.
D. Quantification of cells with unduplicated centrioles (two per cell, open bars), duplicated centrioles (three or four per cell, grey bars) or overduplicated centrioles (>4 per cell, black bars) in U2OS/centrin-GFP populations transfected either with control siRNA duplexes or siRNA duplexes targeting cyclin A2 and left untreated or treated with 1 mM HU for 72 h. Transfected cells were identified by co-transfection of a DsRED-encoding plasmid as transfection marker. At least four independent experiments were performed with similar results. Each bar represents mean and standard error of triple quantifications of at least 100 cells of a representative experiment.
The modest increase of cells with duplicated or overduplicated centrioles following transfection of cells with siRNA against cyclin A2 in comparison to control siRNA-transfected cells may be related to an accumulation of cells in G2 phase of the cell division cycle (Yam et al., 2002). It has previously been shown that a prolonged G2 arrest can stimulate centriole overduplication (Dodson et al., 2004). We therefore sought to determine whether a prolonged G2 arrest is involved in the changes of centriole numbers in cyclin A2 siRNA-transfected cells in the absence of HU. Such an arrest would have biased our finding that HU has no effect on centriole overduplication in cyclin A2-deficient cells because cells may have undergone centriole overduplication prior to HU treatment and this may be the reason why no additional overduplication was detected. The levels of cyclin B rise in G2 phase of the cell division cycle and the protein is imported into the nucleus at the end of G2. It localizes to the mitotic spindle apparatus during metaphase and is degraded by the anaphase-promoting complex (APC) at the metaphase-anaphase transition. Using immunofluorescence for cyclin B1, we found that cells transfected with cyclin A2 siRNA that contain overduplicated centrioles show undetectable levels of cyclin B1 (Fig. 5D). These results indicate that a prolonged G2 arrest does not play a major role in centriole overduplication under our experimental conditions. Another possibility through which depletion of cyclin A2 may interfere with centriole duplication is a cell cycle arrest in G1 or S during the 24 h period before HU was added to the cell culture media. However, no decrease in the expression level of proliferation cell nuclear antigen (PCNA), a widely used S phase marker, was detected in cyclin A2-depleted cells (Fig. 4C). These control experiments show that the transfection of cells with siRNA duplexes targeting cyclin A2 at 24 h before exposure to HU does not lead to a cell cycle arrest. Lastly, depletion of cyclin A2 has been suggested to cause changes in ploidy (Mihaylov et al., 2002). However, there is no indication so far that cells with altered ploidy cannot undergo additional rounds of centriole duplication (Duensing et al., 2001; Guarguaglini et al., 2005; Mantel et al., 1999). We therefore interpret the abrogation of HU-induced centriole overduplication in cyclin A2 siRNA-transfected cells (Fig. 4D) as a result of a depletion of a critical factor for centriole overduplication under conditions of a prolonged S phase.
Figure 5. Changes in centriole numbers in cyclin A2-depleted cells are not due to a G2 arrest.
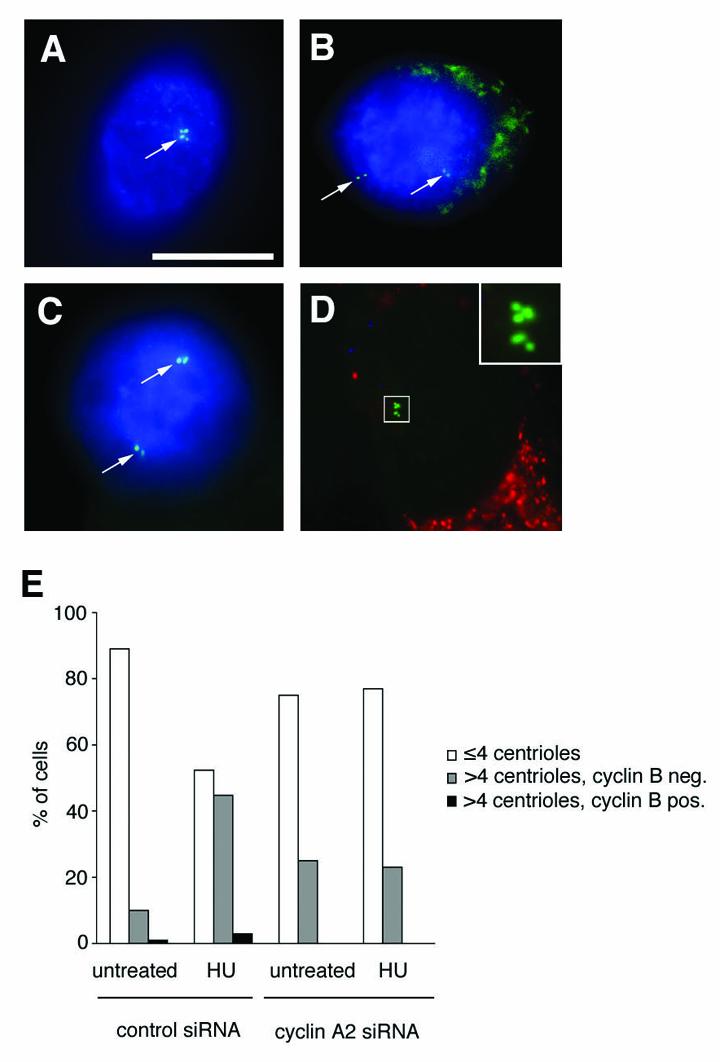
A-D. Immunofluorescence analysis of U2OS/centrin-GFP cells for cyclin B1 (AMCA; blue) either untransfected (A-C) or transfected with cyclin A2 siRNA (D). Note the expression of cyclin B1 in a cell in G2 as indicated by duplicated centrioles (A; arrow), a prometaphase cell with separated pairs of centrioles (B, arrows) and in a metaphase cell with opposing centriole pairs (C; arrows). Note the absence of cyclin B1 expression in a cell showing abnormal centriole numbers (insert) following transfection with cyclin A2 siRNAs. A plasmid encoding DsRED with a mitochondrial target sequence was used as transfection marker. Experiments were performed without a nuclear stain. Scale bar indicates 10 μm.
E. Quantification of cells with normal centriole numbers (≤4 per cell, open bars) in comparison to cells with overduplicated centrioles (> 4 per cell) in the absence of cyclin B1 expression (grey bars) or overduplicated centrioles in the presence of cyclin B1 expression (black bars) in U2OS/centrin-GFP populations transfected either with control siRNA duplexes or siRNA duplexes targeting cyclin A2 and left untreated or treated with 1 mM HU for 72 h. A DsRED-encoding plasmid was used as transfection marker. At least 100 cells were analyzed in a representative experiment.
Discussion
Collectively, the results presented here show that overduplication of centrioles requires active RNA pol II-mediated transcriptional activity. In contrast, normal centriole duplication may not depend on ongoing RNA pol II transcription. Cyclin A2 was identified as a critical transcriptional/translational target in HU-induced centriole overduplication, which is in line with previous results (Meraldi et al., 1999). Although cyclin A2 may not be the only transcriptional target relevant for this process, our results emphasize that ongoing cyclin A2 transcription is critical for centriole overduplication under conditions of an S phase arrest. The fact that cyclin A expression is predominantly affected by α-amanitin in contrast to cyclin E may be related to the time of the HU-induced cell cycle arrest in S phase when cyclin A mRNA is synthesized and does not preclude that cyclin E plays a role in centriole overduplication (Hinchcliffe et al., 1999; Mussman et al., 2000). It is likely that cyclin A promotes centriole overduplication in S phase-arrested cells through a continued stimulation of CDK2 activity since CDK2 has been shown to be essential for this process (Duensing et al., 2006).
Abnormal levels of cyclin A transcripts are frequently detected in human cancers and can arise through transcriptional or post-transcriptional mechanisms (Yam et al., 2002). Our results showing that cyclin A transcription is necessary for abnormal centriole duplication lend support to the notion that unscheduled cyclin A mRNA expression may be a contributing factor to tumorigenesis and is not only a consequence of enhanced proliferation or increased ploidy (Faivre et al., 2002). It is noteworthy that the HPV-16 E7 oncoprotein can disrupt the transcriptional control of cyclin A through multiple mechanisms including aberrant E2F-mediated transcription following degradation of pRB and pRB family members (Munger et al., 2001). In addition, HPV-16 E7 has been found to directly interact with cyclin A (Tommasino et al., 1993) and to activate the cyclin A promoter (Caldeira et al., 2000). The ability of HPV-16 E7 to inactivate the CDK inhibitors p21Cip1 and p27Kip1 may further enhance cyclin A-associated kinase activity (Munger et al., 2001).
Early studies in CHO cells have demonstrated a requirement for the presence of the cell nucleus for the formation of new centrioles and it has been suggested that an RNA component may be critical in this process (Kuriyama & Borisy, 1981). Subsequent experiments in sea urchin eggs found that centrosome reproduction can proceed entirely independent from nuclear transcription (Sluder et al., 1986). Results presented here suggest that transcription is dispensable for the initiation and/or completion of normal centriole duplication but it is required for the formation of supernumerary centrioles in HU-arrested cells. Although structural and regulatory factors may be present in sufficient amounts to promote multiple rounds of duplication, the transcription and translation of cyclin A2 may be a crucial step. Cyclin A stimulates CDK2 activity and this function may be required to overcome the previously proposed centriole-intrinsic block to reduplication (Wong & Stearns, 2003). The finding that HU-arrested cells frequently show excessive numbers of centrioles whereas other cells in the same population arrest after a normal round of duplication supports this notion.
HPV-16 E7-induced centriole overduplication requires CDK2 activity (Duensing et al., 2006; Duensing et al., 2004) and, as shown here, ongoing RNA pol II transcription. These findings highlight that the HPV-16 E7-induced generation of immature daughter centrioles (Guarguaglini et al., 2005) is an active process with specific molecular requirements and not simply a consequence of cytokinesis defects, ploidy alterations or aberrant centriole splitting (Duensing et al., 2001). It has been shown that other events leading to a deregulation of CDK2 activity such as loss of p16IKN4A can also stimulate abnormal centriole numbers following S phase arrest (McDermott et al., 2006). However, the mechanisms may differ fundamentally from that in HPV-16 E7-expressing cells in which a role of aberrant centriole splitting in the formation of supernumerary centrioles could not be substantiated.
Although our results do not prove that increased transcription drives aberrant centriole duplication, they imply that a coordinated downregulation of RNA pol II transcription in normal cells may provide a failsafe mechanism that limits centriole duplication to one round per S phase. RNA pol II-mediated transcription is repressed at mitotic onset involving transcription termination factor 2 (TTF2), the human homologue of Drosophila lodestar (Girdham & Glover, 1991), and it is noteworthy that impaired function of TTF2 can cause multipolar mitoses (Jiang et al., 2004).
Taken together, our results establish RNA pol II-mediated gene transcription as a novel regulatory mechanism involved in aberrant centriole duplication. Many chemotherapeutic agents function through global inhibition of transcription (Derheimer et al., 2005) and findings presented here suggest that a prevention of centrosome overduplication may contribute to the mode of action of these drugs. The use of such compounds as preventive agents to suppress centrosome-mediated cell division errors, for example in pre-neoplastic HPV-positive lesions or in other pre-cancers in which multipolar mitoses are common, warrants further exploration.
Material and Methods
Cell culture, inhibitor treatment and cell transfections
The human U2OS osteosarcoma cell line was obtained from ATCC and cultured in Dulbecco's modified Eagle medium (DMEM; Cambrex) supplemented with 10% fetal bovine serum (FBS; Mediatech), 50 U/ml penicillin and 50 μg/ml streptomycin (both BioWhittaker). Populations of U2OS cells stably expressing centrin-GFP (kindly provided by Michel Bornens, Institut Curie, Paris) were generated as previously described (Duensing et al., 2001). Cells were treated with α-amanitin (Sigma) dissolved in dH2O at the indicated concentrations and time intervals. Hydroxyurea (HU; Calbiochem) was dissolved in dH2O and cells were treated with a 1 mM concentration for 72 h in all experiments. Cycloheximide (CHX; Calbiochem) was dissolved in dH2O and cells were treated at the indicated concentrations for 72 h. In all experiments, cells were plated into 60 mm tissue culture dishes containing four 15 mm round coverslips. After treatment, centrioles were either detected by centrin-GFP signals or following immunofluorescence staining for centrin (see below). For transient transfection experiments, cells were plated into 60 mm plates containing coverslips and grown to 50% confluency before transfection with Fugene 6 (Invitrogen) transfection reagent and 2 μg of plasmid DNA. A pCMVneo-based plasmid containing HPV-16 E7 was kindly provided by Karl Münger (Channing Laboratory, Brigham & Women's Hospital, Boston, MA). A plasmid encoding for DsRED fluorescent protein (Clontech) targeted to mitochondria was used as transfection control for both overexpression experiments and siRNA experiments (see below).
Immunological Methods
For immunoblot analyses, cell lysates were prepared by scraping cells into lysis buffer (1% NP-40, 50 mM Tris-HCl pH 8.0, 100 mM sodium fluoride, 30 mM sodium pyrophosphate, 2 mM sodium molybdate, 5 mM EDTA, 2 mM sodium orthovanadate) containing protease inhibitors (10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 μM phenylmethylsulfonyl fluoride). Lysates were incubated for 1 h with shaking at 4°C and then cleared by centrifugation for 30 min at 13,000 rpm at 4°C. Protein concentrations were determined by the Bradford assay (Biorad). 30 μg of protein were loaded on a 4-12% Bis-Tris gel (Invitrogen) and blotted onto a nitrocellulose membrane. Primary antibodies used were directed against CBP (Santa Cruz), phospho-serine 5 of the CTD of RNA pol II (pRNA pol II S5; Covance), cyclin A (Novocastra), cyclin E (Santa Cruz), CDK2 (Santa Cruz), PCNA (Santa Cruz) and actin (Sigma).
For immunofluoresence analysis, cells grown on coverslips were fixed in 4% paraformaldehyde/PBS for 15 min at room temperature, washed in PBS and permeabilized with 1% Triton-X-100 in PBS for 15 min. After blocking in 10% normal donkey serum (Jackson Immunoresearch), cells were incubated with an anti-centrin monoclonal antibody (clone 20H5; kindly provided by Jeffrey L. Salisbury, Mayo Clinic, Rochester, MN) at a 1:5000 dilution overnight at 4°C. The next morning, cells were warmed up at 37°C for 2 h, washed in PBS and incubated with a FITC-conjugated anti-mouse secondary antibody (Jackson Immunoresearch) at a 1:200 dilution for 2 h and mounted with DAPI. For detection of cyclin B1, U2OS/centrin-GFP cells were fixed and permeabilized as described above, incubated with an anti-cyclin B1 monoclonal antibody (Labvision) overnight at 4°C followed by an AMCA-labeled secondary antibody (Jackson Immunoresearch) at a 1:100 dilution for 3 h at 37°C. Cells were analyzed using an Olympus AX70 epifluorescence microscope equipped with a SpotRT digital camera.
siRNA experiments
Synthetic RNA duplexes targeting CBP or cyclin A2 and control siRNAs were obtained commercially (SmartPool, Dharmacon). For each experiment, U2OS/centrin-GFP cells were grown on coverslips in 60 mm tissue culture dishes with 2 ml DMEM free of antibiotics. Cells were transfected with 12 μl of 20 μM annealed RNA duplexes using Oligofectamine (Life Technologies) transfection reagent.
UTP incorporation assay
U2OS cells grown on coverslips were overlaid with ChromaTide Texas Red-5-UTP (Molecular Probes) and permeabilized with glass beads as previously described (Hoffelder et al., 2004). After incubation for 30 min at 37°C, cells were fixed in 95% ethanol, counterstained with DAPI (Vector) and analyzed by fluorescence microscopy.
RT-PCR
Total RNA was isolated using the RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions. One-step RT-PCR was performed using the Titan OneTube RT-PCR System (Roche) according to the manufacturer's protocol. Cyclin A2 transcripts were amplified from 500 ng of total RNA using cyclin A2-specific PCR primers (Operon) as previously described (Bai et al., 2005). Cycling conditions included an initial denaturation at 94°C for 2 minutes, followed by 35 cycles at 94°C for 10 seconds, 63°C for 30 seconds, 68°C for 45 seconds, and a final extension of 68°C for 7 minutes. β-actin was amplified as control. PCR products were visualized on a 2% agarose gel stained with ethidium bromide.
5-Bromo-2'-deoxy-uridine (BrdU) labeling
S phase progression was determined using a BrdU incorporation assay according to the manufacturer's protocol (Roche Molecular Biochemicals) followed by analysis of the mean fluorescence intensity per cell using NIH ImageJ software.
Statistical analysis
Statistical significance was assessed using Student's t test for independent samples. P values ≤0.05 were considered statistically significant.
Acknowledgements
We are grateful to Michel Bornens, Karl Münger and Jeffrey L. Salisbury for sharing important reagents and William Saunders for helpful suggestions. This work was supported by Public Health Service Grant NIH/NCI R01 CA112598 (to S.D.).
References
- Bai VU, Cifuentes E, Menon M, Barrack ER, Reddy GP. J. Cell Physiol. 2005;204:381–387. doi: 10.1002/jcp.20422. [DOI] [PubMed] [Google Scholar]
- Balczon R, Bao L, Zimmer WE, Brown K, Zinkowski RP, Brinkley BR. J. Cell Biol. 1995;130:105–115. doi: 10.1083/jcb.130.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornens M. Curr. Opin. Cell Biol. 2002;14:25–34. doi: 10.1016/s0955-0674(01)00290-3. [DOI] [PubMed] [Google Scholar]
- Braude PR. Dev. Biol. 1979;68:440–452. doi: 10.1016/0012-1606(79)90216-1. [DOI] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- Bushnell DA, Cramer P, Kornberg RD. Proc. Natl. Acad. Sci. USA. 2002;99:1218–1222. doi: 10.1073/pnas.251664698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldeira S, de Villiers EM, Tommasino M. Oncogene. 2000;19:821–826. doi: 10.1038/sj.onc.1203375. [DOI] [PubMed] [Google Scholar]
- D'Assoro AB, Busby R, Suino K, Delva E, Almodovar-Mercado GJ, Johnson H, Folk C, Farrugia DJ, Vasile V, Stivala F, Salisbury JL. Oncogene. 2004;23:4068–4075. doi: 10.1038/sj.onc.1207568. [DOI] [PubMed] [Google Scholar]
- Derheimer FA, Chang CW, Ljungman M. Eur. J. Cancer. 2005;41:2569–2576. doi: 10.1016/j.ejca.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Dodson H, Bourke E, Jeffers LJ, Vagnarelli P, Sonoda E, Takeda S, Earnshaw WC, Merdes A, Morrison C. EMBO J. 2004;23:3864–3873. doi: 10.1038/sj.emboj.7600393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doxsey S. Mol. Cell. 2002;10:439–440. doi: 10.1016/s1097-2765(02)00654-8. [DOI] [PubMed] [Google Scholar]
- Duensing A, Liu Y, Tseng M, Malumbres M, Barbacid M, Duensing S. Oncogene. 2006 Jan 9; doi: 10.1038/sj.onc.1209310. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duensing S. Cell Biol. Int. 2005;29:352–359. doi: 10.1016/j.cellbi.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Duensing S, Duensing A, Crum CP, Munger K. Cancer Res. 2001;61:2356–2360. [PubMed] [Google Scholar]
- Duensing S, Duensing A, Lee DC, Edwards KM, Piboonniyom S, Manuel E, Skaltsounis L, Meijer L, Munger K. Oncogene. 2004;23:8206–8215. doi: 10.1038/sj.onc.1208012. [DOI] [PubMed] [Google Scholar]
- Duensing S, Lee LY, Duensing A, Basile J, Piboonniyom S, Gonzalez S, Crum CP, Munger K. Proc. Natl. Acad. Sci. USA. 2000;97:10002–10007. doi: 10.1073/pnas.170093297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duensing S, Munger K. Biochim. Biophys. Acta. 2001;2:M81–88. doi: 10.1016/s0304-419x(00)00025-1. [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Faivre J, Frank-Vaillant M, Poulhe R, Mouly H, Jessus C, Brechot C, Sobczak-Thepot J. Oncogene. 2002;21:1493–1500. doi: 10.1038/sj.onc.1205215. [DOI] [PubMed] [Google Scholar]
- Fukasawa K. Cancer Lett. 2005;230:6–19. doi: 10.1016/j.canlet.2004.12.028. [DOI] [PubMed] [Google Scholar]
- Girdham CH, Glover DM. Genes Dev. 1991;5:1786–1799. doi: 10.1101/gad.5.10.1786. [DOI] [PubMed] [Google Scholar]
- Gong XQ, Nedialkov YA, Burton ZF. J. Biol. Chem. 2004;279:27422–27427. doi: 10.1074/jbc.M402163200. [DOI] [PubMed] [Google Scholar]
- Guarguaglini G, Duncan PI, Stierhof YD, Holmstrom T, Duensing S, Nigg EA. Mol. Biol. Cell. 2005;16:1095–1107. doi: 10.1091/mbc.E04-10-0939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn S. Nat. Struct. Mol. Biol. 2004;11:394–403. doi: 10.1038/nsmb763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinchcliffe EH, Li C, Thompson EA, Maller JL, Sluder G. Science. 1999;283:851–854. doi: 10.1126/science.283.5403.851. [DOI] [PubMed] [Google Scholar]
- Hinchcliffe EH, Sluder G. Genes Dev. 2001;15:1167–1181. doi: 10.1101/gad.894001. [DOI] [PubMed] [Google Scholar]
- Hoffelder DR, Luo L, Burke NA, Watkins SC, Gollin SM, Saunders WS. Chromosoma. 2004;112:389–97. doi: 10.1007/s00412-004-0284-6. [DOI] [PubMed] [Google Scholar]
- Janknecht R, Hunter T. Nature. 1996;383:22–23. doi: 10.1038/383022a0. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Liu M, Spencer CA, Price DH. Mol. Cell. 2004;14:375–385. doi: 10.1016/s1097-2765(04)00234-5. [DOI] [PubMed] [Google Scholar]
- Kuriyama R, Borisy GG. J. Cell Biol. 1981;91:814–821. doi: 10.1083/jcb.91.3.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M, Barbacid M. Nat. Rev. Cancer. 2001;1:222–231. doi: 10.1038/35106065. [DOI] [PubMed] [Google Scholar]
- Mantel C, Braun SE, Reid S, Henegariu O, Liu L, Hangoc G, Broxmeyer HE. Blood. 1999;93:1390–1398. [PubMed] [Google Scholar]
- Martin LG, Demers GW, Galloway DA. J. Virol. 1998;72:975–985. doi: 10.1128/jvi.72.2.975-985.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto Y, Hayashi K, Nishida E. Curr. Biol. 1999;9:429–432. doi: 10.1016/s0960-9822(99)80191-2. [DOI] [PubMed] [Google Scholar]
- McDermott KM, Zhang J, Holst CR, Kozakiewicz BK, Singla V, Tlsty TD. PLoS Biol. 2006;4:e51. doi: 10.1371/journal.pbio.0040051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraldi P, Lukas J, Fry AM, Bartek J, Nigg EA. Nat. Cell Biol. 1999;1:88–93. doi: 10.1038/10054. [DOI] [PubMed] [Google Scholar]
- Mihaylov IS, Kondo T, Jones L, Ryzhikov S, Tanaka J, Zheng J, Higa LA, Minamino N, Cooley L, Zhang H. Mol. Cell. Biol. 2002;22:1868–1880. doi: 10.1128/MCB.22.6.1868-1880.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger K, Basile JR, Duensing S, Eichten A, Gonzalez SL, Grace M, Zacny VL. Oncogene. 2001;20:7888–7898. doi: 10.1038/sj.onc.1204860. [DOI] [PubMed] [Google Scholar]
- Munger K, Howley PM. Virus Res. 2002;89:213–228. doi: 10.1016/s0168-1702(02)00190-9. [DOI] [PubMed] [Google Scholar]
- Mussman JG, Horn HF, Carroll PE, Okuda M, Tarapore P, Donehower LA, Fukasawa K. Oncogene. 2000;19:1635–1646. doi: 10.1038/sj.onc.1203460. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Nat. Rev. Cancer. 2002;2:1–11. doi: 10.1038/nrc924. [DOI] [PubMed] [Google Scholar]
- Parvin JD, Young RA. Curr. Opin. Genet. Dev. 1998;8:565–570. doi: 10.1016/s0959-437x(98)80012-9. [DOI] [PubMed] [Google Scholar]
- Phillips SG, Rattner JB. J. Cell Biol. 1976;70:9–19. doi: 10.1083/jcb.70.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salisbury JL, Whitehead CM, Lingle WL, Barrett SL. Biol. Cell. 1999;91:451–460. [PubMed] [Google Scholar]
- Sluder G, Miller FJ, Rieder CL. J. Cell Biol. 1986;103:1873–1881. doi: 10.1083/jcb.103.5.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tommasino M, Adamczewski JP, Carlotti F, Barth CF, Manetti R, Contorni M, Cavalieri F, Hunt T, Crawford L. Oncogene. 1993;8:195–202. [PubMed] [Google Scholar]
- Urbani L, Stearns T. Curr. Biol. 1999;9:R315–317. doi: 10.1016/s0960-9822(99)80201-2. [DOI] [PubMed] [Google Scholar]
- Uzawa M, Grams J, Madden B, Toft D, Salisbury JL. Dev. Biol. 1995;171:51–59. doi: 10.1006/dbio.1995.1259. [DOI] [PubMed] [Google Scholar]
- Wong C, Stearns T. Nat. Cell Biol. 2003;5:539–544. doi: 10.1038/ncb993. [DOI] [PubMed] [Google Scholar]
- Yam CH, Fung TK, Poon RY. Cell. Mol. Life Sci. 2002;59:1317–1326. doi: 10.1007/s00018-002-8510-y. [DOI] [PMC free article] [PubMed] [Google Scholar]