

Abstract
Neurotrophic factors (NTF) are small, versatile proteins that maintain survival and function to specific neuronal populations. In general, the axonal transport of NTF is important as not all of them are synthesized at the site of its action. Nerve growth factor (NGF), for instance, is produced in the neocortex and the hippocampus and then retrogradely transported to the cholinergic neurons of the basal forebrain. Neurodegenerative dementias like Alzheimer’s disease (AD) are linked to deficits in axonal transport. Furthermore, they are also associated with imbalanced distribution and dysregulation of NTF. In particular, brain-derived neurotrophic factor (BDNF) plays a crucial role in cognition, learning and memory formation by modulating synaptic plasticity and is, therefore, a critical molecule in dementia and neurodegenerative diseases. Here, we review the changes of NTF expression and distribution (NGF, BDNF, neurotrophin-3, neurotrophin-4/5 and fibroblast growth factor-2) and their receptors [tropomyosin-related kinase (Trk)A, TrkB, TrkC and p75NTR] in AD and AD models. In addition, we focus on the interaction with neuropathological hallmarks Tau/neurofibrillary tangle and amyloid-β (Abeta)/amyloid plaque pathology and their influence on axonal transport processes in order to unify AD-specific cholinergic degeneration and Tau and Abeta misfolding through NTF pathophysiology.
Keywords: Abeta, APP, BDNF, cholinergic neurons, dementia, neurodegeneration, NGF, NT-3, NT-4/5, Tau
From ‘healthy’ aging to Alzheimer’s disease
Alzheimer’s disease (AD) is a neurodegenerative disorder that is characterized by global cognitive decline including a progressive loss of memory, orientation and reasoning. The neurologist and psychiatrist Alois Alzheimer extensively described a dementia syndrome of his patient D. Auguste, whom he treated in Frankfurt am Main, Germany at the beginning of the past century (Jarvik & Greenson 1987). He recorded a rapidly progressing memory loss of the 52-year-old woman. After her death, he examined her brain and found histological changes that are specific for AD.
Age-associated dementias like AD are becoming more and more important in industrialized countries as life expectancy increased by 2 years per decade during the recent 20 years (Klenk et al.2007). The incidence of age-associated dementias is about 1.3% of the total population of Western Europe; among them, AD is the most common, affecting 50% of all demented patients (Ferri et al.2005; Hofman et al.1991). This is likely to increase dramatically in the next 35 years. According to recent estimations, the number of people with dementia over the age of 60 will be approximately doubled in 2040. An irreversible loss of cognitive and mental abilities is the prognosis of this disorder. In later stages, demented patients are helpless and require full-time nursing care. Besides the personal and familial tragedies that are an aspect of dementia, AD and other dementias are a financial problem for the health service and, thereby, a burden for the whole social community. And this cost will rise in future as more and more persons are aging and becoming older.
Neuropathological changes in the AD brain
Histologically, the neurodegeneration is distinguished by neuropathological changes and deposits of misfolded proteins, mainly consisting of hyperphosphorylated Tau in neurofibrillary tangles and amyloid-β (Abeta) in the form of senile plaques and deposits in cerebral blood vessels.
Neurofibrillary tangles
Neurofibrillary tangles consist of hyperphosphorylated Tau proteins that aggregate inside neurons along neurites – observed as neuropil threads – and finally in the soma. Tau proteins belong to the microtubule-associated protein family. They are mainly found in neurons. Nonneuronal cells usually display trace amounts, but in some diseases, accumulation of tau in glial cells is detected (Bergeron et al.1997).
The human Tau gene is located on chromosome 17 and contains 16 exons. Alternative splicing of three of these exons (exons 2, 3 and 10) allows for six combinations (2−3−10−; 2+3−10−; 2+3+10−; 2−3−10+; 2+3−10+ and 2+3+10+) in the human brain. Tau proteins constitute a family of six isoforms, which range from 352 to 441 amino acids and have a high number of phosphorylation sites. Tau proteins bind microtubules through repetitive regions in their C-terminal part. These repetitive regions are the repeat domains (R1–R4) encoded by exons 9–12. The three (3R) or four copies (4R) are made of a highly conserved 18-amino acid repeat separated from each other by less conserved 13- or 14-amino acid interrepeat domains. Furthermore, the six Tau isoforms appear not to be equally expressed in neurons (for detailed review, see Sergeant et al.2005). Tau proteins are known to act as promoters of tubulin polymerization in vitroand are involved in axonal transport.
A couple of evidences support a role for the microtubule-binding domain in the modulation of the phosphorylation state of Tau proteins. In a low phosphorylated state, Tau binds to microtubules through the microtubule-binding domains and stabilizes their polymerization and assembly. However, microtubule assembly depends partially upon the phosphorylation state as phosphorylated Tau proteins are less effective than nonphosphorylated Tau proteins on microtubule polymerization. Phosphorylation inside and outside the microtubule-binding domains can strongly influence tubulin assembly by modifying the affinity between Tau and microtubules. However, properly assembled microtubules are essential to maintain axonal transport processes.
Most of the kinases involved in Tau phosphorylation include mitogen-activated protein kinase (MAPK), Tau-tubulin kinase and cyclin-dependent kinase. Stress-activated protein kinases have also been recently linked to Tau phosphorylation. Glycogen synthase kinase-3β (GSK-3β) is a Tau kinase that is able to phosphorylate both non-Ser/Thr-Pro sites and Ser/Thr-Pro sites.
In numerous neurodegenerative disorders, Tau proteins aggregate into intraneuronal filamentous inclusions. In AD, these filaments are named paired helical filaments (PHF).
Few phosphorylation-dependent antibodies such as AT100, AP422 or TG3/MC1 antibodies only detect PHF-tau, demonstrating the presence of abnormal phosphorylated sites. With the exception of Ser422, these phosphorylated sites found in PHF-tau are in addition conformation-dependent epitopes (Sergeant et al.2005). There is a direct relationship between hyperphosphorylation, abnormal phosphorylation and Tau aggregation, but it remains to be determined whether phosphorylation is a cause or a consequence in the aggregation process.
During normal aging, Tau hyperphosphorylation occurs in the transentorhinal cortex and spreads from here through the entorhinal cortex to the hippocampus (Braak & Braak 1991; Delacourte et al.2002). Once the hippocampus is reached, amyloid plaques may occur, and then the Tau pathology spreads over to the basal forebrain and several cortical areas in a distinct pattern along neuronal projections. Only the coexistence of Tau and amyloid pathologies is determined as AD.
To comprehend the role and mechanism of Tau pathology in AD, it is important to understand the normal function and processing of the Tau protein and the abnormal posttranslational processing of Tau in tauopathies. Mutations in the Tau gene have been found in several non-AD tauopathies and autosomal-dominant neurodegenerative disorders that exhibit extensive neurofibrillary pathology. However, Tau pathology observed in aging and AD is sporadic and not related to any mutation.
Amyloid plaques
A major feature of both sporadic and familial forms of AD is the accumulation and deposition of Abeta – a peptide of 39–43 residues – within the brain tissue of AD sufferers. The accumulation of Abeta is thought to play a pivotal role in neuronal loss or dysfunction through a cascade of events that include the generation of free radicals, mitochondrial oxidative damage and inflammatory processes. The primary event that results in the abnormal accumulation of Abeta is thought to be the dysregulated proteolytic processing of its parent molecule, the amyloid precursor protein (APP) located on chromosome 21 (Selkoe 2001). The APP molecule is a transmembrane glycoprotein that is proteolytically processed by two competing pathways, the nonamyloidogenic and the amyloidogenic (Abeta-forming) pathways. How these pathways are regulated remain unclear. Three major secretases are postulated to be involved in the proteolytic cleavage of APP. These include α-secretase (of which the metalloproteases a disintegrin and metalloprotease (ADAM)17/TNF-alpha converting enzyme (TACE) and ADAM10 are likely candidates), beta APP cleaving enzyme (BACE, formally known as β-secretase) and the γ-secretase. The α-secretase cleaves within the Abeta domain of APP, thus precluding the formation of Abeta and generating nonamyloidogenic fragments and a secreted form of APP (α-APPs). In the amyloidogenic pathway, BACE cleaves near the N-terminus of the Abeta domain on the APP molecule, liberating another soluble form of APP, β-APP, and a C-terminal fragment (C99) containing the whole Abeta domain. The last step in the amyloidogenic pathway is the intramembranous cleavage of the C99 fragment by γ-secretase, to liberate a number of Abeta isoforms of 39- to 43-amino acid residues in length (Verdile et al. 2004). The same γ-secretase complex that generates Abeta may also generate the APP intracellular domain. The most common isoforms are Abeta40 and Abeta42; the shorter form is typically produced by cleavage that occurs in the endoplasmic reticulum, while the longer form is produced by cleavage in the trans-Golgi network. The Abeta40 form is the more common of the two, but Abeta42 is the more fibrillogenic because of its more hydrophobic nature and is, thus, associated with disease states. The γ-secretase enzyme is thought to be an aspartyl protease that has the unusual ability to regulate intramembrane proteolysis (for review, see Wolfe & Kopan 2004). The mechanism of γ-secretase activity is not yet known. Four components of the γ-secretase complex, presenilins, nicastrin, anterior pharynx defective (aph-1) and presenilin enhancer 2 (pen-2), have been identified.
Recently, it was shown that Abeta42 aggregates into oligomers within endosomal vesicles and along microtubules of neuronal processes, in cultured neurons, in APP transgenic mice and in human AD brain (Takahashi et al.2004). The oligomers that form on the amyloid pathway may be the cytotoxic species rather than the mature fibrils (Kayed et al.2003). Subsequently, anterograde axonal transport delivers Abeta to plaques (Lazarov et al.2002; Stokin et al.2005).
The sites of APP processing and Abeta release have yet remained unclear. Some studies speculate that the axon is the site of Abeta production (Muresan and Muresan, 2006). According to this, amyloid deposition would increase if poor axonal transport delays the progress of APP and its processing enzymes through the axon (Stokin et al.2005) but decreases when overexpression of BACE shifts Abeta generation away from the axon and synapse into the cell soma (Lee et al.2005a). But not all reports can reproduce part of this model, in which APP is cotransported with its processing enzymes (Goldsbury et al.2006; Lazarov et al.2005). Some Abeta release occurs at synapses (Lazarov et al.2005; Sheng et al.2002) and appears to be dependent on synaptic activity (Cirrito et al.2005). However, the occurrence of plaques in white matter tracts that lack synaptic input and the release of Abeta in primary neuronal cultures that lack synapses suggest that Abeta might be released from more proximal sites too (Qiu et al.2001; Wirths et al.2007). Indeed, if all Abeta release were at presynaptic endings, impairing axonal transport should decrease amyloid deposition instead of increasing it.
Autosomal-dominant mutations in APP cause hereditary early-onset AD, likely as a result of altered proteolytic processing. Increase in either the total Abeta levels or the relative concentrations of both Abeta40 and Abeta42 has been implicated in the pathogenesis of both familial and sporadic AD (Lue et al.1999).
Three hypotheses for the pathogenesis of AD
The underlying molecular mechanisms of AD pathogenesis have not yet been identified; therefore, three major hypotheses have been advanced regarding the primary cause. The earliest hypothesis suggests that deficiency in cholinergic signaling initiates the progression of the disease. Two alternative misfolding hypotheses instead propose that either Tau protein or Abeta initiates the cascade.
The oldest hypothesis is the ‘cholinergic hypothesis’. A particular hallmark of AD is the specific neurodegeneration of cholinergic neurons leading to a loss of the neurotransmitter acetylcholine (ACh). Loss of cholinergic neurons seems to be specifically associated with typical clinical symptoms, like memory deficits, impaired attention, cognitive decline and reduced learning abilities (Hasselmo & Stern 2006; Kar et al.2004). All the first-generation therapeutics against AD were based on this hypothesis and work to preserve ACh by inhibiting its degrading enzyme acetylcholine esterase (AChE). These medications have not led to a cure. In all cases, they have served to only treat symptoms of the disease and can delay the progression of AD by 1–2 years but failed to reverse it. Therefore, it was concluded that ACh deficiencies may not be directly causal. More recently, cholinergic effects have been proposed as a potential causative agent for the formation of plaques and tangles (Shen 2004).
Later theories center on the effects of the misfolded and aggregated proteins Tau and Abeta. The hypothesis that Tau is the primary causative factor has been grounded on the fact that AD neuropathology starts in most individuals with hyperphosphorylated Tau and neurofibrillary tangles long before the first signs of Abeta occur (Braak & Braak 1991; Delacourte et al.2002). Nevertheless, accumulations of amyloid are frequently found in the cortex of nondemented individuals in the absence of neurofibrillary changes. A mechanism for neurotoxicity could be that hyperphosphorylated and aggregated Tau impairs axonal transport in murine Tau transgenic models (Ishihara et al.1999; Lewis et al.2000; Probst et al.2000), invertebrate models (Chee et al.2005; Kraemer et al.2003; Mudher et al.2004) and cellular models (Mandelkow et al.2004; Seitz et al.2002; Stamer et al.2002). Problems with axonal transport are believed to be a major cause leading to the symptoms and pathology observed in AD and other neurodegenerative dementias (Adalbert et al.2007). However, up to now, the preexistence of Tau pathology before the occurrence of Abeta pathology has not been shown in any experimental Tau model.
Abeta protein is a key molecule in the pathogenesis of AD. The tendency of Abeta to aggregate, its reported neurotoxicity and genetic linkage studies has led to the amyloid cascade hypothesis (Hardy & Allsop 1991). In this hypothesis, an increased production of Abeta results in neurodegeneration and ultimately dementia through a cascade of events (Verdile et al.2004). Amyloidogenic mouse models have established that overproduction of Abeta leads to dystrophic axons and dendrites around amyloid plaques (Brendza et al.2003; Tsai et al.2004a). Treatment of cultured neurons with fibrillar Abeta results in an increase of Tau phosphorylation, leading to a loss of microtubule-binding capacity and accumulation of Tau in the somatodendritic compartment (Busciglio et al.1995). Moreover, apolipoprotein E4 (ApoE4), the major genetic risk factor for AD, leads to excess amyloid build up in the brain before AD symptoms arise. Thus, Abeta deposition precedes clinical AD (Polvikoski et al.1995).
Advances in the understanding of AD pathogenesis provide strong support for a modified version of the amyloid hypothesis, which is now often referred to as the Abeta cascade hypothesis. The basic tenant of this modified hypothesis is that an intermediate misfolded form of Abeta, neither a soluble monomer nor a mature aggregated polymer but an oligomeric species, triggers a complex pathological cascade leading to neurodegeneration (Barghorn et al.2005; Kokubo et al.2005).
The relationship between APP, axonal transport and aberrant Abeta processing is not as easy as for Tau. Axonopathy and transport deficit can be detected long before extracellular Abeta deposition in AD patients and in a mutant APP mouse model (Stokin et al.2005). Overexpression of human APP695 also impairs specific components of axonal transport in Drosophila and mice (Gunawardena & Goldstein 2001; Salehi et al.2006). In mice, this leads to degeneration of basal forebrain cholinergic neurons (BFCN). Conversely, Abeta itself might impair axonal transport, possibly as oligomeric Abeta42 in microtubule-associated endosomal vesicles (Hiruma et al.2003; Maloney et al.2005; Takahashi et al.2004). In conclusion, impairment of axonal transport might be a cause or an effect of aberrant Abeta production or, in some cases, result from APP overexpression (Adalbert et al.2007).
The latter two theories point out the relevance of axonal transport for proper neuronal function. Finally, ApoE4, the major risk factor for sporadic AD, may directly disrupt the cytoskeleton and hence impair axonal transport also (Mahley et al.2006). Here, we give some insights into how neurotrophins may be the actors allowing to link between cholinergic degeneration, amyloid and Tau pathologies and axonal transport.
Neurotrophins: the NGF family
The most prominent members of the mammalian neurotrophin family are nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3) and neurotrophin-4/5 (NT-4/5). They activate various cell signaling pathways by activating two types of membrane-bound receptors, Trk (actually ‘tropomyosin-related kinase’ but recently ‘tyrosine receptor kinase’ is also used: TrkA, TrkB and TrkC) and p75NTR. These neurotrophins are synthesized as proneurotrophins that all bind to the p75NTR. In their active cleaved form, each neurotrophin selectively activates one of three types of Trk receptors (Fig. 1), NGF activates TrkA, NT-3 activates TrkC, while both BDNF and NT-4 activate TrkB receptors (Patapoutian & Reichardt 2001). The role of proneurotrophins and neurotrophins appears to be contradictory: while neurotrophins maintain survival and function, to certain neuronal populations, proneurotrophins trigger cell death through p75NTR (Friedman 2000).
Figure 1.
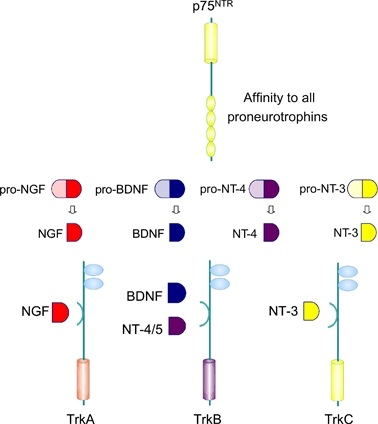
The neurotrophins and their receptors.
These neurotrophic factors (NTF) are small, versatile proteins that maintain neuronal survival, axonal guidance, cell morphology and play key roles in cognition and memory formation. During embryonic development, NTF are essential for the proper architecture and function of the brain. Knockout mice for NGF, BDNF and NT-3 are all fatal and exhibit severe neural defects. Subsequent to neuronal injury and lesions (like cerebral ischemia), NTFs are upregulated and are involved in healing and neurogenesis.
Axonal transport processes are essential for proper NTF signaling. Nerve growth factor, for example, is synthesized far away from its site of action. Vesicles containing NTF and their relevant receptors are shipped along neuronal projections throughout the brain as summarized in Table 1. However, most neurodegenerative dementias are linked to failures in axonal transport and – not surprisingly – the majority of them are associated with impaired regulation and imbalance of NTF.
Table 1.
Axonal transport and function of NFT
| Neurotrophin | Site of synthesis | Transported to (neuronal population) | Transport | Function |
|---|---|---|---|---|
| NGF | Neocortex | ChAT-positive neurons of the nbM | Retrograde | Survival and maintenance of cholinergic, sensory and sympathetic neurons |
| Hippocampus | ChAT-positive neurons of the MS, VDB, HDB and sum | |||
| BDNF | Frontal cortex | Parietal, cingulate, infralimbic, orbital, perilimbic and occipital cortices, contralateral frontal cortex, nbM, hypothalamus, locus coeruleus, thalamus and HDB | Retrograde | Survival and maintenance (of dopaminergic neurons), synaptic plasticity (long-term potentiation, neuronal firing rate, neurotransmitter release and synaptic transmission) and metabolic effects |
| Occipital cortex | Retrosplenial, perirhinal, temporal, entorhinal and frontal cortices, Raphe nucleus, VDB (HDB), thalamus, lateral geniculate nucleus and hypothalamus | |||
| Hippocampus | Ipsi- and contralateral subfields of hippocampus, MS, sum and VDB (HDB) | |||
| Entorhinal cortex | Subiculum, CA1 and CA3 hippocampal subfields, amygdala, MS and VDB | |||
| Amygdala | Temporal, parietal and occipital, entorhinal, cingulate, infralimbic, insular piriform and perirhinal cortices, thalamus, dorsal Raphe, (pre)subiculum and CA1 subfields of hippocampus, medulla, HDB, hypothalamus, nbM and substantia nigra pars compacta | |||
| Striatum | Frontoparietal cortex, TH-positive neurons of the substantia nigra, Raphe and thalamus | |||
| Amygdala | Stria terminalis | Anterograde | ||
| Neocortex | Striatum | |||
| Dentate gyrus | CA3 subfield of hippocampus (through mossy fibers) | |||
| Pons | Amygdala | |||
| NT-3 | Hippocampus | MS, VDB, thalamus and sum of hypothalamus | Retrograde | Survival and maintenance |
HDB, horizontal limb of diagonal band of Broca; MS, medial septum; sum, supramammilary nucleus; TH, tyrosine hydroxylase; VDB, vertical limb of diagonal band of Broca.
Neurotrophins and their receptors in AD
Nerve growth factor
Pro-NGF is the predominant form of NGF in the human and rodent brain, whereas mature NGF can be hardly detected. In AD, pro-NGF is increased in frontal and occipital cortex (Crutcher et al.1993; Fahnestock et al.2001; Hellweg et al.1998; Peng et al.2004) and in hippocampus (Hock et al.2000a; Narisawa-Saito et al.1996; Scott et al.1995), while a loss is observed in the basal forebrain (Mufson et al.1995; Scott et al.1995). The amount of NGF messenger RNA (mRNA) is not altered in AD (Fahnestock et al.1996; Goedert et al.1986, 1989; Jette et al.1994). A decrease of retrograde transport could explain this observation, leading to an accumulation of NGF at the sites of its production (hippocampus and neocortical areas) and a deficiency at the NGF transport terminus, the BFCN.
In the absence of NGF, cholinergic neurons show cell shrinkage, reduction in fiber density and downregulation of transmitter-associated enzymes [e.g. choline-acetyl transferase (ChAT) and AChE], resulting in a decrease of cholinergic transmission (Svendsen et al.1991). In parallel, rats show a decrease in ChAT and TrkA mRNA after fimbria transection that can be restored by NGF treatment (Venero et al.1994).
In AD, a reduction of ChAT and AChE activity and BFCN size and number was observed (Arendt et al.1983; Kasa et al.1997; Loy et al.1990; Perry et al.1992; Treanor et al.1991), implicating a severe cholinergic degeneration. Therefore, the classical AD therapy was treatment with AChE inhibitors that enhance neuronal transmission by increasing the availability of ACh at the receptors. This effect is beneficial to stabilize cognitive function and to improve or stabilize many behavioral symptoms of AD at a steady level during a 1-year period of treatment (Giacobini 2003; Wynn & Cummings 2004). Currently, there is an ongoing gene therapy trial using NGF-grafted autologous fibroblasts that were injected into the basal nucleus of Meynert (nbM) (Tuszynski et al.2005) with the aim to rescue the BFCN of AD patients.
Moreover, a loss of the NGF receptor TrkA was found in the basal forebrain (Boissiere et al.1997; Chu et al.2001; Ginsberg et al.2006a; Mufson et al.1997, 2000; Salehi et al.1996) and in the cortex (Counts et al.2004; Hock et al.1998; Savaskan et al.2000) of AD brains.
The reports about p75NTR in AD are not that clear: one study observed an upregulation of p75NTR in hippocampal tangle-bearing neurons (Hu et al.2002), another unchanged cortical levels without referring to tangle pathology (Counts et al.2004; Hock et al.1998; Perry et al.1993), while in nbM, p75NTR appears to be unchanged (Ginsberg et al.2006b; Mufson et al.2003) or decreased (Kerwin et al.1992; Mufson et al.2002; Salehi et al.2000). Moreover, during aging, a switch from TrkA to p75NTR occurs, resulting in increased amyloidogenic processing of APP (Costantini et al.2005, 2006).
However, there is another interesting link between NGF and APP: neuronal cell cultures upregulate APP expression when treated with NGF (Mobley et al.1988; Robakis et al.1991; Villa et al.2001). In fact, it was shown that NGF acts on the APP promoter mediated by p75NTR and upregulates APP transcription and the secretion of secreted amyloid precursor protein (sAPP) (Ge & Lahiri 2002; Rossner et al.1998), although intraparenchymal NGF delivery did not significantly increase Abeta deposition in monkeys (Tuszynski et al.1998).
However, neuronal cell models secrete more NGF and downregulate TrkA and p75NTR when treated with Abeta or H2O2 (Olivieri et al.2002). Excitingly, the receptor levels of p75NTR increase initially, indicating that vesicular stores of p75NTR appear to fuse to the plasma membrane. The toxicity of Abeta is mediated by p75NTR through p75-like apoptosis-inducing death domain (PLAIDD), inhibitory G protein, C-Jun N-terminal kinases (JNK), reduced nicotinamide adenine dinucleotide phosphate oxidase and caspase-9 and caspase-3 (Costantini et al.2005; Hashimoto et al.2004; Tsukamoto et al.2003). Moreover, NGF potentiates Abeta toxicity shifting the half maximal effective concentration (EC50) from 0.1 μm to 1 pm (Yankner et al.1990).
The interaction between NGF and Tau in AD or tauopathies is less clear: NGF-induced neuronal differentiation of the neuroblastoma cell line pheochromocytoma celline-12 (PC-12) exhibits an increase in Tau promoter activity and subsequently elevated Tau protein levels (Sadot et al.1996). In addition, NGF also regulates Tau phosphorylation: stimulation of differentiated PC-12 with NGF caused a dephosphorylation of Tau proteins (Fisher et al.1996), and NGF deprivation induced hyperphosphorylation of Tau (Nuydens et al.1997; Shelton & Johnson 2001). Moreover, NGF induces ubiquitination of Tau in cultured cells (Babu et al.2005), indicating that NGF may regulate Tau protein levels by inducing proteasomal degradation of Tau.
According to the hypothesis that NGF deprivation is one of the factors involved in the etiology of sporadic forms of AD, a mouse model (AD11 anti-NGF mice) had been developed, based on the expression of transgenic antibodies neutralizing NGF. The model is characterized by a progressive neurodegenerative phenotype defined by the deposition of amyloid peptide, by intracellular neurofibrillary tangles and by a marked cholinergic depletion (Capsoni et al.2002). In addition, spatial memory and neocortical long-term potentiation are impaired in AD11 mice at an age corresponding to early neurodegenerative stage characterized by the first observed decrease in the number of BFCNs without overt cortical neurodegeneration. Acute pharmacological treatment with NGF, ACh or an AChE inhibitor can rescue these symptoms (De Rosa et al.2005; Origlia et al.2006).
Nerve growth factor expression is regulated by cholinergic innervation from the basal forebrain (da Penha Berzaghi et al.1993) and by hippocampal N-methyl-d-aspartate (NMDA) receptors (Thoenen et al.1991) to maintain the normal levels. Kainic acid induces an increase of NGF transcription that can be blocked by benzodiazepine. In that light, it is exciting that treatment with the NMDA antagonist memantine had no effect on the regulation of NGF in a lesion model (Lang et al.2004).
But NGF is not found in neuronal cells only in the AD brain. Astrocytes and microglia show high levels of NGF (Siegel & Chauhan 2000). Inflammatory signals (cytokines and complement factors) as well as Abeta25-35 are potent stimulators of human microglial NGF synthesis (Heese et al.1998). In addition, hippocampal astrocytes incubated with Abeta upregulate NGF expression and its release to the culture medium. Moreover, these astrocytes display increased Tau phosphorylation and reduce the survival of cocultured hippocampal neurons (Saez et al.2006).
Brain-derived neurotrophic factor
Brain-derived neurotrophic factor regulates synaptic plasticity and thus plays a key role in memory formation and storage (Hellweg & Jockers-Scherubl 1994). Therefore, the involvement of BDNF in dementia has been discussed extensively. In that light, it is not surprising that mRNA (Connor et al.1997; Garzon et al.2002; Holsinger et al.2000; Phillips et al.1991) and protein (Ferrer et al.1999; Hock et al.2000a; Michalski & Fahnestock 2003; Peng et al.2005) levels of BDNF are decreased in hippocampus and neocortex of AD brains (for review, see Murer et al.2001; Siegel & Chauhan 2000).
Three out of six transcripts, which code for BDNF, are downregulated (Garzon et al.2002). Excitingly, two of these are controlled by a cyclic adenosine 5′-phosphate response element-binding protein (CREB) responsive promoter. However, CREB deregulation appears to be involved in the pathogenesis of AD (Yamada et al.1997; Yamamoto-Sasaki et al.1999).
Not only is BDNF diminished, but also its full-length receptor TrkB is analogously reduced in hippocampus and frontal cortex in AD (Allen et al.1999; Ferrer et al.1999). The fate of TrkB in BFCN remains to be elucidated: there are two studies reporting a decrease (Ginsberg et al.2006b; Salehi et al.1996) and another indicating no changes (Boissiere et al.1997).
Alzheimer’s disease is tightly associated to neuroimmunological processes (Heneka & O’Banion 2007). Regulation of TrkB in glia differs from that in neurons. Upregulation of truncated TrkB receptors has been found in association with senile plaques (Allen et al.1999; Connor et al.1996; Ferrer et al.1999). In addition, increase of full-length TrkB was observed in glial-like cells in hippocampus and increase of BDNF in dystrophic neurons surrounding senile plaques (Ferrer et al.1999). This was confirmed in the APP23 mouse model and shown to be related to neuronal sprouting (Burbach et al.2004).
Only a few studies do not support the loss of BDNF or TrkB in AD (Durany et al.2000; Hock et al.1998; Savaskan et al.2000). However, most data above refer to mRNA or protein levels in neurons. In activated glia, the regulation of BDNF and truncated TrkB is induced. One of these studies reporting an increase of BDNF in AD was performed using an enzyme-linked immunosorbent assay and so, no data were available concerning plaque densities. Possibly, this study population presented a rather high plaque concentration in hippocampus, resulting in high glial BDNF reactivity. Other brain areas that were examined with the same method showed the reported loss of BDNF (Durany et al.2000).
The role of single-nucleotide polymorphism in AD is still a matter of debate. Polymorphism of the BDNF has been implicated with higher risk for AD. Especially for non-ApoE4 carriers and in specific ethnic groups, this effect is well documented (Akatsu et al.2006; Desai et al.2005; Forero et al.2006; Huang et al.2007; Kunugi et al.2001; Matsushita et al.2005; Nishimura et al.2005; Olin et al.2005; Riemenschneider et al.2002; Tsai et al.2004b,2006).
Other studies observed no association with BDNF polymorphism (Bian et al.2005; Chuu et al.2006; Combarros et al.2004; Lee et al.2005b; Li et al.2005; Nacmias et al.2004; Saarela et al.2006; Vepsalainen et al.2005); so, it remains to be elucidated whether or not this effect is mainly restricted to the Asian population. Compared with wild-type populations, the polymorphisms C270T and V66M appear to be overrepresented in AD. The first is located in a noncoding region and is responsible for the transcription of BDNF mRNA transcript 4, the latter affects BDNF transport and secretion.
But there are more interactions of AD and BDNF: a specific loss of BDNF was found in tangle-bearing neurons (Ferrer et al.1999; Murer et al.1999), and BDNF dephosphorylates Tau including the most crucial sites for microtubule binding through TrkB activation and a PI3-kinase/Akt-dependent mechanism in a cellular model (Elliott et al.2005), implicating a direct Tau–BDNF interaction.
A very interesting link is the fact that during aging and in AD, Tau pathology starts in the entorhinal cortex and proceeds along the retrograde transport pathways of BDNF to the subiculum and the CA1 subfield and then to the basal forebrain, amygdala and finally to several cortical regions.
The interaction of BDNF and the APP promoter is still not that clear as it is for NGF: one study denies an upregulation of APP mRNA after BDNF treatment (Rossner et al.1998), while other reports state upregulation in SH-SY5Y cells mediated by MAPK/Ras and PI3/Akt (Ruiz-Leon & Pascual 2004) or promoter activity in PC12 cells (Ge & Lahiri 2002). In addition, the latter group showed in a neurologic disorder associated with increased cerebral BDNF-enhanced plasma levels of full-length APP and nonamyloidogenic APP (Sokol et al.2006).
Oligomeric Abeta but not fibrillar Abeta42 decreases specifically phospho-CREB and the BDNF transcripts IV and V in differentiated SH-SY5Y neuroblastoma cells (Garzon & Fahnestock 2007), confirming the data that sublethal doses of Abeta without specifying the aggregation form downregulate BDNF and CREB in cultured cortical neurons (Tong et al.2001b, 2004). In contrast, another study found out that differentiated SH-SY5Y cells treated with Abeta upregulate full-length TrkB and BDNF and downregulate truncated TrkB. This effect can be reversed with an antioxidant, indicating that this is mediated by oxidative stress (Olivieri et al.2003).
Another link combining BDNF and AD pathogenesis is BDNF as a regulator of GSK-3β: BDNF increases the phosphorylation of S9-GSK-3β, which turns the kinase activity off (Mai et al.2002).
Physical and cognitive activity and housing mice in an enriched environment increases BDNF and other neurotrophin levels (Chen & Russo-Neustadt 2005; Tong et al.2001a). However, the effect of this on amyloid pathology in murine APP transgenic models remains to be elucidated: two studies report a decrease of amyloid burden (Ambree et al.2006; Lazarov et al.2005), one reports no changes (Wolf et al.2006) although demonstrating a raise of BDNF and NT3 and finally, one study observes even an increase of amyloid pathology (Jankowsky et al.2003). Curiously, a decrease of BDNF regulation was observed during training on spatial navigation in the APP23 mouse, whereas wild-type mice show an increase (Hellweg et al.2006). Nevertheless, it should be kept in mind that enriched housing, cognitive training and wheel running act also on many factors other than BDNF only, so the outcome can be additionally related to aspects other than BDNF.
BDNF regulation is maintained through cholinergic innervation and through NMDA receptors (da Penha Berzaghi et al.1993; Thoenen et al.1991). The maintenance of normal BDNF mRNA levels appears to be mediated predominantly by NMDA receptors, whereas the increases in BDNF above normal levels are mediated by non-NMDA receptors. Interestingly, the NMDA receptor antagonist memantine used as treatment against AD increases the levels of BDNF and TrkB in rats (Marvanova et al.2001).
NT-3 and NT-4/5
Neurotrophin-3 mRNA and protein levels are unchanged in the AD brain (Durany et al.2000; Hock et al.1998, 2000a; Murase et al.1994; Phillips et al.1991), besides a minor reduction of NT-3 in the motor cortex of AD patients, a brain structure often preserved in AD (Narisawa-Saito et al.1996). In addition, cerebrospinal fluid (CSF) levels of NT-3 are not changed either (Hock et al.2000b).
A possible association of missense mutation (G63E) of the NT-3 gene with AD was found in a Japanese cohort. This association was more prominent among those who did not carry the ApoE4 allele than those who carried the ApoE4 allele (Kunugi et al.1998).
PC12 cells show increased APP promoter activity subsequent to NT-3 treatment; however, compared with NGF, this effect is rather mild (Ge & Lahiri 2002). In primary cultures of cortical neurons, NT-3 protects neurons against Abeta toxicity by limiting caspase-8, caspase-9 and caspase-3 cleavage. This neuroprotective effect of NT-3 was concomitant to an increased level of Akt phosphorylation and mediated through phosphoinositide 3-kinase (PI-3K). Moreover, NT-3 induces an upregulation of neuronal apoptosis inhibitory protein-1 expression in neurons that promote the inhibition of Abeta-induced neuronal apoptosis (Lesne 2005). In contrast to NGF, NT-3 does not induce apoptosis through p75NTR in neuroblastoma cells (Kuner & Hertel 1998). Finally, NT-3 prevents the degeneration of noradrenergic neurons of the locus coeruleus in a lesion model that resembles the pattern of cell loss found in AD (Arenas & Persson 1994).
Protein levels of NT-4/5 appear to be slightly decreased in hippocampus and cerebellum, but mRNA levels are not altered in the parietal cortex of AD patients (Hock et al.1998, 2000a).
Neurotrophin-4/5 induces Tau dephosphorylation through TrkB, while NT-3 mediated by TrkC does not show the same effect (Elliott & Ginzburg 2006). Therefore, one can speculate that a lack of endogenous TrkB or impaired BDNF/NT-4/5 signaling may lead to Tau hyperphosphorylation.
Curiously, differentiated SH-SY5Y cells treated with Abeta upregulate NT-4/5 (Olivieri et al.2003).
Fibroblast growth factor-2
Although not belonging to the neurotrophin family, fibroblast growth factor-2 (FGF-2 or formally known as basic FGF) shares many similarities with the classical neurotrophins. Fibroblast growth factor-2 is important in neuronal development and neuroprotection after neuronal lesions (Cheng & Mattson 1992). Interestingly, it regulates BDNF and vice versa(Johnson-Farley et al.2007; Kiprianova et al.2004; Soto et al.2006).
Increased levels and enhanced binding of FGF-2 were detected in senile plaques and neurofibrillary tangles in AD brains (Gomez-Pinilla et al.1990; Kato et al.1991; Siedlak et al.1991; Stieber et al.1996) and in CSF from AD patients (Hanneken et al.1995). Moreover, it was shown that FGF-2 increases the neuritic involvement of plaques (Cummings et al.1993). Immunoreactivity of the FGF receptor-1 that binds FGF-1 and FGF-2 is increased in AD in reactive astrocytes surrounding senile plaques (Ferrer & Marti 1998; Takami et al.1998).
Incubation of neuronal cultures with FGF-2 results in increased Tau phosphorylation (Burack & Halpain 1996) by increasing the levels of the Tau kinase GSK-3β and Tau itself (Butt & Dinsdale 2005; Jin et al.2003; Tatebayashi et al.1999, 2003, 2004).
Fibroblast growth factor-2 acts also on the APP promoter mediated by p75NTR, upregulates APP transcription and the secretion of sAPP (Ge & Lahiri 2002; Rossner et al.1998; Villa et al.2001), but somewhat weaker than NGF does. Glial cells exposed to Abeta produce more FGF-2 (Araujo & Cotman 1992; Pike et al.1994). Double transgenic mice overexpressing APP and FGF-2 display a higher mortality than mice expressing APP alone (Carlson et al.1997). But FGF-2 expression does not act by increasing the amyloidogenic processing of APP to Abeta peptides. In contrast, FGF-2 inhibits Abeta-induced neurotoxicity mediated by p75NTR in neuronal cultures (Costantini et al.2005; Hashimoto et al.2004; Tsukamoto et al.2003).
Conclusions
Neurotrophic factors are key regulators not only for development, maintenance and survival but also for cognition, formation and storage of memory. In AD, NTF are dysregulated and because of impaired axonal transport, unevenly distributed.
In aging, Tau proteins are becoming increasingly hyperphosphorylated, leading to the formation of neurofibrillary tangles in the transentorhinal and entorhinal cortex. As not only Tau but also APP and ApoE4 play a key role in axonal transport (Adalbert et al.2007), it would not be surprising that even at this early stage, deficits in transport processes can occur. Fascinatingly, the progression of neurofibrillary pathology in aging and in AD is identical to the retrograde transport pathways of BDNF in this neuroanatomical region. Under physiological conditions, BDNF is produced in the entorhinal cortex and shipped from here through the CA3 to the CA1–subiculum area, basal forebrain and amygdala, the next stations of neurofibrillary degeneration through the AD brain. One cannot exclude impaired transport of BDNF or downregulation of BDNF in tangle-bearing neurons in the aged brain, both leading to deficits in BDNF levels associated with possibly subclinical insufficiency in cognition and memory. Moreover, Tau pathology is the first visible occurrence of brain aging, but APP or low doses of Abeta or ApoE4 pathology may also influence the axonal transport of NTF at this stage. Furthermore, once the neurofibrillary pathology reaches the basal forebrain (occasionally already in Braak stage I), impaired retrograde transport of NGF could be the consequence, leading to an accumulation of NGF where it is synthesized (hippocampus and neocortex) and to a loss of NGF in the basal forebrain (Fig. 2). The well-known degeneration of BFCN in AD could be the outcome of this scenario. Additionally, cholinergic degeneration leads to a decrease in cholinergic innervation from fibers projecting from the basal forebrain to hippocampus and neocortex and thereby, to a decline of basal levels of BDNF expression with all its possible consequence on Tau phosphorylation. But what is more, NGF accumulation in the target regions may upregulate APP, but also may lead to increased signaling of pro-NGF through p75NTR, which is increasingly expressed in the aged brain, and thus mediates cell death. Tau could function upstream to Abeta to modify APP transport. Blocking APP transport in vivoincreases Abeta generation and deposition. Some studies implicate that tau is required for Abeta toxicity, suggesting that tau lies downstream of Abeta.
Figure 2. Retrograde transport of NGF from the hippocampus to the basal forebrain.
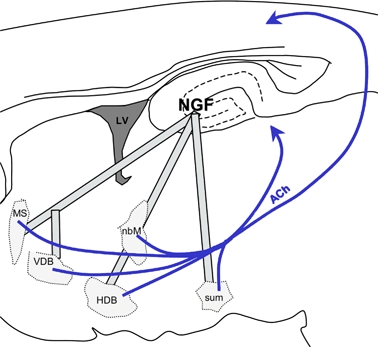
Nerve growth factor maintains survival and function of BFCN. Cholinergic projections (in blue) innervate the neocortex and hippocampus and regulate transcription of BDNF. HDB, horizontal limb of diagonal band of Broca; LV, lateral ventricle; MS, medial septum; sum, supramammilary nucleus; VDB, vertical limb of diagonal band of Broca.
Not surprisingly, all major proteins involved with AD pathology (APP and Tau) or risk for sporadic AD (ApoE4) are associated somehow with axonal transport. However, using this knowledge for the development of therapy is not as simple.
The most important concern regarding a future therapy with NTF is the mode of delivery. Being small proteins with roughly no penetration of the blood–brain barrier, new avenues for therapy need to be found. An ongoing gene therapy focusing on NGF-grafted autologous fibroblasts that are implanted into the basal forebrain of AD patients predicts a slower progression of the dementia, some cognitive improvement and sprouting of axons on the site of injection (Tuszynski et al.2005). Nevertheless, this therapy includes brain surgery and gene therapy and does not appear to be suitable as prophylactic cheap treatment for millions of aging people worldwide. Probably, NTF signaling is more likely a target for AD therapy than the NTFs themselves.
More data and support are needed to elucidate the mechanisms of NTF imbalance and dysregulation in AD. With this knowledge, we will be able to target pathways upstream NTF deregulation or deficits in axonal transport, thus starting the therapy before pathological imbalance of NTF occurs. This could include inhibitors of Tau kinases to avoid pathological Tau hyperphosphorylation that interferes with axonal transport processes and BDNF regulation. Unfortunately, chronic GSK-3β inhibition with lithium ions, which are used in therapy against bipolar disorder, appears not to have the predicted protective effect against AD (Bauer et al.2003; Chuang 2004; Manji et al.1999), although it had been shown to regulate endogenous BDNF and NGF levels (Frey et al.2006a, b).
Nevertheless, the potentials of neurofibrillary tangles (NFT) or drugs that act on their distribution or signaling should be considered carefully as future AD therapy.
Acknowledgments
This work was supported by a Marie Curie fellowship from the European Community and grants from the Deutsche Forschungsgemeinschaft (DFG) and Ligue Européenne Contre La Maladie d’Alzheimer (LECMA) to K.S., a scholarship cosponsored by Région Nord/Pas-de-Calais and CHRU-Lille to K.B., grants from Centre National de la Recherche Scientifique (CNRS), Fédération pour la Recherche sur le Cerveau, Gis-Longévité, Institut National de la Santé Et de la Recherche Médicale (INSERM) and integrated projects from the European Community (APOPIS and NEURAD) and ADERMA (Association d’Etudes et de Recherche sur la maladie d’Alzheimer) to L.B.
Conflicts of interest
This article was presented at a symposium on Alzheimer’s disease – new insights from animal models and molecular pathways, to be translated into human pathology, which took place at the Genes, Brain and Behavior 2007 Society Annual Meeting, 21–25 May 2007, Doorwerth, the Netherlands. The symposium was sponsored by the European Commission [Marie Curie Early Stage Training, MEST-CT-2005-020013 (NEURAD), Alzheimer Ph.D. Graduate School].
The authors declare no conflicts of interest.
References
- Adalbert R, Gilley J, Coleman MP. Abeta, tau and ApoE4 in Alzheimer’s disease: the axonal connection. Trends Mol Med. 2007;13:135–142. doi: 10.1016/j.molmed.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Akatsu H, Yamagata HD, Kawamata J, Kamino K, Takeda M, Yamamoto T, Miki T, Tooyama I, Shimohama S, Kosaka K. Variations in the BDNF gene in autopsy-confirmed Alzheimer’s disease and dementia with Lewy bodies in Japan. Dement Geriatr Cogn Disord. 2006;22:216–222. doi: 10.1159/000094933. [DOI] [PubMed] [Google Scholar]
- Allen SJ, Wilcock GK, Dawbarn D. Profound and selective loss of catalytic TrkB immunoreactivity in Alzheimer’s disease. Biochem Biophys Res Commun. 1999;264:648–651. doi: 10.1006/bbrc.1999.1561. [DOI] [PubMed] [Google Scholar]
- Ambree O, Leimer U, Herring A, Gortz N, Sachser N, Heneka MT, Paulus W, Keyvani K. Reduction of amyloid angiopathy and Abeta plaque burden after enriched housing in TgCRND8 mice: involvement of multiple pathways. Am J Pathol. 2006;169:544–552. doi: 10.2353/ajpath.2006.051107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo DM, Cotman CW. Basic FGF in astroglial, microglial, and neuronal cultures: characterization of binding sites and modulation of release by lymphokines and trophic factors. J Neurosci. 1992;12:1668–1678. doi: 10.1523/JNEUROSCI.12-05-01668.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenas E, Persson H. Neurotrophin-3 prevents the death of adult central noradrenergic neurons in vivo. Nature. 1994;367:368–371. doi: 10.1038/367368a0. [DOI] [PubMed] [Google Scholar]
- Arendt T, Bigl V, Arendt A, Tennstedt A. Loss of neurons in the nucleus basalis of Meynert in Alzheimer’s disease, paralysis agitans and Korsakoff’s disease. Acta Neuropathol (Berl) 1983;61:101–108. doi: 10.1007/BF00697388. [DOI] [PubMed] [Google Scholar]
- Babu JR, Geetha T, Wooten MW. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J Neurochem. 2005;94:192–203. doi: 10.1111/j.1471-4159.2005.03181.x. [DOI] [PubMed] [Google Scholar]
- Barghorn S, Nimmrich V, Striebinger A, Krantz C, Keller P, Janson B, Bahr M, Schmidt M, Bitner RS, Harlan J, Barlow E, Ebert U, Hillen H. Globular amyloid beta-peptide oligomer – a homogenous and stable neuropathological protein in Alzheimer’s disease. J Neurochem. 2005;95:834–847. doi: 10.1111/j.1471-4159.2005.03407.x. [DOI] [PubMed] [Google Scholar]
- Bauer M, Alda M, Priller J, Young LT. Implications of the neuroprotective effects of lithium for the treatment of bipolar and neurodegenerative disorders. Pharmacopsychiatry. 2003;36(Suppl. 3):S250–S254. doi: 10.1055/s-2003-45138. [DOI] [PubMed] [Google Scholar]
- Bergeron C, Pollanen MS, Weyer L, Lang AE. Cortical degeneration in progressive supranuclear palsy. A comparison with cortical-basal ganglionic degeneration. J Neuropathol Exp Neurol. 1997;56:726–734. [PubMed] [Google Scholar]
- Bian JT, Zhang JW, Zhang ZX, Zhao HL. Association analysis of brain-derived neurotrophic factor (BDNF) gene 196 A/G polymorphism with Alzheimer’s disease (AD) in mainland Chinese. Neurosci Lett. 2005;387:11–16. doi: 10.1016/j.neulet.2005.07.009. [DOI] [PubMed] [Google Scholar]
- Boissiere F, Hunot S, Faucheux B, Hersh LB, Agid Y, Hirsch EC. Trk neurotrophin receptors in cholinergic neurons of patients with Alzheimer’s disease. Dement Geriatr Cogn Disord. 1997;8:1–8. doi: 10.1159/000106594. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Brendza RP, Simmons K, Bales KR, Paul SM, Goldberg MP, Holtzman DM. Use of YFP to study amyloid-beta associated neurite alterations in live brain slices. Neurobiol Aging. 2003;24:1071–1077. doi: 10.1016/j.neurobiolaging.2003.04.008. [DOI] [PubMed] [Google Scholar]
- Burack MA, Halpain S. Site-specific regulation of Alzheimer-like tau phosphorylation in living neurons. Neuroscience. 1996;72:167–184. doi: 10.1016/0306-4522(95)00546-3. [DOI] [PubMed] [Google Scholar]
- Burbach GJ, Hellweg R, Haas CA, Del Turco D, Deicke U, Abramowski D, Jucker M, Staufenbiel M, Deller T. Induction of brain-derived neurotrophic factor in plaque-associated glial cells of aged APP23 transgenic mice. J Neurosci. 2004;24:2421–2430. doi: 10.1523/JNEUROSCI.5599-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busciglio J, Lorenzo A, Yeh J, Yankner BA. Beta-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron. 1995;14:879–888. doi: 10.1016/0896-6273(95)90232-5. [DOI] [PubMed] [Google Scholar]
- Butt AM, Dinsdale J. Fibroblast growth factor 2 mediated disruption of myelin-forming oligodendrocytes in vivo is associated with increased tau immunoreactivity. Neurosci Lett. 2005;375:28–32. doi: 10.1016/j.neulet.2004.10.060. [DOI] [PubMed] [Google Scholar]
- Capsoni S, Giannotta S, Cattaneo A. Nerve growth factor and galantamine ameliorate early signs of neurodegeneration in anti-nerve growth factor mice. Proc Natl Acad Sci U S A. 2002;99:12432–12437. doi: 10.1073/pnas.192442999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson GA, Borchelt DR, Dake A, Turner S, Danielson V, Coffin JD, Eckman C, Meiners J, Nilsen SP, Younkin SG, Hsiao KK. Genetic modification of the phenotypes produced by amyloid precursor protein overexpression in transgenic mice. Hum Mol Genet. 1997;6:1951–1959. doi: 10.1093/hmg/6.11.1951. [DOI] [PubMed] [Google Scholar]
- Chee FC, Mudher A, Cuttle MF, Newman TA, MacKay D, Lovestone S, Shepherd D. Over-expression of tau results in defective synaptic transmission in Drosophila neuromuscular junctions. Neurobiol Dis. 2005;20:918–928. doi: 10.1016/j.nbd.2005.05.029. [DOI] [PubMed] [Google Scholar]
- Chen MJ, Russo-Neustadt AA. Exercise activates the phosphatidylinositol 3-kinase pathway. Brain Res Mol Brain Res. 2005;135:181–193. doi: 10.1016/j.molbrainres.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Cheng B, Mattson MP. Glucose deprivation elicits neurofibrillary tangle-like antigenic changes in hippocampal neurons: prevention by NGF and bFGF. Exp Neurol. 1992;117:114–123. doi: 10.1016/0014-4886(92)90120-f. [DOI] [PubMed] [Google Scholar]
- Chu Y, Cochran EJ, Bennett DA, Mufson EJ, Kordower JH. Down-regulation of trkA mRNA within nucleus basalis neurons in individuals with mild cognitive impairment and Alzheimer’s disease. J Comp Neurol. 2001;437:296–307. doi: 10.1002/cne.1284. [DOI] [PubMed] [Google Scholar]
- Chuang DM. Neuroprotective and neurotrophic actions of the mood stabilizer lithium: can it be used to treat neurodegenerative diseases? Crit Rev Neurobiol. 2004;16:83–90. doi: 10.1615/critrevneurobiol.v16.i12.90. [DOI] [PubMed] [Google Scholar]
- Chuu JY, Taylor JL, Tinklenberg J, Noda A, Yesavage J, Murphy GM., Jr The brain-derived neurotrophic factor Val66Met polymorphism and rate of decline in Alzheimer’s disease. J Alzheimers Dis. 2006;9:43–49. doi: 10.3233/jad-2006-9104. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-b levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Combarros O, Infante J, Llorca J, Berciano J. Polymorphism at codon 66 of the brain-derived neurotrophic factor gene is not associated with sporadic Alzheimer’s disease. Dement Geriatr Cogn Disord. 2004;18:55–58. doi: 10.1159/000077736. [DOI] [PubMed] [Google Scholar]
- Connor B, Young D, Lawlor P, Gai W, Waldvogel H, Faull RL, Dragunow M. Trk receptor alterations in Alzheimer’s disease. Brain Res Mol Brain Res. 1996;42:1–17. doi: 10.1016/s0169-328x(96)00040-x. [DOI] [PubMed] [Google Scholar]
- Connor B, Young D, Yan Q, Faull RL, Synek B, Dragunow M. Brain-derived neurotrophic factor is reduced in Alzheimer’s disease. Brain Res Mol Brain Res. 1997;49:71–81. doi: 10.1016/s0169-328x(97)00125-3. [DOI] [PubMed] [Google Scholar]
- Costantini C, Weindruch R, Della Valle G, Puglielli L. A TrkA-to-p75NTR molecular switch activates amyloid beta-peptide generation during aging. Biochem J. 2005;391:59–67. doi: 10.1042/BJ20050700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini C, Scrable H, Puglielli L. An aging pathway controls the TrkA to p75NTR receptor switch and amyloid beta-peptide generation. EMBO J. 2006;25:1997–2006. doi: 10.1038/sj.emboj.7601062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counts SE, Nadeem M, Wuu J, Ginsberg SD, Saragovi HU, Mufson EJ. Reduction of cortical TrkA but not p75(NTR) protein in early-stage Alzheimer’s disease. Ann Neurol. 2004;56:520–531. doi: 10.1002/ana.20233. [DOI] [PubMed] [Google Scholar]
- Crutcher KA, Scott SA, Liang S, Everson WV, Weingartner J. Detection of NGF-like activity in human brain tissue: increased levels in Alzheimer’s disease. J Neurosci. 1993;13:2540–2550. doi: 10.1523/JNEUROSCI.13-06-02540.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings BJ, Su JH, Cotman CW. Neuritic involvement within bFGF immunopositive plaques of Alzheimer’s disease. Exp Neurol. 1993;124:315–325. doi: 10.1006/exnr.1993.1202. [DOI] [PubMed] [Google Scholar]
- da Penha Berzaghi M, Cooper J, Castrén E, Zafra F, Sofroniew M, Thoenen H, Lindholm D. Cholinergic regulation of brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) but not neurotrophin-3 (NT-3) mRNA levels in the developing rat hippocampus. J Neurosci. 1993;13:3813–3826. doi: 10.1523/JNEUROSCI.13-09-03818.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delacourte A, Sergeant N, Champain D, Wattez A, Maurage CA, Lebert F, Pasquier F, David JP. Nonoverlapping but synergetic tau and APP pathologies in sporadic Alzheimer’s disease. Neurology. 2002;59:398–407. doi: 10.1212/wnl.59.3.398. [DOI] [PubMed] [Google Scholar]
- De Rosa R, Garcia AA, Braschi C, Capsoni S, Maffei L, Berardi N, Cattaneo A. Intranasal administration of nerve growth factor (NGF) rescues recognition memory deficits in AD11 anti-NGF transgenic mice. Proc Natl Acad Sci U S A. 2005;102:3811–3816. doi: 10.1073/pnas.0500195102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai P, Nebes R, DeKosky ST, Kamboh MI. Investigation of the effect of brain-derived neurotrophic factor (BDNF) polymorphisms on the risk of late-onset Alzheimer’s disease (AD) and quantitative measures of AD progression. Neurosci Lett. 2005;379:229–234. doi: 10.1016/j.neulet.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Durany N, Michel T, Kurt J, Cruz-Sanchez FF, Cervas-Navarro J, Riederer P. Brain-derived neurotrophic factor and neurotrophin-3 levels in Alzheimer’s disease brains. Int J Dev Neurosci. 2000;18:807–813. [PubMed] [Google Scholar]
- Elliott E, Ginzburg I. The role of neurotrophins and insulin on tau pathology in Alzheimer’s disease. Rev Neurosci. 2006;17:635–642. doi: 10.1515/revneuro.2006.17.6.635. [DOI] [PubMed] [Google Scholar]
- Elliott E, Atlas R, Lange A, Ginzburg I. Brain-derived neurotrophic factor induces a rapid dephosphorylation of tau protein through a PI-3 kinase signalling mechanism. Eur J Neurosci. 2005;22:1081–1089. doi: 10.1111/j.1460-9568.2005.04290.x. [DOI] [PubMed] [Google Scholar]
- Fahnestock M, Scott SA, Jette N, Weingartner JA, Crutcher KA. Nerve growth factor mRNA and protein levels measured in the same tissue from normal and Alzheimer’s disease parietal cortex. Brain Res Mol Brain Res. 1996;42:175–178. doi: 10.1016/s0169-328x(96)00193-3. [DOI] [PubMed] [Google Scholar]
- Fahnestock M, Michalski B, Xu B, Coughlin MD. The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in Alzheimer’s disease. Mol Cell Neurosci. 2001;18:210–220. doi: 10.1006/mcne.2001.1016. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Marti E. Distribution of fibroblast growth factor receptor-1 (FGFR-1) and FGFR-3 in the hippocampus of patients with Alzheimer’s disease. Neurosci Lett. 1998;240:139–142. doi: 10.1016/s0304-3940(97)00948-8. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Marin C, Rey MJ, Ribalta T, Goutan E, Blanco R, Tolosa E, Marti E. BDNF and full-length and truncated TrkB expression in Alzheimer disease. Implications in therapeutic strategies. J Neuropathol Exp Neurol. 1999;58:729–739. doi: 10.1097/00005072-199907000-00007. [DOI] [PubMed] [Google Scholar]
- Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, Hall K, Hasegawa K, Hendrie H, Huang Y, Jorm A, Mathers C, Menezes PR, Rimmer E, Scazufca M. Global prevalence of dementia: a Delphi consensus study. Lancet. 2005;366:2112–2117. doi: 10.1016/S0140-6736(05)67889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher A, Heldman E, Gurwitz D, Haring R, Karton Y, Meshulam H, Pittel Z, Marciano D, Brandeis R, Sadot E, Barg Y, Pinkas-Kramarski R, Vogel Z, Ginzburg I, Treves TA, Verchovsky R, Klimowsky S, Korczyn AD. M1 agonists for the treatment of Alzheimer’s disease. Novel properties and clinical update. Ann N Y Acad Sci. 1996;777:189–196. doi: 10.1111/j.1749-6632.1996.tb34418.x. [DOI] [PubMed] [Google Scholar]
- Forero DA, Benitez B, Arboleda G, Yunis JJ, Pardo R, Arboleda H. Analysis of functional polymorphisms in three synaptic plasticity-related genes (BDNF, COMT AND UCHL1) in Alzheimer’s disease in Colombia. Neurosci Res. 2006;55:334–341. doi: 10.1016/j.neures.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Frey BN, Andreazza AC, Cereser KM, Martins MR, Valvassori SS, Reus GZ, Quevedo J, Kapczinski F. Effects of mood stabilizers on hippocampus BDNF levels in an animal model of mania. Life Sci. 2006a;79:281–286. doi: 10.1016/j.lfs.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Frey BN, Andreazza AC, Rosa AR, Martins MR, Valvassori SS, Reus GZ, Hatch JP, Quevedo J, Kapczinski F. Lithium increases nerve growth factor levels in the rat hippocampus in an animal model of mania. Behav Pharmacol. 2006b;17:311–318. doi: 10.1097/01.fbp.0000205013.59455.09. [DOI] [PubMed] [Google Scholar]
- Friedman WJ. Neurotrophins induce death of hippocampal neurons via the p75 receptor. J Neurosci. 2000;20:6340–6346. doi: 10.1523/JNEUROSCI.20-17-06340.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzon D, Yu G, Fahnestock M. A new brain-derived neurotrophic factor transcript and decrease in brain-derived neurotrophic factor transcripts 1, 2 and 3 in Alzheimer’s disease parietal cortex. J Neurochem. 2002;82:1058–1064. doi: 10.1046/j.1471-4159.2002.01030.x. [DOI] [PubMed] [Google Scholar]
- Garzon DJ, Fahnestock M. Oligomeric amyloid decreases basal levels of brain-derived neurotrophic factor (BDNF) mRNA via specific downregulation of BDNF transcripts IV and V in differentiated human neuroblastoma cells. J Neurosci. 2007;27:2628–2635. doi: 10.1523/JNEUROSCI.5053-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge YW, Lahiri DK. Regulation of promoter activity of the APP gene by cytokines and growth factors: implications in Alzheimer’s disease. Ann N Y Acad Sci. 2002;973:463–467. doi: 10.1111/j.1749-6632.2002.tb04684.x. [DOI] [PubMed] [Google Scholar]
- Giacobini E. Cholinesterases: new roles in brain function and in Alzheimer’s disease. Neurochem Res. 2003;28:515–522. doi: 10.1023/a:1022869222652. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Che S, Counts SE, Mufson EJ. Shift in the ratio of three-repeat tau and four-repeat tau mRNAs in individual cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer’s disease. J Neurochem. 2006a;96:1401–1408. doi: 10.1111/j.1471-4159.2005.03641.x. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Che S, Wuu J, Counts SE, Mufson EJ. Down regulation of trk but not p75NTR gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer’s disease. J Neurochem. 2006b;97:475–487. doi: 10.1111/j.1471-4159.2006.03764.x. [DOI] [PubMed] [Google Scholar]
- Goedert M, Fine A, Hunt SP, Ullrich A. Nerve growth factor mRNA in peripheral and central rat tissues and in the human central nervous system: lesion effects in the rat brain and levels in Alzheimer’s disease. Brain Res. 1986;387:85–92. doi: 10.1016/0169-328x(86)90023-9. [DOI] [PubMed] [Google Scholar]
- Goedert M, Fine A, Dawbarn D, Wilcock GK, Chao MV. Nerve growth factor receptor mRNA distribution in human brain: normal levels in basal forebrain in Alzheimer’s disease. Brain Res Mol Brain Res. 1989;5:1–7. doi: 10.1016/0169-328x(89)90011-9. [DOI] [PubMed] [Google Scholar]
- Goldsbury C, Mocanu MM, Thies E, Kaether C, Haass C, Keller P, Biernat J, Mandelkow E, Mandelkow EM. Inhibition of APP trafficking by tau protein does not increase the generation of amyloid-beta peptides. Traffic. 2006;7:873–888. doi: 10.1111/j.1600-0854.2006.00434.x. [DOI] [PubMed] [Google Scholar]
- Gomez-Pinilla F, Cummings BJ, Cotman CW. Induction of basic fibroblast growth factor in Alzheimer’s disease pathology. Neuroreport. 1990;1:211–214. doi: 10.1097/00001756-199011000-00009. [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Goldstein LS. Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron. 2001;32:389–401. doi: 10.1016/s0896-6273(01)00496-2. [DOI] [PubMed] [Google Scholar]
- Hanneken A, Frautschy S, Galasko D, Baird A. A fibroblast growth factor binding protein in human cerebral spinal fluid. Neuroreport. 1995;6:886–888. doi: 10.1097/00001756-199504190-00015. [DOI] [PubMed] [Google Scholar]
- Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Kaneko Y, Tsukamoto E, Frankowski H, Kouyama K, Kita Y, Niikura T, Aiso S, Bredesen DE, Matsuoka M, Nishimoto I. Molecular characterization of neurohybrid cell death induced by Alzheimer’s amyloid-beta peptides via p75NTR/PLAIDD. J Neurochem. 2004;90:549–558. doi: 10.1111/j.1471-4159.2004.02513.x. [DOI] [PubMed] [Google Scholar]
- Hasselmo ME, Stern CE. Mechanisms underlying working memory for novel information. Trends Cogn Sci. 2006;10:487–493. doi: 10.1016/j.tics.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heese K, Hock C, Otten U. Inflammatory signals induce neurotrophin expression in human microglial cells. J Neurochem. 1998;70:699–707. doi: 10.1046/j.1471-4159.1998.70020699.x. [DOI] [PubMed] [Google Scholar]
- Hellweg R, Jockers-Scherubl M. Neurotrophic factors in memory disorders. Life Sci. 1994;55:2165–2169. doi: 10.1016/0024-3205(94)00397-1. [DOI] [PubMed] [Google Scholar]
- Hellweg R, Gericke CA, Jendroska K, Hartung HD, Cervos-Navarro J. NGF content in the cerebral cortex of non-demented patients with amyloid-plaques and in symptomatic Alzheimer’s disease. Int J Dev Neurosci. 1998;16:787–794. doi: 10.1016/s0736-5748(98)00088-4. [DOI] [PubMed] [Google Scholar]
- Hellweg R, Lohmann P, Huber R, Kuhl A, Riepe MW. Spatial navigation in complex and radial mazes in APP23 animals and neurotrophin signaling as a biological marker of early impairment. Learn Mem. 2006;13:63–71. doi: 10.1101/lm.2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, O’Banion MK. Inflammatory processes in Alzheimer’s disease. J Neuroimmunol. 2007;184:69–91. doi: 10.1016/j.jneuroim.2006.11.017. [DOI] [PubMed] [Google Scholar]
- Hiruma H, Katakura T, Takahashi S, Ichikawa T, Kawakami T. Glutamate and amyloid beta-protein rapidly inhibit fast axonal transport in cultured rat hippocampal neurons by different mechanisms. J Neurosci. 2003;23:8967–8977. doi: 10.1523/JNEUROSCI.23-26-08967.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock C, Heese K, Muller-Spahn F, Hulette C, Rosenberg C, Otten U. Decreased trkA neurotrophin receptor expression in the parietal cortex of patients with Alzheimer’s disease. Neurosci Lett. 1998;241:151–154. doi: 10.1016/s0304-3940(98)00019-6. [DOI] [PubMed] [Google Scholar]
- Hock C, Heese K, Hulette C, Rosenberg C, Otten U. Region-specific neurotrophin imbalances in Alzheimer disease: decreased levels of brain-derived neurotrophic factor and increased levels of nerve growth factor in hippocampus and cortical areas. Arch Neurol. 2000a;57:846–851. doi: 10.1001/archneur.57.6.846. [DOI] [PubMed] [Google Scholar]
- Hock C, Heese K, Muller-Spahn F, Huber P, Riesen W, Nitsch RM, Otten U. Increased cerebrospinal fluid levels of neurotrophin 3 (NT-3) in elderly patients with major depression. Mol Psychiatry. 2000b;5:510–513. doi: 10.1038/sj.mp.4000743. [DOI] [PubMed] [Google Scholar]
- Hofman A, Rocca WA, Brayne C, Breteler MM, Clarke M, Cooper B, Copeland JR, Dartigues JF, da Silva Droux A, Hagnell O, Heere TJ, Engedal K, Jonker C, Lindsay J, Lobo A, Mann AH, Mölsä PK, Morgan K, O’Connor DW, Sulkava R, Kay DWK, Amaducci L, The Euroderm Prevalence Research Group The prevalence of dementia in Europe: a collaborative study of 1980-1990 findings. Eurodem Prevalence Research Group. Int J Epidemiol. 1991;20:736–748. doi: 10.1093/ije/20.3.736. [DOI] [PubMed] [Google Scholar]
- Holsinger RM, Schnarr J, Henry P, Castelo VT, Fahnestock M. Quantitation of BDNF mRNA in human parietal cortex by competitive reverse transcription-polymerase chain reaction: decreased levels in Alzheimer’s disease. Brain Res Mol Brain Res. 2000;76:347–354. doi: 10.1016/s0169-328x(00)00023-1. [DOI] [PubMed] [Google Scholar]
- Hu XY, Zhang HY, Qin S, Xu H, Swaab DF, Zhou JN. Increased p75(NTR) expression in hippocampal neurons containing hyperphosphorylated tau in Alzheimer patients. Exp Neurol. 2002;178:104–111. doi: 10.1006/exnr.2002.8018. [DOI] [PubMed] [Google Scholar]
- Huang R, Huang J, Cathcart H, Smith S, Poduslo SE. Genetic variants in brain-derived neurotrophic factor associated with Alzheimer’s disease. J Med Genet. 2007;44 doi: 10.1136/jmg.2006.044883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, Trojanowski JQ, Lee VM. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron. 1999;24:751–762. doi: 10.1016/s0896-6273(00)81127-7. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Xu G, Fromholt D, Gonzales V, Borchelt DR. Environmental enrichment exacerbates amyloid plaque formation in a transgenic mouse model of Alzheimer disease. J Neuropathol Exp Neurol. 2003;62:1220–1227. doi: 10.1093/jnen/62.12.1220. [DOI] [PubMed] [Google Scholar]
- Jarvik L, Greenson H. About a peculiar disease of the cerebral cortex. By Alois Alzheimer, 1907 (Translated by L. Jarvik and H. Greenson) Alzheimer Dis Assoc Disord. 1987;1:3–8. [PubMed] [Google Scholar]
- Jette N, Cole MS, Fahnestock M. NGF mRNA is not decreased in frontal cortex from Alzheimer’s disease patients. Brain Res Mol Brain Res. 1994;25:242–250. doi: 10.1016/0169-328x(94)90159-7. [DOI] [PubMed] [Google Scholar]
- Jin K, Sun Y, Xie L, Batteur S, Mao XO, Smelick C, Logvinova A, Greenberg DA. Neurogenesis and aging: FGF-2 and HB-EGF restore neurogenesis in hippocampus and subventricular zone of aged mice. Aging Cell. 2003;2:175–183. doi: 10.1046/j.1474-9728.2003.00046.x. [DOI] [PubMed] [Google Scholar]
- Johnson-Farley NN, Patel K, Kim D, Cowen DS. Interaction of FGF-2 with IGF-1 and BDNF in stimulating Akt, ERK, and neuronal survival in hippocampal cultures. Brain Res. 2007;1154:40–49. doi: 10.1016/j.brainres.2007.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar S, Slowikowski SP, Westaway D, Mount HT. Interactions between beta-amyloid and central cholinergic neurons: implications for Alzheimer’s disease. J Psychiatry Neurosci. 2004;29:427–441. [PMC free article] [PubMed] [Google Scholar]
- Kasa P, Rakonczay Z, Gulya K. The cholinergic system in Alzheimer’s disease. Prog Neurobiol. 1997;52:511–535. doi: 10.1016/s0301-0082(97)00028-2. [DOI] [PubMed] [Google Scholar]
- Kato T, Sasaki H, Katagiri T, Sasaki H, Koiwai K, Youki H, Totsuka S, Ishii T. The binding of basic fibroblast growth factor to Alzheimer’s neurofibrillary tangles and senile plaques. Neurosci Lett. 1991;122:33–36. doi: 10.1016/0304-3940(91)90186-w. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Kerwin JM, Morris CM, Perry RH, Perry EK. Hippocampal nerve growth factor receptor immunoreactivity in patients with Alzheimer’s and Parkinson’s disease. Neurosci Lett. 1992;143:101–104. doi: 10.1016/0304-3940(92)90242-y. [DOI] [PubMed] [Google Scholar]
- Kiprianova I, Schindowski K, von Bohlen und Halbach O, Krause S, Dono R, Schwaninger M, Unsicker K. Enlarged infarct volume and loss of BDNF mRNA induction following brain ischemia in mice lacking FGF-2. Exp Neurol. 2004;189:252–260. doi: 10.1016/j.expneurol.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Klenk J, Rapp K, Buchele G, Keil U, Weiland SK. Increasing life expectancy in Germany: quantitative contributions from changes in age- and disease-specific mortality. Eur J Public Health. 2007 doi: 10.1093/eurpub/ckm024. [DOI] [PubMed] [Google Scholar]
- Kokubo H, Kayed R, Glabe CG, Yamaguchi H. Soluble Abeta oligomers ultrastructurally localize to cell processes and might be related to synaptic dysfunction in Alzheimer’s disease brain. Brain Res. 2005;1031:222–228. doi: 10.1016/j.brainres.2004.10.041. [DOI] [PubMed] [Google Scholar]
- Kraemer BC, Zhang B, Leverenz JB, Thomas JH, Trojanowski JQ, Schellenberg GD. Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc Natl Acad Sci U S A. 2003;100:9980–9985. doi: 10.1073/pnas.1533448100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuner P, Hertel C. NGF induces apoptosis in a human neuroblastoma cell line expressing the neurotrophin receptor p75NTR. J Neurosci Res. 1998;54:465–474. doi: 10.1002/(SICI)1097-4547(19981115)54:4<465::AID-JNR4>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Kunugi H, Hattori M, Ueki A, Isse K, Hirasawa H, Nanko S. Possible association of missense mutation (Gly[-63]Glu) of the neurotrophin-3 gene with Alzheimer’s disease in Japanese. Neurosci Lett. 1998;241:65–67. doi: 10.1016/s0304-3940(97)00953-1. [DOI] [PubMed] [Google Scholar]
- Kunugi H, Ueki A, Otsuka M, Isse K, Hirasawa H, Kato N, Nabika T, Kobayashi S, Nanko S. A novel polymorphism of the brain-derived neurotrophic factor (BDNF) gene associated with late-onset Alzheimer’s disease. Mol Psychiatry. 2001;6:83–86. doi: 10.1038/sj.mp.4000792. [DOI] [PubMed] [Google Scholar]
- Lang UE, Muhlbacher M, Hesselink MB, Zajaczkowski W, Danysz W, Danker-Hopfe H, Hellweg R. No nerve growth factor response to treatment with memantine in adult rats. J Neural Transm. 2004;111:181–190. doi: 10.1007/s00702-003-0090-y. [DOI] [PubMed] [Google Scholar]
- Lazarov O, Lee M, Peterson DA, Sisodia SS. Evidence that synaptically released beta-amyloid accumulates as extracellular deposits in the hippocampus of transgenic mice. J Neurosci. 2002;22:9785–9793. doi: 10.1523/JNEUROSCI.22-22-09785.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarov O, Robinson J, Tang YP, Hairston IS, Korade-Mirnics Z, Lee VM, Hersh LB, Sapolsky RM, Mirnics K, Sisodia SS. Environmental enrichment reduces Abeta levels and amyloid deposition in transgenic mice. Cell. 2005;120:701–713. doi: 10.1016/j.cell.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Lee EB, Zhang B, Liu K, Greenbaum EA, Doms RW, Trojanowski JQ, Lee VM. BACE overexpression alters the subcellular processing of APP and inhibits Ab deposition in vivo. J Cell Biol. 2005a;168:291–302. doi: 10.1083/jcb.200407070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Fukumoto H, Orne J, Klucken J, Raju S, Vanderburg CR, Irizarry MC, Hyman BT, Ingelsson M. Decreased levels of BDNF protein in Alzheimer temporal cortex are independent of BDNF polymorphisms. Exp Neurol. 2005b;194:91–96. doi: 10.1016/j.expneurol.2005.01.026. [DOI] [PubMed] [Google Scholar]
- Lesné S, Gabriel C, Nelson DA, White E, Mackenzie ET, Vivien D, Buisson A. Akt-dependent expression of NAIP-1 protects neurons against amyloid-β toxicity. J Biol Chem. 2005;280:24941–24947. doi: 10.1074/jbc.M413495200. [DOI] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Li Y, Rowland C, Tacey K, Catanese J, Sninsky J, Hardy J, Powell J, Lovestone S, Morris JC, Thal L, Goate A, Owen M, Williams J, Grupe A. The BDNF Val66Met polymorphism is not associated with late onset Alzheimer’s disease in three case-control samples. Mol Psychiatry. 2005;10:809–810. doi: 10.1038/sj.mp.4001702. [DOI] [PubMed] [Google Scholar]
- Loy R, Heyer D, Clagett-Dame M, DiStefano PS. Localization of NGF receptors in normal and Alzheimer’s basal forebrain with monoclonal antibodies against the truncated form of the receptor. J Neurosci Res. 1990;27:651–664. doi: 10.1002/jnr.490270426. [DOI] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci U S A. 2006;103:5644–5651. doi: 10.1073/pnas.0600549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai L, Jope RS, Li X. BDNF-mediated signal transduction is modulated by GSK3beta and mood stabilizing agents. J Neurochem. 2002;82:75–83. doi: 10.1046/j.1471-4159.2002.00939.x. [DOI] [PubMed] [Google Scholar]
- Maloney MT, Minamide LS, Kinley AW, Boyle JA, Bamburg JR. Beta-secretase-cleaved amyloid precursor protein accumulates at actin inclusions induced in neurons by stress or amyloid beta: a feedforward mechanism for Alzheimer’s disease. J Neurosci. 2005;25:11313–11321. doi: 10.1523/JNEUROSCI.3711-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow EM, Thies E, Trinczek B, Biernat J, Mandelkow E. MARK/PAR1 kinase is a regulator of microtubule-dependent transport in axons. J Cell Biol. 2004;167:99–110. doi: 10.1083/jcb.200401085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manji HK, Moore GJ, Chen G. Lithium at 50: have the neuroprotective effects of this unique cation been overlooked? Biol Psychiatry. 1999;46:929–940. doi: 10.1016/s0006-3223(99)00165-1. [DOI] [PubMed] [Google Scholar]
- Marvanova M, Lakso M, Pirhonen J, Nawa H, Wong G, Castren E. The neuroprotective agent memantine induces brain-derived neurotrophic factor and trkB receptor expression in rat brain. Mol Cell Neurosci. 2001;18:247–258. doi: 10.1006/mcne.2001.1027. [DOI] [PubMed] [Google Scholar]
- Matsushita S, Arai H, Matsui T, Yuzuriha T, Urakami K, Masaki T, Higuchi S. Brain-derived neurotrophic factor gene polymorphisms and Alzheimer’s disease. J Neural Transm. 2005;112:703–711. doi: 10.1007/s00702-004-0210-3. [DOI] [PubMed] [Google Scholar]
- Michalski B, Fahnestock M. Pro-brain-derived neurotrophic factor is decreased in parietal cortex in Alzheimer’s disease. Brain Res Mol Brain Res. 2003;111:148–154. doi: 10.1016/s0169-328x(03)00003-2. [DOI] [PubMed] [Google Scholar]
- Mobley WC, Neve RL, Prusiner SB, McKinley MP. Nerve growth factor increases mRNA levels for the prion protein and the beta-amyloid protein precursor in developing hamster brain. Proc Natl Acad Sci U S A. 1988;85:9811–9815. doi: 10.1073/pnas.85.24.9811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudher A, Shepherd D, Newman TA, Mildren P, Jukes JP, Squire A, Mears A, Drummond JA, Berg S, MacKay D, Asuni AA, Bhat R, Lovestone S. GSK-3beta inhibition reverses axonal transport defects and behavioural phenotypes in Drosophila. Mol Psychiatry. 2004;9:522–530. doi: 10.1038/sj.mp.4001483. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Conner JM, Kordower JH. Nerve growth factor in Alzheimer’s disease: defective retrograde transport to nucleus basalis. Neuroreport. 1995;6:1063–1066. doi: 10.1097/00001756-199505090-00028. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Lavine N, Jaffar S, Kordower JH, Quirion R, Saragovi HU. Reduction in p140-TrkA receptor protein within the nucleus basalis and cortex in Alzheimer’s disease. Exp Neurol. 1997;146:91–103. doi: 10.1006/exnr.1997.6504. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Ma SY, Cochran EJ, Bennett DA, Beckett LA, Jaffar S, Saragovi HU, Kordower JH. Loss of nucleus basalis neurons containing trkA immunoreactivity in individuals with mild cognitive impairment and early Alzheimer’s disease. J Comp Neurol. 2000;427:19–30. doi: 10.1002/1096-9861(20001106)427:1<19::aid-cne2>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Ma SY, Dills J, Cochran EJ, Leurgans S, Wuu J, Bennett DA, Jaffar S, Gilmor ML, Levey AI, Kordower JH. Loss of basal forebrain P75(NTR) immunoreactivity in subjects with mild cognitive impairment and Alzheimer’s disease. J Comp Neurol. 2002;443:136–153. doi: 10.1002/cne.10122. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Ginsberg SD, Ikonomovic MD, DeKosky ST. Human cholinergic basal forebrain: chemoanatomy and neurologic dysfunction. J Chem Neuroanat. 2003;26:233–242. doi: 10.1016/s0891-0618(03)00068-1. [DOI] [PubMed] [Google Scholar]
- Murase K, Igarashi K, Hayashi K. Neurotrophin-3 (NT-3) levels in the developing rat nervous system and in human samples. Clin Chim Acta. 1994;227:23–36. doi: 10.1016/0009-8981(94)90132-5. [DOI] [PubMed] [Google Scholar]
- Murer MG, Boissiere F, Yan Q, Hunot S, Villares J, Faucheux B, Agid Y, Hirsch E, Raisman-Vozari R. An immunohistochemical study of the distribution of brain-derived neurotrophic factor in the adult human brain, with particular reference to Alzheimer’s disease. Neuroscience. 1999;88:1015–1032. doi: 10.1016/s0306-4522(98)00219-x. [DOI] [PubMed] [Google Scholar]
- Murer MG, Yan Q, Raisman-Vozari R. Brain-derived neurotrophic factor in the control human brain, and in Alzheimer’s disease and Parkinson’s disease. Prog Neurobiol. 2001;63:71–124. doi: 10.1016/s0301-0082(00)00014-9. [DOI] [PubMed] [Google Scholar]
- Muresan Z, Muresan V. Neuritic deposits of amyloid-b peptide in a subpopulation of central nervous system-derived neuronal cells. Mol Cell Biol. 2006;26:4982–4997. doi: 10.1128/MCB.00371-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacmias B, Piccini C, Bagnoli S, Tedde A, Cellini E, Bracco L, Sorbi S. Brain-derived neurotrophic factor, apolipoprotein E genetic variants and cognitive performance in Alzheimer’s disease. Neurosci Lett. 2004;367:379–383. doi: 10.1016/j.neulet.2004.06.039. [DOI] [PubMed] [Google Scholar]
- Narisawa-Saito M, Wakabayashi K, Tsuji S, Takahashi H, Nawa H. Regional specificity of alterations in NGF, BDNF and NT-3 levels in Alzheimer’s disease. Neuroreport. 1996;7:2925–2928. doi: 10.1097/00001756-199611250-00024. [DOI] [PubMed] [Google Scholar]
- Nishimura M, Kuno S, Kaji R, Kawakami H. Brain-derived neurotrophic factor gene polymorphisms in Japanese patients with sporadic Alzheimer’s disease, Parkinson’s disease, and multiple system atrophy. Mov Disord. 2005;20:1031–1033. doi: 10.1002/mds.20491. [DOI] [PubMed] [Google Scholar]
- Nuydens R, Dispersyn G, de Jong M, van den Kieboom G, Borgers M, Geerts H. Aberrant tau phosphorylation and neurite retraction during NGF deprivation in PC12 cells. Biochem Biophys Res Commun. 1997;240:687–691. doi: 10.1006/bbrc.1997.7721. [DOI] [PubMed] [Google Scholar]
- Olin D, MacMurray J, Comings DE. Risk of late-onset Alzheimer’s disease associated with BDNF C270T polymorphism. Neurosci Lett. 2005;381:275–278. doi: 10.1016/j.neulet.2005.02.017. [DOI] [PubMed] [Google Scholar]
- Olivieri G, Novakovic M, Savaskan E, Meier F, Baysang G, Brockhaus M, Muller-Spahn F. The effects of beta-estradiol on SHSY5Y neuroblastoma cells during heavy metal induced oxidative stress, neurotoxicity and beta-amyloid secretion. Neuroscience. 2002;113:849–855. doi: 10.1016/s0306-4522(02)00211-7. [DOI] [PubMed] [Google Scholar]
- Olivieri G, Otten U, Meier F, Baysang G, Dimitriades-Schmutz B, Muller-Spahn F, Savaskan E. Beta-amyloid modulates tyrosine kinase B receptor expression in SHSY5Y neuroblastoma cells: influence of the antioxidant melatonin. Neuroscience. 2003;120:659–665. doi: 10.1016/s0306-4522(03)00342-7. [DOI] [PubMed] [Google Scholar]
- Origlia N, Capsoni S, Domenici L, Cattaneo A. Time window in cholinomimetic ability to rescue long-term potentiation in neurodegenerating anti-nerve growth factor mice. J Alzheimers Dis. 2006;9:59–68. doi: 10.3233/jad-2006-9106. [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Reichardt LF. Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol. 2001;11:272–280. doi: 10.1016/s0959-4388(00)00208-7. [DOI] [PubMed] [Google Scholar]
- Peng S, Wuu J, Mufson EJ, Fahnestock M. Increased proNGF levels in subjects with mild cognitive impairment and mild Alzheimer disease. J Neuropathol Exp Neurol. 2004;63:641–649. doi: 10.1093/jnen/63.6.641. [DOI] [PubMed] [Google Scholar]
- Peng S, Wuu J, Mufson EJ, Fahnestock M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J Neurochem. 2005;93:1412–1421. doi: 10.1111/j.1471-4159.2005.03135.x. [DOI] [PubMed] [Google Scholar]
- Perry EK, Johnson M, Kerwin JM, Piggott MA, Court JA, Shaw PJ, Ince PG, Brown A, Perry RH. Convergent cholinergic activities in aging and Alzheimer’s disease. Neurobiol Aging. 1992;13:393–400. doi: 10.1016/0197-4580(92)90113-c. [DOI] [PubMed] [Google Scholar]
- Perry EK, Irving D, Kerwin JM, McKeith IG, Thompson P, Collerton D, Fairbairn AF, Ince PG, Morris CM, Cheng AV, Perry RH. Cholinergic transmitter and neurotrophic activities in Lewy body dementia: similarity to Parkinson’s and distinction from Alzheimer disease. Alzheimer Dis Assoc Disord. 1993;7:69–79. doi: 10.1097/00002093-199307020-00002. [DOI] [PubMed] [Google Scholar]
- Phillips HS, Hains JM, Armanini M, Laramee GR, Johnson SA, Winslow JW. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron. 1991;7:695–702. doi: 10.1016/0896-6273(91)90273-3. [DOI] [PubMed] [Google Scholar]
- Pike CJ, Cummings BJ, Monzavi R, Cotman CW. Beta-amyloid-induced changes in cultured astrocytes parallel reactive astrocytosis associated with senile plaques in Alzheimer’s disease. Neuroscience. 1994;63:517–531. doi: 10.1016/0306-4522(94)90547-9. [DOI] [PubMed] [Google Scholar]
- Polvikoski T, Sulkava R, Haltia M, Kainulainen K, Vuorio A, Verkkoniemi A, Niinisto L, Halonen P, Kontula K. Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. N Engl J Med. 1995;333:1242–1247. doi: 10.1056/NEJM199511093331902. [DOI] [PubMed] [Google Scholar]
- Probst A, Gotz J, Wiederhold KH, Tolnay M, Mistl C, Jaton AL, Hong M, Ishihara T, Lee VM, Trojanowski JQ, Jakes R, Crowther RA, Spillantini MG, Burki K, Goedert M. Axonopathy and amyotrophy in mice transgenic for human four-repeat tau protein. Acta Neuropathol (Berl) 2000;99:469–481. doi: 10.1007/s004010051148. [DOI] [PubMed] [Google Scholar]
- Qiu Z, Naten DL, Liston JC, Yess J, Rebeck GW. A novel approach for studying endogenous ab processing using cultured primary neurons isolated from APP transgenic mice. Exp Neurol. 2001;170:186–194. doi: 10.1006/exnr.2001.7703. [DOI] [PubMed] [Google Scholar]
- Riemenschneider M, Schwarz S, Wagenpfeil S, Diehl J, Muller U, Forstl H, Kurz A. A polymorphism of the brain-derived neurotrophic factor (BDNF) is associated with Alzheimer’s disease in patients lacking the Apolipoprotein E epsilon4 allele. Mol Psychiatry. 2002;7:782–785. doi: 10.1038/sj.mp.4001073. [DOI] [PubMed] [Google Scholar]
- Robakis NK, Anderson JP, Refolo LM, Wallace W. Expression of the Alzheimer amyloid precursor in brain tissue and effects of NGF and EGF on its metabolism. Clin Neuropharmacol. 1991;14(Suppl. 1):S15–S23. doi: 10.1097/00002826-199114001-00004. [DOI] [PubMed] [Google Scholar]
- Rossner S, Ueberham U, Schliebs R, Perez-Polo JR, Bigl V. p75 and TrkA receptor signaling independently regulate amyloid precursor protein mRNA expression, isoform composition, and protein secretion in PC12 cells. J Neurochem. 1998;71:757–766. doi: 10.1046/j.1471-4159.1998.71020757.x. [DOI] [PubMed] [Google Scholar]
- Ruiz-Leon Y, Pascual A. Regulation of beta-amyloid precursor protein expression by brain-derived neurotrophic factor involves activation of both the Ras and phosphatidylinositide 3-kinase signalling pathways. J Neurochem. 2004;88:1010–1018. doi: 10.1046/j.1471-4159.2003.02226.x. [DOI] [PubMed] [Google Scholar]
- Saarela MS, Lehtimaki T, Rinne JO, Huhtala H, Rontu R, Hervonen A, Roytta M, Ahonen JP, Mattila KM. No association between the brain-derived neurotrophic factor 196 G>A or 270 C>T polymorphisms and Alzheimer’s or Parkinson’s disease. Folia Neuropathol. 2006;44:12–16. [PubMed] [Google Scholar]
- Sadot E, Heicklen-Klein A, Barg J, Lazarovici P, Ginzburg I. Identification of a tau promoter region mediating tissue-specific-regulated expression in PC12 cells. J Mol Biol. 1996;256:805–812. doi: 10.1006/jmbi.1996.0126. [DOI] [PubMed] [Google Scholar]
- Saez ET, Pehar M, Vargas MR, Barbeito L, Maccioni RB. Production of nerve growth factor by beta-amyloid-stimulated astrocytes induces p75(NTR)-dependent tau hyperphosphorylation in cultured hippocampal neurons. J Neurosci Res. 2006;84:1098–1106. doi: 10.1002/jnr.20996. [DOI] [PubMed] [Google Scholar]
- Salehi A, Verhaagen J, Dijkhuizen PA, Swaab DF. Co-localization of high-affinity neurotrophin receptors in nucleus basalis of Meynert neurons and their differential reduction in Alzheimer’s disease. Neuroscience. 1996;75:373–387. doi: 10.1016/0306-4522(96)00273-4. [DOI] [PubMed] [Google Scholar]
- Salehi A, Ocampo M, Verhaagen J, Swaab DF. P75 neurotrophin receptor in the nucleus basalis of meynert in relation to age, sex, and Alzheimer’s disease. Exp Neurol. 2000;161:245–258. doi: 10.1006/exnr.1999.7252. [DOI] [PubMed] [Google Scholar]
- Salehi A, Delcroix JD, Belichenko PV, Zhan K, Wu C, Valletta JS, Takimoto-Kimura R, Kleschevnikov AM, Sambamurti K, Chung PP, Xia W, Villar A, Campbell WA, Kulnane LS, Nixon RA, Lamb BT, Epstein CJ, Stokin GB, Goldstein LS, Mobley WC. Increased App expression in a mouse model of Down’s syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006;51:29–42. doi: 10.1016/j.neuron.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Savaskan E, Muller-Spahn F, Olivieri G, Bruttel S, Otten U, Rosenberg C, Hulette C, Hock C. Alterations in trk A, trk B and trk C receptor immunoreactivities in parietal cortex and cerebellum in Alzheimer’s disease. Eur Neurol. 2000;44:172–180. doi: 10.1159/000008229. [DOI] [PubMed] [Google Scholar]
- Scott SA, Mufson EJ, Weingartner JA, Skau KA, Crutcher KA. Nerve growth factor in Alzheimer’s disease: increased levels throughout the brain coupled with declines in nucleus basalis. J Neurosci. 1995;15:6213–6221. doi: 10.1523/JNEUROSCI.15-09-06213.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz A, Kojima H, Oiwa K, Mandelkow EM, Song YH, Mandelkow E. Single-molecule investigation of the interference between kinesin, tau and MAP2c. EMBO J. 2002;21:4896–4905. doi: 10.1093/emboj/cdf503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Sergeant N, Delacourte A, Buee L. Tau protein as a differential biomarker of tauopathies. Biochim Biophys Acta. 2005;1739:179–197. doi: 10.1016/j.bbadis.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Shelton SB, Johnson GV. Tau and HMW tau phosphorylation and compartmentalization in apoptotic neuronal PC12 cells. J Neurosci Res. 2001;66:203–213. doi: 10.1002/jnr.1212. [DOI] [PubMed] [Google Scholar]
- Shen ZX. Brain cholinesterases: III. Future perspectives of AD research and clinical practice. Med Hypotheses. 2004;63:298–307. doi: 10.1016/j.mehy.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Sheng JG, Price DL, Koliatsos VE. Disruption of corticocortical connections ameliorates amyloid burden in terminal fields in a transgenic model of Ab amyloidosis. J Neurosci. 2002;22:9794–9799. doi: 10.1523/JNEUROSCI.22-22-09794.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siedlak SL, Cras P, Kawai M, Richey P, Perry G. Basic fibroblast growth factor binding is a marker for extracellular neurofibrillary tangles in Alzheimer disease. J Histochem Cytochem. 1991;39:899–904. doi: 10.1177/39.7.1865106. [DOI] [PubMed] [Google Scholar]
- Siegel GJ, Chauhan NB. Neurotrophic factors in Alzheimer’s and Parkinson’s disease brain. Brain Res Brain Res Rev. 2000;33:199–227. doi: 10.1016/s0165-0173(00)00030-8. [DOI] [PubMed] [Google Scholar]
- Sokol DK, Chen D, Farlow MR, Dunn DW, Maloney B, Zimmer JA, Lahiri DK. High levels of Alzheimer beta-amyloid precursor protein (APP) in children with severely autistic behavior and aggression. J Child Neurol. 2006;21:444–449. doi: 10.1177/08830738060210062201. [DOI] [PubMed] [Google Scholar]
- Soto I, Rosenthal JJ, Blagburn JM, Blanco RE. Fibroblast growth factor 2 applied to the optic nerve after axotomy up-regulates BDNF and TrkB in ganglion cells by activating the ERK and PKA signaling pathways. J Neurochem. 2006;96:82–96. doi: 10.1111/j.1471-4159.2005.03510.x. [DOI] [PubMed] [Google Scholar]
- Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol. 2002;156:1051–1063. doi: 10.1083/jcb.200108057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stieber A, Mourelatos Z, Gonatas NK. In Alzheimer’s disease the Golgi apparatus of a population of neurons without neurofibrillary tangles is fragmented and atrophic. Am J Pathol. 1996;148:415–426. [PMC free article] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- Svendsen CN, Cooper JD, Sofroniew MV. Trophic factor effects on septal cholinergic neurons. Ann N Y Acad Sci. 1991;640:91–94. doi: 10.1111/j.1749-6632.1991.tb00197.x. [DOI] [PubMed] [Google Scholar]
- Takahashi RH, Almeida CG, Kearney PF, Yu F, Lin MT, Milner TA, Gouras GK. Oligomerization of Alzheimer’s beta-amyloid within processes and synapses of cultured neurons and brain. J Neurosci. 2004;24:3592–3599. doi: 10.1523/JNEUROSCI.5167-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takami K, Matsuo A, Terai K, Walker DG, McGeer EG, McGeer PL. Fibroblast growth factor receptor-1 expression in the cortex and hippocampus in Alzheimer’s disease. Brain Res. 1998;802:89–97. doi: 10.1016/s0006-8993(98)00552-6. [DOI] [PubMed] [Google Scholar]
- Tatebayashi Y, Iqbal K, Grundke-Iqbal I. Dynamic regulation of expression and phosphorylation of tau by fibroblast growth factor-2 in neural progenitor cells from adult rat hippocampus. J Neurosci. 1999;19:5245–5254. doi: 10.1523/JNEUROSCI.19-13-05245.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatebayashi Y, Lee MH, Li L, Iqbal K, Grundke-Iqbal I. The dentate gyrus neurogenesis: a therapeutic target for Alzheimer’s disease. Acta Neuropathol (Berl) 2003;105:225–232. doi: 10.1007/s00401-002-0636-3. [DOI] [PubMed] [Google Scholar]
- Tatebayashi Y, Haque N, Tung YC, Iqbal K, Grundke-Iqbal I. Role of tau phosphorylation by glycogen synthase kinase-3beta in the regulation of organelle transport. J Cell Sci. 2004;117:1653–1663. doi: 10.1242/jcs.01018. [DOI] [PubMed] [Google Scholar]
- Thoenen H, Zafra F, Hengerer B, Lindholm D. The synthesis of nerve growth factor and brain-derived neurotrophic factor in hippocampal and cortical neurons is regulated by specific transmitter systems. Ann N Y Acad Sci. 1991;640:86–90. doi: 10.1111/j.1749-6632.1991.tb00196.x. [DOI] [PubMed] [Google Scholar]
- Tong L, Shen H, Perreau VM, Balazs R, Cotman CW. Effects of exercise on gene-expression profile in the rat hippocampus. Neurobiol Dis. 2001a;8:1046–1056. doi: 10.1006/nbdi.2001.0427. [DOI] [PubMed] [Google Scholar]
- Tong L, Thornton PL, Balazs R, Cotman CW. Beta-amyloid-(1-42) impairs activity-dependent cAMP-response element-binding protein signaling in neurons at concentrations in which cell survival is not compromised. J Biol Chem. 2001b;276:17301–17306. doi: 10.1074/jbc.M010450200. [DOI] [PubMed] [Google Scholar]
- Tong L, Balazs R, Thornton PL, Cotman CW. Beta-amyloid peptide at sublethal concentrations downregulates brain-derived neurotrophic factor functions in cultured cortical neurons. J Neurosci. 2004;24:6799–6809. doi: 10.1523/JNEUROSCI.5463-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treanor JJ, Dawbarn D, Allen SJ, MacGowan SH, Wilcock GK. Low affinity nerve growth factor receptor binding in normal and Alzheimer’s disease basal forebrain. Neurosci Lett. 1991;121:73–76. doi: 10.1016/0304-3940(91)90652-a. [DOI] [PubMed] [Google Scholar]
- Tsai J, Grutzendler J, Duff K, Gan WB. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat Neurosci. 2004a;7:1181–1183. doi: 10.1038/nn1335. [DOI] [PubMed] [Google Scholar]
- Tsai SJ, Hong CJ, Liu HC, Liu TY, Hsu LE, Lin CH. Association analysis of brain-derived neurotrophic factor Val66Met polymorphisms with Alzheimer’s disease and age of onset. Neuropsychobiology. 2004b;49:10–12. doi: 10.1159/000075332. [DOI] [PubMed] [Google Scholar]
- Tsai SJ, Hong CJ, Liu HC, Liu TY, Liou YJ. The brain-derived neurotrophic factor gene as a possible susceptibility candidate for Alzheimer’s disease in a Chinese population. Dement Geriatr Cogn Disord. 2006;21:139–143. doi: 10.1159/000090673. [DOI] [PubMed] [Google Scholar]
- Tsukamoto E, Hashimoto Y, Kanekura K, Niikura T, Aiso S, Nishimoto I. Characterization of the toxic mechanism triggered by Alzheimer’s amyloid-beta peptides via p75 neurotrophin receptor in neuronal hybrid cells. J Neurosci Res. 2003;73:627–636. doi: 10.1002/jnr.10703. [DOI] [PubMed] [Google Scholar]
- Tuszynski MH, Smith DE, Roberts J, McKay H, Mufson E. Targeted intraparenchymal delivery of human NGF by gene transfer to the primate basal forebrain for 3 months does not accelerate beta-amyloid plaque deposition. Exp Neurol. 1998;154:573–582. doi: 10.1006/exnr.1998.6956. [DOI] [PubMed] [Google Scholar]
- Tuszynski MH, Thal L, Pay M, Salmon DP, U HS, Bakay R, Patel P, Blesch A, Vahlsing HL, Ho G, Tong G, Potkin SG, Fallon J, Hansen L, Mufson EJ, Kordower JH, Gall C, Conner J. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat Med. 2005;11:551–555. doi: 10.1038/nm1239. [DOI] [PubMed] [Google Scholar]
- Venero JL, Knusel B, Beck KD, Hefti F. Expression of neurotrophin and trk receptor genes in adult rats with fimbria transections: effect of intraventricular nerve growth factor and brain-derived neurotrophic factor administration. Neuroscience. 1994;59:797–815. doi: 10.1016/0306-4522(94)90285-2. [DOI] [PubMed] [Google Scholar]
- Vepsalainen S, Castren E, Helisalmi S, Iivonen S, Mannermaa A, Lehtovirta M, Hanninen T, Soininen H, Hiltunen M. Genetic analysis of BDNF and TrkB gene polymorphisms in Alzheimer’s disease. J Neurol. 2005;252:423–428. doi: 10.1007/s00415-005-0667-5. [DOI] [PubMed] [Google Scholar]
- Verdile G, Fuller S, Atwood CS, Laws SM, Gandy SE, Martins RN. The role of beta amyloid in Alzheimer’s disease: still a cause of everything or the only one who got caught? Pharmacol Res. 2004;50:397–409. doi: 10.1016/j.phrs.2003.12.028. [DOI] [PubMed] [Google Scholar]
- Villa A, Latasa MJ, Pascual A. Nerve growth factor modulates the expression and secretion of beta-amyloid precursor protein through different mechanisms in PC12 cells. J Neurochem. 2001;77:1077–1084. doi: 10.1046/j.1471-4159.2001.00315.x. [DOI] [PubMed] [Google Scholar]
- Wirths O, Weis J, Kayed R, Saido TC, Bayer TA. Age-dependent axonal degeneration in an Alzheimer mouse model. Neurobiol Aging. 2006;8:1689–1699. doi: 10.1016/j.neurobiolaging.2006.07.021. [DOI] [PubMed] [Google Scholar]
- Wolf SA, Kronenberg G, Lehmann K, Blankenship A, Overall R, Staufenbiel M, Kempermann G. Cognitive and physical activity differently modulate disease progression in the amyloid precursor protein (APP)-23 model of Alzheimer’s disease. Biol Psychiatry. 2006;60:1314–1323. doi: 10.1016/j.biopsych.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Wolfe MS, Kopan R. Intramembrane proteolysis: theme and variations. Science. 2004;305:1119–1123. doi: 10.1126/science.1096187. [DOI] [PubMed] [Google Scholar]
- Wynn ZJ, Cummings JL. Cholinesterase inhibitor therapies and neuropsychiatric manifestations of Alzheimer’s disease. Dement Geriatr Cogn Disord. 2004;17:100–108. doi: 10.1159/000074281. [DOI] [PubMed] [Google Scholar]
- Yamada T, Yoshiyama Y, Kawaguchi N. Expression of activating transcription factor-2 (ATF-2), one of the cyclic AMP response element (CRE) binding proteins, in Alzheimer disease and non-neurological brain tissues. Brain Res. 1997;749:329–334. doi: 10.1016/S0006-8993(96)01356-X. [DOI] [PubMed] [Google Scholar]
- Yamamoto-Sasaki M, Ozawa H, Saito T, Rosler M, Riederer P. Impaired phosphorylation of cyclic AMP response element binding protein in the hippocampus of dementia of the Alzheimer type. Brain Res. 1999;824:300–303. doi: 10.1016/s0006-8993(99)01220-2. [DOI] [PubMed] [Google Scholar]
- Yankner BA, Caceres A, Duffy LK. Nerve growth factor potentiates the neurotoxicity of beta amyloid. Proc Natl Acad Sci U S A. 1990;87:9020–9023. doi: 10.1073/pnas.87.22.9020. [DOI] [PMC free article] [PubMed] [Google Scholar]