

Abstract
Expression of the chemokine receptor CXCR4 by tumor cells promotes metastasis, possibly by activating pro-survival signals that render cancer cells resistant to immune attack. Inhibition of CXCR4 with a peptide antagonist, T22, blocks metastatic implantation of CXCR4-transduced B16 (CXCR4-luc-B16) melanoma cells in lung, but not the outgrowth of established metastases, raising the question of how T22 can best be used in a clinical setting. Herein, whereas the treatment of CXCR4-luc-B16 cells in vitro with the CXCR4 ligand CXCL12 did not reduce killing induced by cisplatin or cyclophosphamide, CXCL12 markedly reduced Fas-dependent killing by gp100-specific (pmel-1) CD8+ T cells. T22 pretreatment restored sensitivity of CXCR4-luc-B16 cells to pmel-1 killing, even in the presence of CXCL12. Two immune-augmenting regimens were used in combination with T22 to treat experimental lung metastases. First, low-dose cyclophosphamide treatment (100 mg/kg) on day 5 in combination with T22 (days 4–7) yielded a ~70% reduction of B16 metastatic tumor burden in the lungs compared with cyclophosphamide treatment alone (P < 0.001). Furthermore, whereas anti–CTL antigen 4 (CTLA4) monoclonal antibody (mAb; or T22 treatment) alone had little effect on established B16 metastases, pretreatment with T22 (in combination with anti-CTLA4 mAb) resulted in a 50% reduction in lung tumor burden (P = 0.02). Thus, in vitro, CXCR4 antagonism with T22 renders B16 cells susceptible to killing by antigen-specific T cells. In vivo, T22 synergizes with cyclophosphamide or anti-CTLA4 mAb in the treatment of established lung metastases, suggesting a novel strategy for augmenting the efficacy of immunotherapy.
Introduction
Whereas great strides have been made in the clinical immunotherapy for advanced melanoma (1, 2), the majority of patients obtain only partial remissions (3). In animals, experimental immunotherapy for cancer is somewhat better, but a combination of immunotherapeutic treatments, sometimes accompanied by autoimmune toxicity (4, 5), must often be initiated to order to achieve complete (or near complete) response. For example, targeting the T-cell inhibitory receptor CTL antigen 4 (CTLA4; ref. 6) with monoclonal antibodies (mAb) is relatively ineffective against established B16 tumors although it is much more active in combination with tumor vaccine therapy (4, 7–9). Thus, targeting key survival pathways in cancer cells, particularly those that allow tumor cells to survive host immunologic attack, is an attractive approach for increasing the efficacy of immunotherapy without subjecting patients to a greater risk of autoimmunity.
Chemokine receptor–mediated activation of prosurvival pathways such as Akt has been implicated as a mechanism by which cancer cells can evade host immunity (10, 11) and increase their metastatic properties (12). CXCR4 is the chemokine receptor most frequently upregulated by hematopoietic and solid malignancies, including those of the breast, prostate, lung, and colon (13). We showed that CXCR4 is expressed by primary human melanoma in situ and by melanoma metastases in the lung (14), whereas others showed that the expression of CXCR4 correlates with poor patient prognosis (15). Overexpression of CXCR4 increased murine B16 melanoma lung metastases by >6-fold (14) and inhibition of CXCR4 with blocking antibodies resulted in markedly decreased experimental lung metastases following i.v. inoculation of breast cancer cells (12).
Activation of CXCR4 via CXCL12 leads to enhancement of melanoma survival and to up-regulation of integrin adhesion (16, 17). Whereas the latter function may increase the ability of circulating tumor cells to attach and possibly extravasate at sites of metastasis (17), CXCR4 may also be critical for the outgrowth of tumor cells into micrometastases or macrometastases following extravasation from the blood vessel through mechanisms that are currently unclear (18). Our studies indicated that treatment of CXCR4-luc-B16 cells with the CXCR4 antagonist T22 (14, 19, 20) markedly reduced metastatic implantation but did not alter the outgrowth of established metastases in the lung (17).
Another chemokine receptor, CCR10, may enhance melanoma metastasis to the skin (10). Our prior studies showed that, in the presence of the skin-derived chemokine CCL27, CCR10-expressing B16 murine melanoma cells were resistant in vitro to killing induced by Fas cross-linking and by gp100 peptide–specific CTL (10). Furthermore, mice inoculated in skin with B16 murine melanoma cells expressing both luciferase and CCR10 formed tumors, whereas mice inoculated with tumor cells expressing luciferase alone did not. Of note, antibodies that neutralized CCL27 (synthesized constitutively in skin; ref. 21) blocked tumor formation by CCR10-luciferase-B16 cells, suggesting that activation of CCR10 may contribute to the ability of melanoma cells to evade the immune system and, thus, form tumors in skin (10).
These studies raised a series of questions that we now address. First, does activation through CXCR4 also lead to increased resistance of B16 melanoma cells to apoptosis? If so, is CXCR4-mediated enhancement in survival specific to killing mechanisms used by cytolytic T cells (i.e., Fas death pathway) or does CXCR4 activation lead to resistance to killing that is mediated through other means, including chemotherapy agents? Lastly, if CXCR4 activation through its ligand CXCL12 mediates resistance to immune-mediated killing, would inhibition of CXCR4 with a specific peptide antagonist render cancer cells more susceptible to gp100-specific CTL or to systemic therapy that augments host antitumor responses?
Our current in vitro results indicate that CXCR4 activation protects B16 cells from antigen-specific CTL and that inhibition of CXCR4 with T22 results in the restoration of the sensitivity of B16 cells to CTL-mediated killing. In addition, we find that CXCR4 blockade with T22 enhances the efficacy of both anti-CTLA4 mAb and cyclophosphamide in the treatment of established B16 lung metastases. In neither model did T22 treatment alone result in significant a significant reduction of tumor burden in the lung, suggesting that T22 sensitizes the B16 cells for more efficient killing by appropriately triggered host immune cells. Targeted approaches that selectively render tumor cells more sensitive to killing in the setting of immunotherapy may be of great use in treating cancer patients.
Materials and Methods
Animals, Cell Lines, and Reagents
Female C57BL/6, severe combined immunodeficient/beige (SCID/bge), and SCID mice (8–12 weeks old) were obtained from The Jackson Laboratory (Bar Harbor, ME) or the NIH Animal Production Area (Frederick, MD). Pmel-1 T-cell receptor (TCR), pmel-1+/Thy1.1+ transgenic mice (now deposited at The Jackson Laboratory) were generated as previously described (22). All mice were housed in specific pathogen-free National Cancer Institute animal facilities and used in experiments approved by the Animal Care and Use Committee of the National Cancer Institute. Transgenes (pmel-1 mice) were confirmed by PCR analysis for the pmel-1-1 TCR α- and β-chains, at 100 pmol/L of each of the following primers: TCRα sense, 5′-GGTCCTGTGGCTCCAGTTTAAT-3′; TCRα antisense, 5′-CTGCTTAACCTGTCCCTCATGT-3′; TCRβ sense, 5′-CTGGGCAGTGTTCTGTCTCC-3′; TCRβ antisense, 5′-ACCATGGTCATCCAACACAG-3′; and gold Taq (Applied Biosystems, Branchburg, NJ), with 1 × PCR buffer, 3 mmol/L MgCl, and 10 mmol/L deoxynucleotide triphosphates. PCR was done at 94°C for 13 minutes, then 94°C for 1 minute, 61°C for 1 minute, 72°C for 1.5 minutes for 35 cycles, and at 72°C for 15 minutes. Perforin- and Fas ligand (FasL)–deficient mice were obtained from The Jackson Laboratory (strain nos. 2407 and 1021, respectively) and bred to pmel-1 mice to obtain TCR transgenic and homozygous knockouts, which were confirmed by PCR.
Murine B16/F1 melanoma cells (23) were sequentially transduced with cDNA encoding either human CXCR4 in the pLNCX2 retroviral vector (Clontech, Palo Alto, CA) or pLNCX2 alone (empty vector) and then with cDNA encoding firefly (Photinus puralis) luciferase as previously described (14). For convenience, the CXCR4-luciferase-B16 cell line is hereafter designated as ‘‘CXCR4-luc-B16,’’ whereas the control vector-luciferase-transduced cell line is called ‘‘luc-B16.’’ Transduced B16 cells were cultured in complete DMEM (Life Technologies, Inc., Gaithersburg, MD) with 10% heat-inactivated FCS and supplements, including puromycin and G418.
The human CD8+ T-cell line (JR6C12) with reactivity against human gp100209–217 peptide was a kind gift from Dr. Mark Dudley (Surgery Branch, National Cancer Institute, Bethesda, MD). JR6C12 was originally obtained by limiting dilution from in vitro sensitized peripheral blood mononuclear cells of a melanoma patient (now deceased) who had been treated with a gp100 peptide vaccine as described (24) and was cultured in complete RPMI 1640 with 10% FCS and interleukin 2 (1,000 units/mL). The human melanoma cell line MEL501A (also known as MEL624; ref. 25) was cultured in complete DMEM and was a gift of Dr. Susan Topalian (Surgery Branch, National Cancer Institute).
For production of anti-CTLA4 mAb, 9H10 hybridoma cells (26) were cultured in hybridoma serum-free medium containing interleukin-6 (10 units/mL; Invitrogen, Carlsbad, CA) in a BD CELLine (culture) device (BD Biosciences, Franklin Lakes, NJ) according to the instructions of the manufacturer.
The CXCR4 antagonist peptide T22 (NH2-RRWCYRK-CYKGYCYRKCR-COOH) and an inactive control peptide (ALA) were synthesized by Synpep (Dublin, CA; ref. 14). Chemokines were purchased from Peprotech (Rocky Hill, NJ). Cyclophosphamide, cisplatin, and camptothecin were purchased from Sigma-Aldrich (St. Louis, MO). The pancaspase inhibitor zVAD-fmk (Clontech) was used at 1 μmol/L (10). Anti-FasL antibody (R&D Systems, Minneapolis, MN) was used at 100 μg/mL.
In vivo Metastasis Assays
CXCR4-luc-B16 and luc-B16 cells in exponential growth phase were harvested by trypsinization and washed twice with PBS before injection. Cell viability was >95% as determined by trypan blue dye exclusion. CXCR4-luc-B16 (4 × 105 cells in 400 μL of PBS) or luc-B16 cells (8 × 105 cells in 400 μL of PBS) were injected into the tail veins of mice (n = 5 mice per treatment group unless otherwise noted; ref. 14). T22 and ALA peptides in sterile PBS were administered i.p. using 40 μg peptide per mouse daily on days 4 to 7 after tumor cell injection. Mice were euthanized after 12 to 14 days for gross inspection of lungs, including counting of surface tumor nodules and quantification of tumor burden in the lungs using an in vitro luciferase activity as previously described (14). Whereas the number of surface nodules was highly correlated with in vitro luciferase activity, the latter assay was deemed more objective and, hence, all relevant figures show luciferase activity measurements in relative light units.
Apoptosis Assay
CXCR4-luc-B16 cells (1.5 × 105 per well) were seeded in six-well plates. On reaching 30% confluence, cells were treated with cyclophosphamide, cisplatin, or camptothecin at various concentrations with or without pretreatment of CXCL12 (1 μg/mL) for 3 hours. After 24 hours of incubation, the cells were collected, washed twice, and stained with Annexin V-FITC (BD PharMingen, San Jose, CA) for assessment of apoptosis. Flow cytometry was done with a FACScan flow cytometer (BD Biosciences, San Jose, CA) using FloJo analysis software (Treestar, San Carlos, CA).
Cytotoxicity Assays
To increase Fas expression, CXCR4-luc-B16 cells were treated with recombinant murine IFN-γ (200 ng/mL; Peprotech) for 12 to 16 hours in DMEM/0.5% FCS. To confirm Fas expression, cells were harvested and washed and then stained with phycoeryhthrin-conjugated anti-mouse Fas mAb (BD Biosciences) for flow cytometric analysis. Pmel-1 T cells from transgenic mice that expressed T-cell antigen receptors specific for a H-2Db-restricted, mouse gp100–specific peptide were activated for 5 to 10 days by incubation in complete RPMI 1640 (10% FCS) with 30 IU/mL recombinant human interleukin 2 and 1 μmol/L human gp10025–33 peptide. T cells from pmel-1 mice bred on FasL-deficient or perforin-deficient backgrounds were harvested and activated in a similar manner. CXCR4-luc-B16 target cells were treated with IFN-γ with or without CXCL12 (1 μg/mL), serum starved (0.5% FCS) overnight, labeled with calcein-AM (Molecular Probes, Portland, OR) at 1 μmol/L final concentration, washed, and added to round-bottomed microtiter plates (1.5 × 104 per well) with variable numbers of effector pmel-1 cells for 3 hours at 37°C. For human melanoma cytotoxicity assays, MEL501A cells (as described above) were treated with human IFNγ (200 ng/mL) for 12 to 16 hours in DMEM/0.5% FCS and then labeled with calcein-AM before exposure to cultured, interleukin 2–treated JR6C12 CD8+ T cells for 3 hours at 37°C at various effector/target ratios. In both cases, supernatants were recovered, and calcein release was measured using a CytoFluor 2350 plate reader (Millipore). Specific lysis = [(experimental × spontaneous) /(maximal spontaneous)] × 100. Maximal lysis was achieved with 0.1% Triton X-100, whereas spontaneous lysis was obtained by incubating target cells alone. Cytotoxicity (%) was calculated as the mean of triplicate assays.
Statistical Methods
P values were based on unpaired, two-sided Student’s t tests unless otherwise specified and were calculated with Instat (GraphPad, San Diego, CA).
Results
CXCR4 Activation by CXCL12 Does Not Protect B16 Cells from Apoptosis Induced by Cyclophosphamide, Cisplatin, or Camptothecin In vitro
Activation of chemokine receptors has been shown to decrease tumor cell death exposed to antigen-specific T cells (10), and others have shown that activation through CXCR4 can render lung cancer cells more resistant to the effects of the chemotherapeutic drug etoposide (27). Thus, we initially hypothesized that CXCR4 activation would have similar prosurvival effects when tumor cells were exposed to most chemotherapeutic agents. Surprisingly, CXCL12 pretreatment did not protect CXCR4-luc-B16 cells from the cytotoxic effects of cyclophosphamide over a range of cytotoxic concentrations (Fig. 1). Moreover, the addition of CXCL12 did not reduce cytotoxicity in the presence of two other cytotoxic agents, cisplatin (1–50 μg/mL) and campothecin (1–50 μmol/L), over a wide range of drug concentrations (data not shown). Lastly, to determine if human melanoma cells behaved similarly, we tested two human cell lines that either expressed CXCR4 endogenously (TC2031-2 cell line) or expressed this receptor following serum deprivation (MEL501A cell line). Both cells lines showed equivalent cell death in response to cisplatin (1–250 μg/mL) whether or not CXCL12 pretreatment was done (data not shown). Thus, CXCL12 exposure did not inhibit drug-induced apoptosis in CXCR4-luc-B16 cells and human melanoma cells that are capable of expressing CXCR4 endogenously.
Figure 1.

CXCR4 activation does not protect CXCR4-B16 from cell death induced by cyclophosphamide. CXCR4-luc-B16 cells were seeded in six-well plates (1.5 × 105 per well) in DMEM with 0.5% FCS. On reaching 30% confluence, the cells were treated with CXCL12 (1 μg/mL) overnight if indicated. Cells were then treated with cyclophosphamide at the indicated concentrations for 24 h and then assessed for viability by the Annexin V assay. Representative of three experiments with similar results.
CXCR4 Activation in B16 Cells by CXCL12 Reduces pmel-1T Cell ^ Induced Cytotoxicity In vitro
We have previously shown (10) that addition of CCL27, a CCR10 ligand, protects CCR10-B16 cells from apoptosis induced by both Fas cross-linking and pmel-1 (gp100 peptide–specific) CD8+ CTL (22). Because the activation of CXCR4 in B16 cells could not protect them from the cytotoxic effects of cyclophosphamide, cisplatin, and camptothecin in vitro, we asked if CXCR4 activation in B16 cells could protect them from death induced by pmel-1 CTL. Furthermore, it had not been clearly established whether pmel-1 CTL killing of B16 cells was primarily mediated by Fas-FasL–dependent pathways or via perforin-granzyme–dependent pathways, or whether both contributed to B16 killing.
To address these questions, we assessed the effect of activating CXCR4 (with CXCL12) in B16 cells before exposing these cells to pmel-1 CD8+ CTL isolated from wild-type (WT), FasL-deficient, and perforin-deficient mice. To induce Fas expression in CXCR4-luc-B16, we treated the cells with IFN-γ, which resulted in an increase of Fas expression of IFN-γ-treated CXCR4-luc-B16 cells compared with PBS-treated cells (data not shown). To confirm that up-regulation of Fas expression occurs in B16 in vivo, we compared Fas expression in cultured CXCR4-luc-B16 cells and from metastatic lung nodules following i.v. inoculation and clearly observed enhanced Fas expression in the B16 cells within the lungs (data not shown).
After IFN-γ treatment, calcein-labeled CXCR4-luc-B16 cells were mixed with gp100 peptide–activated pmel-1 CTL (see Materials and Methods) at different effector-to-target cell (E/T) ratios to measure pmel-1 cytotoxicity against B16 cells. WT pmel-1 CTL showed efficient killing of IFN-γ-stimulated CXCR4-luc-B16 cells with increasing E/T ratios (Fig. 2A). As expected, killing was inhibited by zVAD-fmk (a pan-caspase inhibitor). Of note, anti-FasL neutralizing antibodies blocked killing to the same degree as zVAD-fmk (Fig. 2A), suggesting that killing was dependent on Fas-FasL interactions. Whereas perforin-deficient pmel-1 CTL showed no difference in killing of target B16 cells compared with WT pmel-1, FasL-deficient pmel-1 CTL showed markedly reduced killing that was similar to that observed with WT pmel-1 cells in the presence of anti-FasL antibodies (Fig. 2A). Thus, the Fas-FasL pathway is critical for pmel-1-mediated killing of B16 cells.
Figure 2.

Activation of CXCR4 in B16 cells by CXCL12 prevents Fas-dependent pmel-1-induced cytotoxicity. A to C, after serum starvation and IFN-γ treatment, CXCR4-luc-B16 cells were labeled with calcein-AM and mixed with different ratios of pmel-1 cells (from WT as well as perforin- and FasL-deficient mice) for 3 h at 37°C in the presence of CXCL12, T22, zVAD-fmk (zVAD), and anti-FasL antibody. The supernatants were recovered and cell lysis fraction was calculated based on calcein release. The lysis fraction (%) was calculated by dividing the observed calcein release by the maximal (total) calcein release of the cells treated with 0.1% Triton X-100. A, pmel-1 cells from WT (◆), WT with 100 μg/mL anti-FasL antibody (●), FasL knockout (■), and perforin knockout mice (▲) were used. B, T22 was added to CXCR4-luc-B16 cells for 3 h at 37°C before treatment with CXCL12 (□). T22 was also added to CXCR4-luc-B16 cells without CXCL12 treatment (■). C, luc-B16 cells were used as target cells in the presence of pmel-1 cells (◆) with 1 μmol/L zVAD-fmk (●) or 1 μg/mL CXCL12 (▲). D, human MEL501A melanoma cells were serum starved overnight to increase CXCR4 expression, then treated with 1 mg/mL CXCL12 (▲), 1 μmol/L zVAD-fmk (●), 1 μg/mL T22 alone (■), T22 + CXCL12 (□), or left untreated (◆) overnight, labeled with calcein-AM, and then added to interleukin 2 (IL-2)–treated melanoma antigen–specific JR6C12 T cells at different E/T ratios for 3 h at 37°C. Percent cytotoxicity was calculated as for B16 cells. A to D, representative experiment from two or more experiments with consistent findings.
To determine if CXCR4 activation would protect B16 cells from killing induced by pmel-1 CTL, we compared killing of CXCR4-luc-B16 cells that had (diamonds) or had not (triangles) been pretreated with CXCL12 and observed a substantial difference in cell death (Fig. 2B). CXCR4 expression by the B16 was required for protection because control luc-B16 cells were effectively killed by pmel-1 CTL despite pretreatment with CXCL12 (Fig. 2C). Next, we asked whether treating CXCR4-luc-B16 cells with T22 abrogated the protective effects of CXCL12 pretreatment. Indeed, treating CXCR4-luc-B16 cells with T22 before exposure to CXCL12 completely restored the sensitivity of the B16 cells to killing induced by pmel-1 T cells (Fig. 2B, open squares). T22, however, by itself did not protect B16 cells from pmel-1-mediated killing, confirming that T22 does not act as a CXCR4 agonist (Fig. 2B, closed squares). Lastly, T22 clearly restored the sensitivity of serum-deprived (CXCR4+) MEL501A cells to killing by melanoma-specific CTL (JR6C12 T-cell line) in the presence of CXCL12 (Fig. 2D), suggesting that CXCR4 antagonism may render human melanoma–specific immunotherapy more efficient. In summary, these results suggest that pmel-1-mediated killing of CXCR4-luc-B16 cells is primarily mediated by Fas-FasL interactions. Furthermore, CXCL12-mediated protection from cell death is reversed by the CXCR4 antagonist T22 in both B16 cells and human melanoma cells.
T22 Synergizes with Cyclophosphamide in the Treatment of Established B16 Lung Metastases
The ability of T22 to restore the sensitivity of CXCR4-B16 cells and human melanoma cells expressing CXCR4 to killing by antigen-specific CTL in vitro prompted us to ask if T22 could be used in vivo to augment antitumor immunity. Cyclophosphamide has been reported to enhance host antitumor immune responses via effects on CD4+CD25+ regulatory T cells (28–30). We first established that cyclophosphamide used in a single dose at 100 mg/kg reduced tumor burden by ~50% (Fig. 3A). In addition to a ~50% reduction in lung tumor burden at the conclusion of this cyclophosphamide treatment regimen, we observed a ~5-fold increase in the number of CD8+ T cells in B16 tumor cell–inoculated lungs compared with PBS-treated mice (data not shown). These results suggest that cyclophosphamide treatment may lead to increases in CD8+ T-cell numbers in tumor-bearing lungs, possibly contributing to B16 killing (see Discussion).
Figure 3.

CXCR4 inhibition by T22 potentiates cyclophosphamide in reducing established lung metastases of CXCR4-luc-B16 melanoma in vivo. CXCR4-luc-B16 cells (4 × 105 per mouse) were injected via tail vein of C57BL/6 mice (n = 5 per treatment group). Mice were euthanized on day 14 to measure luciferase activity in lungs. A, on day 5, the indicated doses of cyclophosphamide were administered i.p. to mice. B, in addition to cyclophosphamide treatment at 100 mg/kg on day 5, some of the mice were also treated with T22 40 μg (or the nonactive control peptide ALA, 40 μg) i.p. daily on day 4 to 7. Representative of three or more independent experiments. Luciferase activity is shown in relative light units.
Following establishment of the cyclophosphamide treatment model, mice were treated with T22 (40 μg/mouse, i.p.) on days 4, 5, 6, and 7, in addition to treatment with cyclophosphamide on day 5. T22 treatment in the absence of cyclophosphamide did not result in reduction of tumor burden in the lungs compared with PBS treatment (data not shown), confirming our prior results showing that T22 had no effect on established B16 lung metastases (17). Whereas treatment with cyclophosphamide alone resulted in ~50% reduction in lung metastases, combination treatment with cyclophosphamide and T22 resulted in an additional 70% reduction in tumor burden compared with cyclophosphamide alone (P < 0.001; Fig. 3B). Combination treatment with cyclophosphamide and ALA (20), a non-active control peptide, resulted in no greater reduction in lung tumor burden from that achieved with cyclophosphamide treatment alone (Fig. 3B). The effect of T22 for CXCR4-expressing B16 cells was specific because T22 did not enhance the efficacy of cyclophosphamide treatment when luc-B16 cells were used instead of CXCR4-luc-B16 cells (P = 0.9; data not shown). Because T22 blocks murine as well as human CXCR4, the latter result also suggests that T22 increases the efficacy of cyclophosphamide treatment by blocking CXCR4 present on the tumor cells (rather than endogenous murine CXCR4 present on other cells in the inoculated mice).
T22 Does Not Synergize with Cyclophosphamide in theAbsence of B Cells,T Cells, and Natural Killer Cells
To determine if the synergy observed with cyclophosphamide and T22 required the interaction of the host immune system, we compared lung tumor burden in severely immunocompromised (SCID/bge) and WT mice following i.v. inoculation of CXCR4-luc-B16 cells. Tumor burden was ~4 times greater in SCID/bge mice compared with WT mice when equivalent numbers of cells were injected (data not shown), confirming that host immune cells markedly inhibit the establishment of B16 lung tumor nodules. Cyclophosphamide clearly diminished tumor burden in inoculated SCID/bge mice (Fig. 4A), suggesting that cyclophosphamide has some direct killing effect on tumor cells. In contrast to WT mice, treatment with T22 and cyclophosphamide did not further reduce lung tumor burden compared with cyclophosphamide treatment alone (Fig. 4B). Thus, whereas cyclophosphamide has direct effects on tumor cells, the enhancement of tumor killing observed in WT mice requires functional T cells, B cells, or natural killer cells.
Figure 4.

An intact immune system is required for T22 to synergize with cyclophosphamide. A and B, CXCR4-luc-B16 cells (4 × 105 per mouse) were injected into SCID/bge mice via tail vein. A, SCID/bge mice were treated with cyclophosphamide (100 mg/kg i.p.) on day 5. On day 14, tumor burden was assessed by measuring individual lung mass. Lungs from noninjected mice served as controls. B, SCID/bge mice were treated with cyclophosphamide (100 mg/kg i.p.) on day 5 with or without T22 (40 μg/mouse i.p.) daily on day 4 to 7. On day 14, lung tumor burden was measured by luciferase assay. The experiments depicted were done on different days with separate preparations of cells.
T22 Pretreatment Is Required for Reduction of Established B16 Lung Tumor Metastases after Treatment with Anti-CTLA4 mAb
Others have shown that anti-CTLA4 mAb as monotherapy has little effect on established B16 tumors whereas the combination of anti-CTLA4 with vaccines has greatly increased therapeutic benefit (4, 9). To determine whether T22 can increase the efficacy of a non-vaccine-based immunotherapy, we next combined T22 with anti-CTLA4 mAb treatment as therapy for established experimental lung metastases.
We first inoculated WT mice with CXCR4-B16 cells via tail vein (as in Fig. 3). Three days later, we pretreated mice with T22, which was followed 2 hours later by initiation of treatment with a function-blocking anti-CTLA4 mAb (see Fig. 5 legend for complete dosing schedules of T22 and anti-CTLA4 mAb). Whereas neither T22 nor anti-CTLA4 mAb treatment alone had a significant effect compared with PBS control (Fig. 5B), pretreatment with T22 followed by anti-CTLA4 mAb resulted in a ~50% reduction in tumor burden in the lungs as measured by counting surface lung nodules [188 ± 16 (PBS) versus 89 ± 26 (anti-CTLA4 + T22), P = 0.02; representative photos shown in Fig. 5A] and by quantitative luciferase assay (Fig. 5B). In a separate duplicate experiment, we showed that combination therapy yielded a 42% decrease (P = 0.03) in lung tumor burden compared with PBS treatment whereas anti-CTLA4 treatment alone was not statistically significantly different from PBS treatment (data not shown). Therefore, T22 treatment is required for the antitumor benefit of anti-CTLA4 therapy to be observed.
Figure 5.
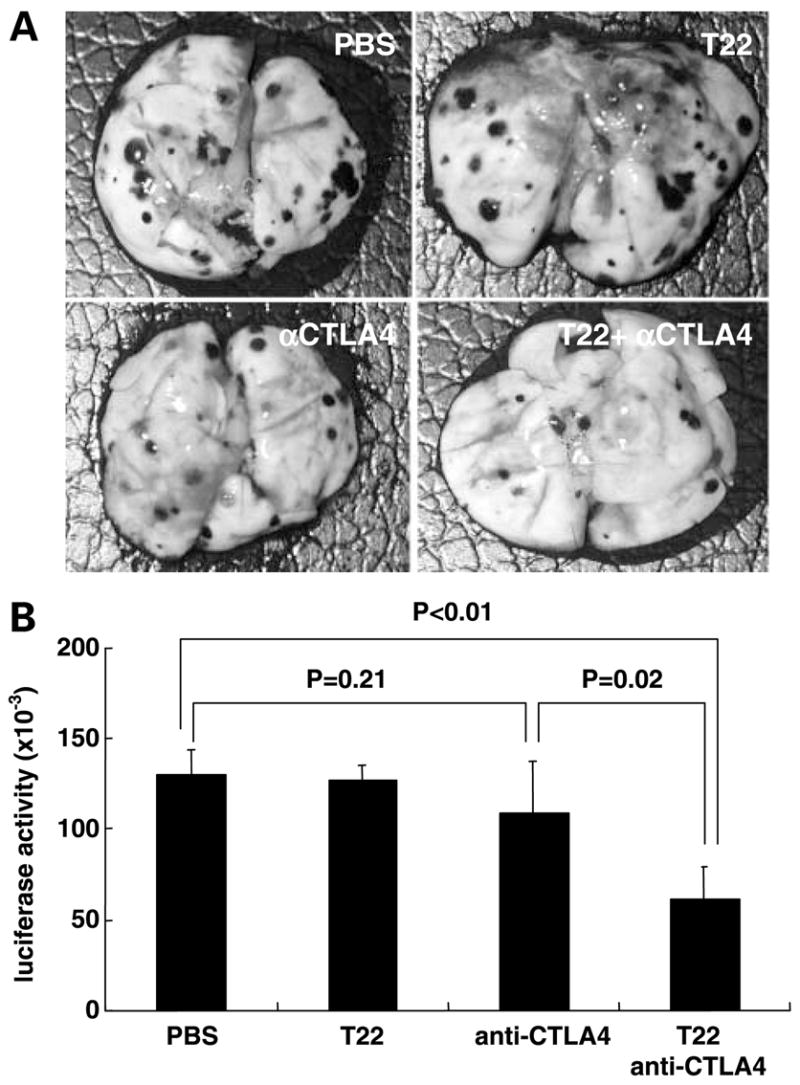
T22 pretreatment is required for anti-CTLA4-mediated reduction of established tumor burden in the lungs. A and B, CXCR4-B16 cells were inoculated via the tail vein in WT mice (n = 4 mice per treatment group, otherwise as described in Fig. 3). T22, when used, was administered i.p. (40 μg/mouse) daily on day 3 to 7 and day 9 to 10. Anti-CTLA4 mAb (clone 9H10), when used, was administered i.p. (250 μg/mouse) on day 3, 6, and 9. When combination treatment was administered, T22 injections were given i.p. 2 h before injection of anti-CTLA4 mAb. Mice were euthanized on day 13 for visual inspection of lung metastases (representative lungs from one of four mice used per group; A) and quantitative in vitro measurement of luciferase activity (B). Columns, mean; bars, SE.
Discussion
A growing body of evidence suggests that chemokine receptors are expressed by a wide variety of solid and hematopoietic cancers and may play critical roles in cancer cell survival and metastasis (11). CXCR4 seems to be the chemokine receptor that is most commonly expressed by cancers, possibly due to increased expression in response to hypoxia (31). Studies have indicated that CXCR4 is up-regulated in melanoma at the mRNA (12) and protein levels (14, 15). Of note, patients whose melanomas were CXCR4 positive had a 3.1-fold increased risk for death due to disease compared with patients whose tumors did not show CXCR4 expression (15), implicating CXCR4 expression as a strong independent prognostic factor.
Multiple studies have shown that inhibition of CXCR4 via antibodies or peptide antagonists expressed by cancer cells reduces metastasis and/or tumor growth (12–14, 18). Most of the studies, however, did not address whether or not CXCR4 antagonists can block established disease, which is the clinical setting facing most cancer patients. A study showing that a CXCR4 antagonist can inhibit the growth of brain tumor xenografts was done in nude mice, eliminating the effect of the immune system (32). Other results suggest that CXCR4 activation may be required for metastatic tumor cells to proliferate beyond the single or paucicellular stage once they arrive at distant sites, although clearly many patients have progressed beyond this point by the time current diagnostic techniques can detect abnormalities (18). Our own studies using CXCR4-overexpressing B16 cells showed that pretreatment of these cells with T22 markedly reduced their ability to initiate lung metastases (17). There was, however, no effect of T22 on established CXCR4-luc-B16 lung metastases, suggesting that tumors had already bypassed the need for CXCR4 at the time the T22 was administered (17). These results suggest that alternative strategies must be pursued (in some cancers) to clinically take advantage of a number of described CXCR4 antagonists (20, 33, 34).
Herein, we show that T22 has no effect on established CXCR4-luc-B16 tumors in the lung, confirming our prior results (17). However, T22 pretreatment significantly increased the efficacy (by an additional 70%) of single-dose cyclophosphamide treatment. T22 treatment did not enhance the efficacy of cyclophosphamide in SCID-bge mice, suggesting that the synergy observed with T22 + cyclophosphamide treatment requires that animals have B cells, natural killer cells, and/or T cells. Because cyclophosphamide alone reduced tumor burden in SCID-bge mice inoculated with CXCR4-luc-B16 cells, we conclude that cyclophosphamide has both direct cytotoxic effects on B16 cells as well as indirect killing effects that are most likely mediated through the immune system.
Although cyclophosphamide at high doses used for chemotherapy frequently causes white cell count depression and immune suppression, treatment with low doses of cyclophosphamide has long been observed to result in enhanced immune responses (35, 36). Recently, a mechanism involving the effects of cyclophosphamide of CD4+CD25+ regulatory T cells has been formulated based on several experimental studies. Cyclophosphamide reduced the numbers and function of CD25+ regulatory T cells in mice, leading to enhancement of contact hypersensitivity (30). Lutsiak et al. (29) showed that low-dose cyclophosphamide enhanced apoptosis and decreased homeostatic proliferation of regulatory T cells, in addition to inhibiting their suppressive function. Ghiringhelli et al. (28) provided striking evidence that cyclophosphamide reduced numbers of CD4+CD25+ T cells in spleen and lymph nodes of rats, which was accompanied by complete cure of PROb (colon carcinoma) tumors when cyclophosphamide treatment was administered with a tumor vaccine plus adjuvant. Our data are consistent with those of others showing that elimination of CD4+CD25+ regulatory T cells can result in rejection of poorly immunogenic B16 tumors under specific conditions (37) and with those indicating that cyclophosphamide treatment can induce concomitant immunity against B16 tumors (38).
T22 pretreatment was also required for us to observe any benefit from treatment with anti-CTLA4 mAb. The finding that anti-CTLA4 mAb treatment by itself had little effect on the poorly immunogenic B16 tumors was not surprising, given the previous finding by others (4, 9) and the general observation that anti-CTLA4 therapy is most effective on immunogenic tumors (6). Anti-CTLA4 therapy has already been used in human clinical trials for advanced melanoma with some evidence of tumor regression, but partial or ineffective responses are still common (5). Moreover, responses have been linked to increased autoimmunity (5, 39). The concomitant use of CXCR4 antagonists such as CXCR4 may also allow clinicians to use lower doses of anti-CTLA4, increase efficacy, and/or reduce the risk of autoimmune toxicity. Thus, inhibiting CXCR4 may be a novel strategy to increase the efficacy of immunotherapeutic approaches to cancer.
CXCR4 activation in our studies, however, did not protect B16 cells from three cytotoxic drugs—cyclophosphamide, camptothecin, and cisplatin—despite contrary data with the use of small-cell lung cancer cells (27). At a variety of doses, we did not observe any protection of CXCR4-luc-B16 cells from cell death induced on direct exposure to these three agents, some of which are commonly used in treating a variety of human cancers. We did not address a mechanism by which CXCR4 activation failed to protect B16 from cisplatin- and camptothecin-induced cell death, but others have shown that the same chemokine receptor (CXCR4) can mediate both proapoptotic and antiapoptotic signals depending on the specific signaling cascades induced in the cells (40). Nonetheless, our results suggest that the combination of T22 treatment with agents such as cisplatin, which has been used in clinical trials for advanced melanoma (41), would unlikely be effective.
In summary, we provide compelling in vitro data that CXCR4 activation in both murine and human melanoma results in protection from immune-mediated cytotoxicity. Furthermore, we show in vivo that the combination of T22 and anti-CTLA4 (or cyclophosphamide) increases the efficacy of immunotherapy for the treatment of established B16 lung metastases. Because CXCR4 is so widely expressed in many human cancers (e.g., breast, colon, and prostate among others), the use of CXCR4 antagonists to increase the efficacy of immunotherapy may be broadly useful in cancer treatment.
Acknowledgments
We thank Dr. Mark C. Udey (National Cancer Institute) and members of the Hwang laboratory for many helpful suggestions.
Grant support: Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research and a Howard Hughes Medical Student Fellowship (J. Wang).
References
- 1.Rosenberg SA. Shedding light on immunotherapy for cancer. N Engl J Med. 2004;350:1461–3. doi: 10.1056/NEJMcibr045001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dudley ME, Wunderlich JR, Yang JC, et al. Adoptive cell transfer therapy following nonmyeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–57. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–15. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and meta-static tumors accompanied by autoimmune depigmentation. J Exp Med. 1999;190:355–66. doi: 10.1084/jem.190.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Attia P, Phan GQ, Maker AV, et al. Autoimmunity correlates with tumor regression in patients with metastatic melanoma treated with anti-CTLA-4. J Clin Oncol. 2005;23:6043–53. doi: 10.1200/JCO.2005.06.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Egen JG, Kuhns MS, Allison JP. CTLA-4: new insights into its biological function and use in tumor immunotherapy. Nat Immunol. 2002;3:611–8. doi: 10.1038/ni0702-611. [DOI] [PubMed] [Google Scholar]
- 7.Sutmuller RP, van Duivenvoorde LM, van Elsas A, et al. Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25(+) regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J Exp Med. 2001;194:823–32. doi: 10.1084/jem.194.6.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Elsas A, Sutmuller RP, Hurwitz AA, et al. Elucidating the autoimmune and antitumor effector mechanisms of a treatment based on cytotoxic T lymphocyte antigen-4 blockade in combination with a B16 melanoma vaccine: comparison of prophylaxis and therapy. J Exp Med. 2001;194:481–9. doi: 10.1084/jem.194.4.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gregor PD, Wolchok JD, Ferrone CR, et al. CTLA-4 blockade in combination with xenogeneic DNA vaccines enhances T-cell responses, tumor immunity and autoimmunity to self antigens in animal and cellular model systems. Vaccine. 2004;22:1700–8. doi: 10.1016/j.vaccine.2003.10.048. [DOI] [PubMed] [Google Scholar]
- 10.Murakami T, Cardones AR, Finkelstein SE, et al. Immune evasion by murine melanoma mediated through CC chemokine receptor-10. J Exp Med. 2003;198:1337–47. doi: 10.1084/jem.20030593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kakinuma T, Hwang ST. Chemokines, chemokine receptors, and cancer metastasis. J Leukoc Biol. 2006;79:639–51. doi: 10.1189/jlb.1105633. [DOI] [PubMed] [Google Scholar]
- 12.Müller A, Homey B, Soto H, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–6. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 13.Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–50. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- 14.Murakami T, Maki W, Cardones AR, et al. Expression of CXC chemokine receptor (CXCR)-4 enhances the pulmonary metastatic potential of murine B16 melanoma cells. Cancer Res. 2002;62:7328–34. [PubMed] [Google Scholar]
- 15.Scala S, Ottaiano A, Ascierto PA, et al. Expression of CXCR4 predicts poor prognosis in patients with malignant melanoma. Clin Cancer Res. 2005;11:1835–41. doi: 10.1158/1078-0432.CCR-04-1887. [DOI] [PubMed] [Google Scholar]
- 16.Robledo MM, Bartolome RA, Longo N, et al. Expression of functional chemokine receptors CXCR3 and CXCR4 on human melanoma cells. J Biol Chem. 2001;276:45098–105. doi: 10.1074/jbc.M106912200. [DOI] [PubMed] [Google Scholar]
- 17.Cardones AR, Murakami T, Hwang ST. CXCR4 enhances adhesion of B16 tumor cells to endothelial cells in vitro and in vivo via β1 integrin. Cancer Res. 2003;63:6751–7. [PubMed] [Google Scholar]
- 18.Zeelenberg IS, Ruuls-Van Stalle L, Roos E. The chemokine receptor CXCR4 is required for outgrowth of colon carcinoma micrometastases. Cancer Res. 2003;63:3833–9. [PubMed] [Google Scholar]
- 19.Nakashima H, Masuda M, Murakami T, et al. Anti-human immunodeficiency virus activity of a novel synthetic peptide, T22 ([Tyr-5,12, Lys-7]polyphemusin II): a possible inhibitor of virus-cell fusion. Antimicrob Agents Chemother. 1992;36:1249–55. doi: 10.1128/aac.36.6.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murakami T, Nakajima T, Koyanagi Y, et al. A small molecule CXCR4 inhibitor that blocks T cell line-trophic HIV-1 infection. J Exp Med. 1997;186:1389–93. doi: 10.1084/jem.186.8.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morales J, Homey B, Vicari AP, et al. CTACK, a skin-associated chemokine that preferentially attracts skin-homing memory T cells. Proc Natl Acad Sci U S A. 1999;96:14470–5. doi: 10.1073/pnas.96.25.14470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Overwijk WW, Theoret MR, Finkelstein SE, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–80. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fidler IJ. Selection of successive tumour lines for metastasis. Nature (New Biol) 1973;242:148–9. doi: 10.1038/newbio242148a0. [DOI] [PubMed] [Google Scholar]
- 24.Dudley ME, Wunderlich J, Nishimura MI, et al. Adoptive transfer of cloned melanoma-reactive T lymphocytes for the treatment of patients with metastatic melanoma. J Immunother. 2001;24:363–73. doi: 10.1097/00002371-200107000-00012. [DOI] [PubMed] [Google Scholar]
- 25.Rivoltini L, Barracchini KC, Viggiano V, et al. Quantitative correlation between HLA class I allele expression and recognition of melanoma cells by antigen-specific cytotoxic T lymphocytes. Cancer Res. 1995;55:3149–57. [PMC free article] [PubMed] [Google Scholar]
- 26.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182:459–65. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hartmann TN, Burger JA, Glodek A, Fujii N, Burger M. CXCR4 chemokine receptor and integrin signaling co-operate in mediating adhesion and chemoresistance in small cell lung cancer (SCLC) cells. Oncogene. 2005;24:4462–71. doi: 10.1038/sj.onc.1208621. [DOI] [PubMed] [Google Scholar]
- 28.Ghiringhelli F, Larmonier N, Schmitt E, et al. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol. 2004;34:336–44. doi: 10.1002/eji.200324181. [DOI] [PubMed] [Google Scholar]
- 29.Lutsiak ME, Semnani RT, De Pascalis R, Kashmiri SV, Schlom J, Sabzevari H. Inhibition of CD4(+)25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood. 2005;105:2862–8. doi: 10.1182/blood-2004-06-2410. [DOI] [PubMed] [Google Scholar]
- 30.Ikezawa Y, Nakazawa M, Tamura C, Takahashi K, Minami M, Ikezawa Z. Cyclophosphamide decreases the number, percentage and the function of CD25+ CD4+ regulatory T cells, which suppress induction of contact hypersensitivity. J Dermatol Sci. 2005;39:105–12. doi: 10.1016/j.jdermsci.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 31.Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W. Chemokine receptor CXCR4 down-regulated by von Hippel-Lindau tumour suppressor pVHL. Nature. 2003;425:307–11. doi: 10.1038/nature01874. [DOI] [PubMed] [Google Scholar]
- 32.Rubin JB, Kung AL, Klein RS, et al. A small-molecule antagonist of CXCR4 inhibits intracranial growth of primary brain tumors. Proc Natl Acad Sci U S A. 2003;100:13513–8. doi: 10.1073/pnas.2235846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arakaki R, Tamamura H, Premanathan M, et al. T134, a small-molecule CXCR4 inhibitor, has no cross-drug resistance with AMD3100, a CXCR4 antagonist with a different structure. J Virol. 1999;73:1719–23. doi: 10.1128/jvi.73.2.1719-1723.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schols D, Struyf S, Damme JV, Este JA, Henson G, De Clerq E. Inhibition of T-trophic HIV strains by selective antagonization of the chemokine receptor CXCR4. J Exp Med. 1997;186:1383–8. doi: 10.1084/jem.186.8.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Polak L, Geleick H, Turk JL. Reversal by cyclophosphamide of tolerance in contact sensitization. Tolerance induced by prior feeding with DNCB. Immunology. 1975;28:939–42. [PMC free article] [PubMed] [Google Scholar]
- 36.Rollinghoff M, Starzinski-Powitz A, Pfizenmaier K, Wagner H. Cyclophosphamide-sensitive T lymphocytes suppress the in vivo generation of antigen-specific cytotoxic T lymphocytes. J Exp Med. 1977;145:455–9. doi: 10.1084/jem.145.2.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nagai H, Horikawa T, Hara I, et al. In vivo elimination of CD25+ regulatory T cells leads to tumor rejection of B16F10 melanoma, when combined with interleukin-12 gene transfer. Exp Dermatol. 2004;13:613–20. doi: 10.1111/j.0906-6705.2004.00198.x. [DOI] [PubMed] [Google Scholar]
- 38.Turk MJ, Guevara-Patino JA, Rizzuto GA, Engelhorn ME, Houghton AN. Concomitant tumor immunity to a poorly immunogenic melanoma is prevented by regulatory T cells. J Exp Med. 2004;200:771–82. doi: 10.1084/jem.20041130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Phan GQ, Yang JC, Sherry RM, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci U S A. 2003;100:8372–7. doi: 10.1073/pnas.1533209100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vlahakis SR, Villasis-Keever A, Gomez T, Vanegas M, Vlahakis N, Paya CV. G protein-coupled chemokine receptors induce both survival and apoptotic signaling pathways. J Immunol. 2002;169:5546–54. doi: 10.4049/jimmunol.169.10.5546. [DOI] [PubMed] [Google Scholar]
- 41.Tas F, Argon A, Camlica H, Topuz E. Temozolomide in combination with cisplatin in patients with metastatic melanoma: a phase II trial. Melanoma Res. 2005;15:543–8. doi: 10.1097/00008390-200512000-00010. [DOI] [PubMed] [Google Scholar]