

Abstract
In the present paper, we characterize an antibody, mAb BV13, directed to mouse vascular endothelial (VE)-cadherin, a major adhesive protein of interendothelial adherens junctions. When added to cultured endothelial cells, BV13 induces a redistribution of VE-cadherin from intercellular junctions. VE-cadherin redistribution did not change the localization of platelet endothelial cell adhesion molecule or tight junction markers such as zonula occludens 1, cingulin, and junctional adhesion molecule. Intravenous administration of mAb BV13 induced a concentration- and time-dependent increase in vascular permeability in heart and lungs. By electron microscopy, interstitial edema and accumulation of mixed types of inflammatory cells in heart and lungs were observed. Injection of (rhodamine-labeled) Ricinus communis I lectin showed focal spots of exposed basement membrane in the alveolar capillaries and in some larger pulmonary vessels. These data indicate that VE-cadherin is required for vascular integrity and normal organ functions.
Keywords: endothelium, permeability, adherens junction
The endothelium regulates vascular permeability to plasma proteins and circulating cells. This specific function is mediated by different systems, which include transcellular and paracellular pathways (1–7). Endothelial cell-to-cell junctions are complex structures formed by different adhesive molecules (8, 9). Endothelial cells have tight junctions (TJ) and adherens junctions (AJ), which present a general organization similar to that described in epithelial cells (10–13). In addition, other adhesive proteins such as platelet endothelial cell adhesion molecule-1 (PECAM-1), S-endo 1/muc 18, endoglin, and CD34 are concentrated at intercellular contacts in the endothelium (14).
AJs are ubiquitous along the vascular tree and are formed by transmembrane proteins belonging to the cadherin superfamily (10, 11). Endothelial cells express a cell-specific cadherin that was called vascular endothelial (VE)-cadherin (8, 15). This protein is linked inside the cells to β-catenin and plakoglobin, which, in turn, through the binding to α-catenin, promote the anchorage to the actin cytoskeleton. Although the extracellular domain of VE-cadherin is necessary for homotypic adhesion and clustering, the intracellular association to catenins and the actin cytoskeleton is required for the stabilization of the complex and a full control of junctional permeability (16).
TJs, in contrast, are formed by different molecular structures. Three types of transmembrane proteins, occludin (17), claudins (18), and junctional adhesion molecule (JAM) (19), have been found to colocalize with TJ. Inside the cells, several cytoskeletal signaling molecules are concentrated in the TJ area, such as zonula occludens 1 (ZO-1), cingulin, and 7H6 (12, 13).
AJ and, in particular VE-cadherin, are targets of the signaling pathway of agents that increase vascular permeability such as vascular endothelial growth factor (20, 21), histamine, and thrombin (22, 23). Endothelial cells that carry a null mutation in the VE-cadherin gene present major alterations in their functional behavior and cannot organize vascular like structures (24). To study the biological role of VE-cadherin in vivo in the adult, we developed an anti-VE-cadherin blocking mAb. The data suggest that VE-cadherin exerts a relevant and specific activity in the maintenance of vascular integrity.
MATERIALS AND METHODS
Cells.
Endothelial cells from mouse lung (1G11) and heart (H5V) microcirculation were kindly provided by A. Vecchi (Istituto Mario Negri, Milan) (25, 26). VE-cadherin-transfected Chinese hamster ovary cells were previously described (27, 28).
Antibodies.
Lewis rats were immunized with the mouse VE-cadherin-Ig fusion protein spanning amino acids 1–486 of the extracellular domain (29). mAbs were produced (30) and screened for reactivity with VE-cadherin-IgG and H5V cells by an ELISA test (30) and by immunofluorescence microscopy (31). Fab fragments of BV13 were prepared by standard procedures (32, 33). By cross-competitive mAb-binding assay (32), BV14 and BV13 appear to recognize a different epitope.
The antibodies used in fluorescence microscopy were rabbit polyclonal antibody to mouse VE-cadherin (rabbit antiserum raised by injecting the recombinant fragment of VE-cadherin extracellular domain, as indicated above); purified rat immunoglobulins anti-PECAM-1, MEC 7.46 (30); rat mAb anti-JAM (19); rat mAb anti-ZO-1 by B. Stevenson (University of Alberta, Canada) (34); rabbit polyclonal antibody to cingulin by S. Citi (Università di Padova, Padova, Italy;) (35, 36). Antibodies to α- and β-catenin and plakoglobin were from Transduction Laboratories (Lexington, KY).
In Vitro Assays.
The methodology for the evaluation of permeability of VE-cadherin-transfected Chinese hamster ovary cell monolayers (21, 28), immunofluorescence analysis (15, 31), immunoprecipitation, and Western blot was as described (16, 21, 31). Biotinylation of cell surface proteins was performed by using sulfo-nitrohydroxysuccinimido-biotin (Pierce) (16).
In Vivo Experiments.
Male BALB/c, DBA/2J, C57BL/6N, and Crl:nu/nu(CD-1)BR mice (Charles River Italia, Calco, Italy) 10–12 weeks old were used. Antibodies were injected into the tail vein (100 μg/mouse). This injection was followed at different times by a second i.v. injection of Evans blue (100 μl/mouse, 1% solution). The antibodies used were purified mAb BV14, purified mAb BV13, and the isotype-matched purified mAb anti-PECAM-1, MEC 7.46 (30). Fifteen minutes after Evans blue injection, animals were killed, and Evans blue was extracted from tissues as described (37, 38). In some experiments, animals were treated with cobra venom factor to obtain C3 complement-depleted mice (39, 40).
Data were expressed as percentage increase in permeability in comparison to animals treated with the control mAb MEC 7.46. In preliminary experiments, MEC 7.46 did not induce any significant change in permeability values at any dose (up to 200 μg/mouse) and any time of treatment (up to 24 hours) as compared with animals treated with comparable doses of nonimmune rat IgG (Sigma).
For immunofluorescence and electron microscopy analysis, BV13 (100 μg/mouse) or nonspecific rat IgG was injected into the tail vein of C57BL/6 mice (Charles River), and the specimens were processed as described (41, 42). In ricin staining assay after BV13 or control antibody i.v. treatment, rhodamine-labeled Ricinus communis I lectin (200 μg/mouse in 100 μl, Vector) was injected into a femoral vein to mark sites of exposed basement membrane as described (43).
RESULTS
Production and Characterization of Monoclonal Antibodies to VE-Cadherin.
mAbs able to bind to the VE-cadherin recombinant fragment and to VE-cadherin-transfected Chinese hamster ovary cells by ELISA assay were further screened for their capacity to increase paracellular permeability in VE-cadherin transfectants (Fig. 1A). mAb BV13 and mAb BV14 were selected and further characterized. By Western blot analysis, both BV13 and BV14 were able to recognize a band of 120 kDa corresponding to the molecular weight of VE-cadherin (27, 28) in transfectants (Fig. 1B) and endothelial cell lines (1G11 and H5V; data not shown).
Figure 1.

Characterization of monoclonal antibodies to VE-cadherin. (A) mAbs BV13 and BV14 increase paracellular permeability in comparison to other anti-VE-cadherin mAbs (13E6 and 13C7). Data are means ± SEM of three different experiments and are expressed as percentage increase in comparison to the values obtained with an irrelevant mAb (26.1 ± 2.4 was the fluorescence unit value of the irrelevant mAb at the same time). (B) mAb BV13 and BV14 specifically recognize a protein of the molecular weight of VE-cadherin. VE-cadherin and control Chinese hamster ovary transfectants were analyzed by Western blot using BV13 and BV14.
Effect of BV13 on VE-Cadherin Organization in Cultured Cells.
As reported in Fig. 2, addition of BV13 to endothelial cell monolayers induced time-dependent redistribution of VE-cadherin from intercellular junctions. As reported in Fig. 3, the total amount of biotin-labeled VE-cadherin was not significantly modified by BV13 treatment. This indicates that the lack of staining of VE-cadherin from intercellular contacts was mostly caused by its diffuse redistribution on the cell surface and not by internalization. As shown in Fig. 4, the distribution of a series of intercellular markers such as PECAM-1, JAM, ZO-1, and cingulin was not modified by BV13 addition. By immunoprecipitation and Western blot analysis, VE-cadherin remained associated to β-catenin, plakoglobin, and α-catenin even after BV13 treatment of the cells (data not shown).
Figure 2.

Effect of BV13 on VE-cadherin organization in endothelial cells. BV13 (50 μg/ml) was added to cultured endothelial monolayers (1G11). VE-cadherin staining was strongly reduced at junctions within 1 hour, and this effect lasted up to 24 hours of incubation with the mAb. When BV13 was removed after 7 hours of incubation and the cells were cultured for additional 17 hours, a partial recovery of VE-cadherin at junctions was detected (white arrowheads). Actin staining shows that reduction of VE-cadherin staining from junctions was not accompanied by cell retraction. Comparable results were obtained when, after fixation of the cells, VE-cadherin was detected by using either mAb BV14 or a VE-cadherin rabbit polyclonal antiserum (data not shown). (Bar = 20 μm.)
Figure 3.
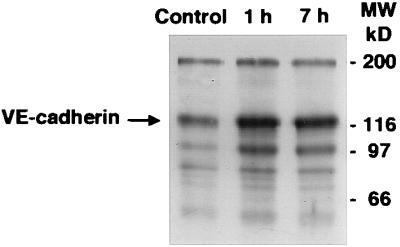
Effect of BV13 on VE-cadherin internalization. Endothelial cells (1G11) were incubated with 50 μg/ml of BV13 for 1 hour and 7 hours (1 h and 7 h lanes) or nonimmune IgG for 7 hours (Control lane). Before extraction, endothelial cells were surface-labeled with biotin by using sulfo-nitrohydroxysuccinimido-biotin (16).
Figure 4.

Effect of BV13 on the organization of other junctional components in the endothelium. Endothelial cell monolayers were incubated with BV13 (50 μg/ml) for 7 hours. Cells then were fixed, and the distribution of junctional proteins was analyzed by immunofluorescence microscopy. PECAM-1, JAM, ZO-1, and cingulin distribution was not significantly affected by addition of BV13. (Bar = 20 μm.)
Effect of BV13 Administration on Vascular Permeability in Vivo.
Seven hours after administration of 100 μg of purified BV13, the mice presented signs of strong dyspnea and hypothermia and died within 24 hours. Fig. 5 reports that administration of BV13 induced a marked time- and concentration-dependent increase of lung and heart permeability (A and B). Administration of another VE-cadherin mAb (BV14), which was found to be less effective than BV13 on permeability in vitro (see Fig. 1A), had only a moderate activity in increasing lung, and not heart, permeability in vivo (Fig. 5C).
Figure 5.

Effect of BV13 administration on vascular permeability in vivo. (A) BV13 (100 μg/mouse) induced a significant and time-dependent increase in Evans blue accumulation in heart (white columns) and lungs (striped columns). (B) BV13 increased vascular permeability in a concentration-dependent fashion. Different doses—10 μg/mouse (white columns), 50 μg/mouse (striped columns), or 100 μg/mouse (black columns) of BV13—were administered, and, after 7 hours, Evans blue extravasation was evaluated. (C) BV13 or BV14, 25 μg/mouse (gray columns) and 50 μg/mouse (stripped columns), was administered, and, after 7 hours, Evans blue leakage was measured. For the animals treated with BV13 and BV14, data are expressed as percentage increase in Evans blue content of the different organs in comparison to mice treated with the same concentration of the control mAb MEC 7.46 for the same time. Data are means ± SEM of at least five experiments, each performed in quadruplicates.
As another control for BV13, we used the isotype-matched (IgG1) anti PECAM-1 mAb (MEC 7.46), which is able to bind endothelial cells and to localize at intercellular junctions. This mAb did not significantly change Evans blue extravasation up to a concentration of 200 μg/mouse and at any time after treatment (up to 24 hours) in comparison to mice treated with comparable concentrations of nonimmune IgG. For instance, the Evans blue values (as nanograms of Evans blue content/mg of tissue) were 89.7 ± 7.3 in the heart and 85.7 ± 1.4 in the lungs for animals treated with 200 μg/mouse of MEC 7.46 at 7 hours and 97.1 ± 10.5 in the heart and 79.0 ± 4.6 in lungs of mice treated with the same concentration of nonimmune rat IgG in the same time interval. Evans blue extravasation was unchanged, either in the brain at any BV13 dose or in the ear skin, trachea, intestine, and quadriceps muscle (data not shown).
Binding of BV13 to Endothelial Cells in Vivo.
To address this question, animals were treated i.v. with BV13 (100 μg/mouse). Twenty minutes and 2 hours after treatment, the animals were anesthetized, vessels were perfused with fixative, and the antibody staining was evidentiated with Cy3-labeled goat anti-rat secondary antibody. The vessels then were examined by fluorescence confocal microscopy. As reported in Fig. 6, in large arterioles and venules, the VE-cadherin mAb decorated interendothelial junctions in a sharp and continuous way at 20 minutes and 2 hours after treatment (A and B, arrows). In contrast, in small pulmonary vessels near arterioles (Fig. 6B, arrowheads), in alveolar capillaries (D, arrow), or in capillaries of cardiac muscle (F, arrow), at 2 hours after BV13 administration, the staining became discontinuous and diffuse.
Figure 6.

Endothelial cell junctions stained by i.v. injection of VE-cadherin antibody. Blood vessels were labeled by i.v. injection of BV13 and ex vivo incubation in Cy3-labeled secondary antibody. Images of vessels in thick tissue sections were obtained by fluorescence confocal microscopy. Shown are intrapulmonary segments of pulmonary arterioles at 20 minutes (A) or 2 hours (B) after i.v. injection of BV13. In the large arterioles and venules, antibody bound continuously along endothelial cell–cell junctions at 20 minutes and 2 hours (arrows in A and B). In small pulmonary vessels near arterioles, antibody also bound in sharp, continuous lines along endothelial cell junctions at 20 minutes (arrowheads in A), but, in contrast, binding was discontinuous and diffuse at 2 hours (arrowheads in B). Similarly, in alveolar capillaries, antibody bound in sharp, continuous lines at 20 minutes (arrow in C), but binding was discontinuous and diffuse at 2 hours (arrow in D). In capillaries of cardiac muscle, antibody bound in sharp, continuous lines at 20 minutes (arrow in E) but was discontinuous and diffuse at 2 hours (arrow in F). (Bar = 25 μm.)
Effect of Complement Activation on BV13 Activity in Vivo.
We further investigated whether the observed in vivo effects could be caused by complement activation and deposition of immune complexes. To this end, we first used C5-deficient mice (strain DBA/2J) (44). As shown in Fig. 7A, BV13 administration caused a comparable increase in lung and heart permeability in C5-deficient and control animals (C57BL/6N).
Figure 7.

Complement requirements for BV13 effects on vascular permeability. BV13 was able to increase heart (white columns) and lung (striped columns) permeability in C5 complement-deficient animals (A). Mice DBA/2J (C5 deficient) and their matched controls C57BL/6N (Control) were injected with BV13 or the control mAb MEC 7.46 (100 μg/mouse), and permeability was evaluated after 7 hours. (B) Mice depleted of C3 complement. Animals were treated with either cobra venom factor as described in Materials and Methods to deplete C3 complement element (C3-depleted) or saline (Control). The mice then were injected i.v. with BV13 or MEC 7.46 (100 μg/mouse), and, after 7 hours, permeability in heart and lungs was measured. (C) Fab fragments of BV13 (200 μg/mouse) or BV13 whole mAb (100 μg/mouse) were injected i.v., and the organs were extracted after 2 hours. Data are expressed as percentage increase in Evans blue content of the different organs and are means ± SEM of three experiments, each performed in triplicates.
In additional experiments, BV13 was administered to mice previously treated with cobra venom factor for complement depletion. This treatment schedule is known to eliminate C3 complement levels (39, 40). Fig. 7B shows that, even in these conditions, the effect of BV13 on organ permeability was not lower than in control animals.
Finally, Fab fragments of BV13 were used. The Fab were able to recognize intercellular junctions in cultured endothelial cells and to induce disappearance of VE-cadherin from cell-to-cell contacts (data not shown). The effect was less marked than with the whole mAb. This difference is likely caused by the fact that the Fab lost some activity during preparation. For instance, compared with the whole BV13 antibody, a two-fold greater concentration of Fab was required to induce VE-cadherin disappearance from junctions of cultured cell monolayers. The data reported in Fig. 7C show that Fab fragments of BV13 at 200 μg/mouse are able to significantly increase lung and heart permeability at 2 hours in a way not significantly different from 100 μg/mouse of the whole mAb.
Histological Features of the Effects of BV13.
Toluidine blue stained sections of the different organs were examined. In the normal heart (time 0), nuclei of myocytes were prominently displayed as light staining rectangular bodies, along with no evidence of interstitial edema or cellular infiltrates (Fig. 8A). At 7 (Fig. 8B) and 9 (C) hours after infusion of BV13, there was interstitial edema as indicated by dilated interstitial areas and interstitial hemorrhage (open arrows), together with collections of neutrophils (black arrows) and mononuclear cells (white arrows).
Figure 8.

Histological analysis of the effects of BV13 on heart and lungs. Morphological features of effects of BV13 on heart (A–C) and lungs (D–F). In the hearts of control animals, the characteristics of normal myocardium are evident (A). At 7 (B) and 9 hours (C) after BV13, there are interstitial edema and hemorrhage (open arrows) and interstitial accumulations of neutrophils (black arrows) and mononuclear cells (white arrows). The features of normal lung, including intact alveolar walls and venules, are apparent in control animals (D) whereas 9 hours after infusion of BV13, endothelial cell bleb formation is evident (open arrowheads) (E), together with intravascular neutrophils (black arrows), bleb formation involving small capillaries and epithelial cell (black arrowheads), and intravascular platelet aggregates (F). (Toluidine blue stain; 100×.)
In the lung at 0 time, venules and alveolar walls and airspaces were morphologically normal (Fig. 8D) 9 hours after the infusion of BV13, endothelial bleb formation in small venules was prominent (E, open arrowhead), and intracapillary platelet plugs in association with neutrophils were detectable (F, black arrows). In addition, there was pronounced bleb formation involving small capillaries and alveolar epithelial cells (Fig. 8F, black arrowhead).
Ultrastructural examination of the lung samples revealed morphological changes as a function of time after infusion of BV13 (Fig. 9). Lungs at 0 hour appeared essentially normal, with intact junction between endothelial cells (Fig. 9A). One hour after infusion of antibody (Fig. 9 B and C), some neutrophils (open arrows) and small aggregates of platelets (P) were observed in lumens of capillaries. At 7 hours (Fig. 9D), endothelial cells present blebbing (black arrows), and some gaps were present in the endothelial layer, revealing bare basement membrane. By 9 hours (Fig. 9 E and F) widespread accumulation of platelet aggregates was found, often revealing degranulation (Fig. 9F). Multiple gaps and breaks in the endothelium occurred (Fig. 9 E and F, black arrows), leaving denuded basement membrane in some areas and allowing neutrophils to be in direct contact with basement membrane (E). Neutrophils were observed migrating through the vascular wall (Fig. 9E). Direct contact between the interstitial compartment and the vascular lumen was also evident (Fig. 9F). In the kidney, the results of infused BV13 were not striking, but some glomerular changes were consistently found (data not shown). Histological analysis showed no evidence of significant damage in brain, liver, adrenal glands, lymph nodes, thymus, skin, and eye (data not shown).
Figure 9.

Electron micrographs of lung samples 0 (A), 1 (B and C) 7 (D), and 9 hours (E and F) after BV13 infusion. In A, normal vascular endothelial cell morphology is present. In B, some neutrophils are evident (open arrows) in lung capillaries. C shows platelet (P) aggregates present in venules. In D, endothelial cells present blebbing (black arrows). In E and F, multiple gaps and breaks are evident (black arrows), and platelets aggregates are present (P). Neutrophils (open arrows) appear to be adhering to denuded basement membrane and passing through the gaps in vascular wall (E). Interstitial cell debris are present in the vascular lumen (F, double-headed arrow), where the endothelial cell barrier has been lost. Sections were stained with uranyl acetate and lead citrate. (×4,800.)
Sites of Plasma Leakage Stained with Ricin Lectin.
As a further analysis of the vascular changes induced by BV13, confocal micrographs of pulmonary vessels stained by injection of rhodamine-labeled Ricinus communis I lectin was performed. Fig. 10 shows that the pulmonary vessels of the BV13-treated animals present focal spots of bright fluorescence in the alveolar capillaries and in some larger pulmonary vessels, indicative of areas of exposed basement membrane. Lung microvessels from both treatment groups had occasional intravascular leukocytes that stain brightly with Ricin lectin.
Figure 10.

Confocal micrographs of pulmonary vessels stained by injection of rhodamine-labeled Ricin lectin. (A) Alveolar capillaries in mouse given rat IgG. Luminal surface of endothelial cells stains uniformly faint whereas adherent leukocytes stain more brightly (arrowhead). (B) Alveolar capillaries in mouse given BV13 (50 μg, 4 hours). Bright focal patches of staining (arrows) indicate exposed basement membrane. (Bar = 25 μm.)
DISCUSSION
The anti-VE-cadherin mAb BV13 was first selected for its capacity to increase permeability in vitro. By immunofluorescence microscopy, this mAb induced the redistribution of VE-cadherin from intercellular contacts. A large part of this effect did not seem to be attributable to either internalization of the molecule or its detachment from catenins. It is likely that BV13 exerts its activity by disrupting VE-cadherin homotypic adhesion and clustering, which then leads to a diffuse pattern of the protein on the cell surface. Calcium chelation (14, 45) resulted in a comparable picture, and, also in this case, neither significant internalization of the protein nor its detachment from catenins was observed. A similar pattern of redistribution of VE-cadherin on endothelial cell surface could be observed in vivo, too. As reported here, at 2 hours after BV13 administration, VE-cadherin showed a diffuse pattern of distribution on the endothelium of heart and lung microvessels. Of interest, in arteriolae, VE-cadherin localization at junctions did not change after BV13 treatment. A possible explanation for this difference is that, in arteriolae, the endothelium presents complex junctions with TJs intermingled between AJs (9, 46, 47). The mAb may therefore have an incomplete access to AJs and disrupt only partially VE-cadherin clusters.
When VE-cadherin was removed from junctions by BV13 treatment, we did not observe cell retraction or intercellular gap formation in vitro. This may be explained by the fact that the cells are not stimulated to retract and pull away from each other. In addition, in vitro, BV13 did not affect the distribution of TJ markers (12, 13, 48) such as occludin (49) ZO-1 (34, 50), and JAM (19). PECAM-1 was also regularly organized at junctions after BV13 addition. These adhesive structures may act in maintaining cell to cell contacts.
When administered in vivo, BV13 induced marked changes in vascular permeability. On the basis of histological features, the effects of BV13 antibody were quite impressive, especially in the myocardium and in the lungs. In the former, interstitial edema and hemorrhage were hallmarks of vascular damage. The transmission electron micrographs demonstrate damage of vascular walls in lungs as early as 1 hour and extensive structural damage of the endothelium by 9 hours. So severe is the damage that there are physical gaps in these walls, leading to a direct communication between the vascular compartment and either the interstitial and/or the alveolar compartments. In these cases, neutrophils are present, as are naked basement membranes that lack any endothelial covering. There are also aggregates of degranulated platelets, a finding that is not surprising in view of exposed basement membranes. The striking bleb formation involving vascular endothelial cells and alveolar epithelial cells indicates that these cells have been damaged.
The data reported here strongly imply that complement activation is not involved in these events because the increase in vascular permeability was not affected by natural deficiency of C5, induced deficiency of C3, or by the use of Fab fragments of BV13. Overall, VE-cadherin inhibition induces a more important damage of the endothelium monolayer in vivo as compared with in vitro conditions. Inhibition of VE-cadherin is particularly deleterious where the vessels are subjected to significant changes in pressure and shear like in the heart or lungs. Previous work indicates that AJ organization is required to maintain endothelial cell to cell contacts in static condition but not under flow, where endothelial cells retracted and exposed the subendothelial matrix (51).
In vivo endothelial cell retraction would induce formation of platelet microthrombi and leukocyte activation, which would further extend endothelial damage (52). Vestweber et al. also reported that neutrophils extravasation was markedly increased by blocking VE-cadherin in a peritoneal model of inflammation (53). An aggravating element in comparison to other permeability increasing agents, such as histamine, is that the effect of VE-cadherin mAb is lasting. After mAb addition, inhibition of VE-cadherin clustering was apparent also after several hours. The irreversible nature of the effect may worsen the vascular damage.
In conclusion, the data reported here indicate that VE-cadherin is an important determinant of vascular integrity in some organs. An important issue is whether the phenotype described after BV13 administration may be reminiscent of any human pathological condition. VE-cadherin is particularly susceptible to proteolytic digestion (15). Leukocyte activation and release of proteolytic enzymes (54) may lead to cleavage of the extracellular domain of VE-cadherin (15), which then would cause increase in permeability in the areas of leukocyte deposition. This may occur in inflammatory conditions in general and in ischemia-reperfusion injury characterized by neutrophil accumulation in the microvasculature and edema (55).
Acknowledgments
We thank Drs. Peter Bohlen, Daniel J. Hicklin, and Fang Liao (ImClone Systems Inc., New York) for their critical comments and support. This study was supported by Associazione Italiana per la Ricerca sul Cancro, Consiglio Nazionale delle Ricerche (CNR Grant 97.01299.PF49), the European Community (Biomed Grants BMH4-CT 950875, BMH4-CT 960669, and BIO4-CT 960036), Human Frontiers Science Program (Grant GR0006/1997M), and National Institutes of Health Grants HL-24136 and HL-59157. I.M.-P. is a recipient of a fellowship from Istituto Mario Negri.
ABBREVIATIONS
- VE
vascular endothelial
- PECAM
platelet endothelial cell adhesion molecule
- ZO-1
zonula occludens 1
- JAM
junctional adhesion molecule
- TJ
tight junction
- AJ
adherens junction
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.van Hinsbergh W M. Arterioscler Thromb Vasc Biol. 1997;17:1018–1023. doi: 10.1161/01.atv.17.6.1018. [DOI] [PubMed] [Google Scholar]
- 2.Lum H, Malik A B. Am J Physiol. 1994;267:L223–L241. doi: 10.1152/ajplung.1994.267.3.L223. [DOI] [PubMed] [Google Scholar]
- 3.Anderson R G. Proc Natl Acad Sci USA. 1993;90:10909–10913. doi: 10.1073/pnas.90.23.10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schnitzer J E. Cardiovasc Med. 1993;3:124–130. doi: 10.1016/1050-1738(93)90012-U. [DOI] [PubMed] [Google Scholar]
- 5.Dvorak A M, Kohn S, Morgan E S, Fox P, Nagy J A, Dvorak H F. J Leukocyte Biol. 1996;59:100–115. [PubMed] [Google Scholar]
- 6.Predescu S A, Predescu D N, Palade G E. Am J Physiol. 1997;272:H937–H949. doi: 10.1152/ajpheart.1997.272.2.H937. [DOI] [PubMed] [Google Scholar]
- 7.Predescu D, Predescu S, Palade G E. Proc Natl Acad Sci USA. 1998;95:6175–6180. doi: 10.1073/pnas.95.11.6175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dejana E. J Clin Invest. 1996;98:1949–1953. doi: 10.1172/JCI118997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lampugnani M G, Dejana E. Curr Opin Cell Biol. 1997;9:674–682. doi: 10.1016/s0955-0674(97)80121-4. [DOI] [PubMed] [Google Scholar]
- 10.Takeichi M. Curr Opin Cell Biol. 1993;5:806–811. doi: 10.1016/0955-0674(93)90029-p. [DOI] [PubMed] [Google Scholar]
- 11.Aberle H, Schwartz H, Kemler R. J Cell Biochem. 1996;61:514–523. doi: 10.1002/(SICI)1097-4644(19960616)61:4%3C514::AID-JCB4%3E3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 12.Gumbiner B M. Cell. 1996;84:345–357. doi: 10.1016/s0092-8674(00)81279-9. [DOI] [PubMed] [Google Scholar]
- 13.Anderson J M, Van Itallie C M. Am J Pathol. 1995;269:G465–G475. [Google Scholar]
- 14.Dejana E, Corada M, Lampugnani M G. FASEB J. 1995;9:910–918. [PubMed] [Google Scholar]
- 15.Lampugnani M G, Resnati M, Raiteri M, Pigott R, Pisacane A, Houen G, Ruco L, Dejana E. J Cell Biol. 1992;118:1511–1522. doi: 10.1083/jcb.118.6.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Navarro P, Caveda L, Breviario F, Lampugnani M G, Dejana E. J Biol Chem. 1995;270:30965–30972. doi: 10.1074/jbc.270.52.30965. [DOI] [PubMed] [Google Scholar]
- 17.Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S. J Cell Biol. 1993;123:1777–1788. doi: 10.1083/jcb.123.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furuse M, Fujita K, Hiiragi T, Fujimoto K, Tsukita S. J Cell Biol. 1998;141:1539–1550. doi: 10.1083/jcb.141.7.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P, Panzeri C, Stoppacciaro A, Ruco L, Villa A, et al. J Cell Biol. 1998;142:117–127. doi: 10.1083/jcb.142.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lampugnani M G, Corada M, Andriopoulou P, Esser S, Risau W, Dejana E. J Cell Sci. 1997;110:2065–2077. doi: 10.1242/jcs.110.17.2065. [DOI] [PubMed] [Google Scholar]
- 21.Esser S, Lampugnani M G, Corada M, Dejana E, Risau W. J Cell Sci. 1998;111:1853–1865. doi: 10.1242/jcs.111.13.1853. [DOI] [PubMed] [Google Scholar]
- 22.Rabiet M J, Plantier J L, Rival Y, Genoux Y, Lampugnani M G, Dejana E. Arterioscler Thromb Vasc Biol. 1996;16:488–496. doi: 10.1161/01.atv.16.3.488. [DOI] [PubMed] [Google Scholar]
- 23.Rabiet M J, Plantier J L, Dejana E. Br Med Bull. 1994;50:936–945. doi: 10.1093/oxfordjournals.bmb.a072935. [DOI] [PubMed] [Google Scholar]
- 24.Vittet D, Buchou T, Schweitzer A, Dejana E, Huber P. Proc Natl Acad Sci USA. 1997;94:6273–6278. doi: 10.1073/pnas.94.12.6273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong Q G, Bernasconi S, Lostaglio S, Wainstok de Calmanovici R, Martin-Padura I, Breviario F, Garlanda C, Ramponi S, Mantovani A, Vecchi A. Arterioscler Thromb Vasc Biol. 1997;17:1599–1604. doi: 10.1161/01.atv.17.8.1599. [DOI] [PubMed] [Google Scholar]
- 26.Garlanda C, Parravicini C, Sironi M, De Rossi M, Wainstok de Calmanovici R, Carozzi F, Bussolino F, Colotta F, Mantovani A, Vecchi A. Proc Natl Acad Sci USA. 1994;91:7291–7295. doi: 10.1073/pnas.91.15.7291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Breier G, Breviario F, Caveda L, Berthier R, Schnurch U, Gotsch D, Vestweber D, Risau W, Dejana E. Blood. 1996;87:630–642. [PubMed] [Google Scholar]
- 28.Breviario F, Caveda L, Corada M, Martin-Padura I, Navarro P, Golay J, Introna M, Gulino D, Lampugnani M G, Dejana E. Arterioscler Thromb Vasc Biol. 1995;15:1229–1239. doi: 10.1161/01.atv.15.8.1229. [DOI] [PubMed] [Google Scholar]
- 29.Simmons D L, Walker C, Power C, Pigott R. J Exp Med. 1990;171:2147–2152. doi: 10.1084/jem.171.6.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vecchi A, Garlanda C, Lampugnani M G, Resnati M, Matteucci C, Stoppacciaro A, Schnurch H, Risau W, Ruco L, Mantovani A, et al. Eur J Cell Biol. 1994;63:247–254. [PubMed] [Google Scholar]
- 31.Lampugnani M G, Corada M, Caveda L, Ayalon O, Geiger B, Dejana E. J Cell Biol. 1995;129:203–217. doi: 10.1083/jcb.129.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin-Padura I, Bazzoni G, Zanetti A, Bernasconi S, Elices M J, Mantovani A, Dejana E. J Biol Chem. 1994;269:6124–6132. [PubMed] [Google Scholar]
- 33.Harlow E, Lane D. Antibodies: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1988. [Google Scholar]
- 34.Stevenson B R, Siciliano J D, Mooseker M S, Goodenough D A. J Cell Biol. 1986;103:755–766. doi: 10.1083/jcb.103.3.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Citi S, Sabanay H, Jakes R, Geiger B, Kendrick-Jones J. Nature (London) 1988;333:272–276. doi: 10.1038/333272a0. [DOI] [PubMed] [Google Scholar]
- 36.Citi S, Sabanay H, Kendrick-Jones J, Geiger B. J Cell Sci. 1989;93:107–122. doi: 10.1242/jcs.93.1.107. [DOI] [PubMed] [Google Scholar]
- 37.Uyama O, Okamura N, Yanase M, Narita M, Kawabata K, Sugita M. J Cereb Blood Flow Metabol. 1988;8:282–284. doi: 10.1038/jcbfm.1988.59. [DOI] [PubMed] [Google Scholar]
- 38.Rossner W, Tempel K. Med Pharmacol Exp Int J Exp Med. 1966;14:169–182. [PubMed] [Google Scholar]
- 39.Cochrane C G, Muller-Eberhard H J, Aikin B S. J Immunol. 1970;105:55–69. [PubMed] [Google Scholar]
- 40.Mulligan M S, Schmid E, Beck-Schimmer B, Till G O, Friedl H P, Brauer R B, Hugli T E, Miyasaka M, Warner R L, Johnson K J, et al. J Clin Invest. 1996;98:503–512. doi: 10.1172/JCI118818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thurston G, McLean J W, Rizen M, Baluk P, Haskell A, Murphy T J, Hanan D, McDonald D M. J Clin Invest. 1998;101:1401–1413. doi: 10.1172/JCI965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eppinger M J, Deeb G M, Bolling S F, Ward P A. Am J Pathol. 1997;150:1773–1784. [PMC free article] [PubMed] [Google Scholar]
- 43.Thurston G, Baluk P, Hirata A, McDonald D M. Am J Physiol. 1996;271:H2547–H2562. doi: 10.1152/ajpheart.1996.271.6.H2547. [DOI] [PubMed] [Google Scholar]
- 44.Sawtell N M, Hartman A L, Weiss M A, Pesce A J, Michael J G. Lab Invest. 1988;58:287–293. [PubMed] [Google Scholar]
- 45.Ayalon O, Sabanai H, Lampugnani M G, Dejana E, Geiger B. J Cell Biol. 1994;126:247–258. doi: 10.1083/jcb.126.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simionescu N, Simionescu N Z. Cell Biol Rev. 1991;25:5–80. [Google Scholar]
- 47.Staddon J M, Rubin L L. Curr Opin Neurobiol. 1996;6:622–627. doi: 10.1016/s0959-4388(96)80094-8. [DOI] [PubMed] [Google Scholar]
- 48.Anderson J M, Balda M S, Fanning A S. Curr Opin Cell Biol. 1993;5:772–776. doi: 10.1016/0955-0674(93)90024-k. [DOI] [PubMed] [Google Scholar]
- 49.Van Itallie C M, Anderson J M. J Cell Sci. 1997;110:1113–1121. doi: 10.1242/jcs.110.9.1113. [DOI] [PubMed] [Google Scholar]
- 50.Anderson J M, Stevenson B R, Jesaitis L A, Goodenough D A, Mooseker M S. J Cell Biol. 1988;106:1141–1149. doi: 10.1083/jcb.106.4.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schnittler H J, Puschel B, Drenckhahn D. Am J Physiol. 1997;273:H2396–H2405. doi: 10.1152/ajpheart.1997.273.5.H2396. [DOI] [PubMed] [Google Scholar]
- 52.Del Maschio A, Dejana E, Bazzoni G. Ann Hematol. 1993;67:23–31. doi: 10.1007/BF01709662. [DOI] [PubMed] [Google Scholar]
- 53.Gotsch U, Borges E, Bosse R, Boggemeyer E, Simon M, Vestweber D. J Cell Sci. 1997;110:583–588. doi: 10.1242/jcs.110.5.583. [DOI] [PubMed] [Google Scholar]
- 54.Weiss S J. N Engl J Med. 1989;320:365–376. doi: 10.1056/NEJM198902093200606. [DOI] [PubMed] [Google Scholar]
- 55.Lucchesi B R. Annu Rev Physiol. 1990;52:561–576. doi: 10.1146/annurev.ph.52.030190.003021. [DOI] [PubMed] [Google Scholar]