

Abstract
Human heart Na+ channels were expressed transiently in both mammalian cells and Xenopus oocytes, and Na+ currents measured using 150 mM intracellular Na+. The kinetics of decaying outward Na+ current in response to 1-s depolarizations in the F1485Q mutant depends on the predominant cation in the extracellular solution, suggesting an effect on slow inactivation. The decay rate is lower for the alkali metal cations Li+, Na+, K+, Rb+, and Cs+ than for the organic cations Tris, tetramethylammonium, N-methylglucamine, and choline. In whole cell recordings, raising [Na+]o from 10 to 150 mM increases the rate of recovery from slow inactivation at −140 mV, decreases the rate of slow inactivation at relatively depolarized voltages, and shifts steady-state slow inactivation in a depolarized direction. Single channel recordings of F1485Q show a decrease in the number of blank (i.e., null) records when [Na+]o is increased. Significant clustering of blank records when depolarizing at a frequency of 0.5 Hz suggests that periods of inactivity represent the sojourn of a channel in a slow-inactivated state. Examination of the single channel kinetics at +60 mV during 90-ms depolarizations shows that neither open time, closed time, nor first latency is significantly affected by [Na+]o. However raising [Na+]o decreases the duration of the last closed interval terminated by the end of the depolarization, leading to an increased number of openings at the depolarized voltage. Analysis of single channel data indicates that at a depolarized voltage a single rate constant for entry into a slow-inactivated state is reduced in high [Na+]o, suggesting that the binding of an alkali metal cation, perhaps in the ion-conducting pore, inhibits the closing of the slow inactivation gate.
Keywords: sodium channels, gating, single channel recording, kinetics
introduction
In the previous paper we reported effects of extracellular cations on the peak open probability (P open)1 of cardiac Na+ currents (Townsend et al., 1997). Low concentrations of permeant cations were associated with a reduction of P open at depolarized voltages with little effect on the kinetics of activation. The process of fast inactivation is also relatively insensitive to extracellular Na+ concentration (Armstrong and Bezanilla, 1974; Oxford and Yeh, 1985; Correa and Bezanilla, 1994; O'Leary et al., 1994; Bezanilla and Correa, 1995; Tang et al., 1996). Fast inactivation is believed to involve a cytoplasmically-located gate (Rojas and Armstrong, 1971; Stühmer et al., 1989; Moorman et al., 1990; Patton et al., 1992; West et al., 1992). Another type of inactivation with much slower kinetics (seconds versus milliseconds) co-exists with fast inactivation in all Na+ channels (Adelman and Palti, 1969; Ruff et al., 1987; Simoncini and Stühmer, 1987; Ruben et al., 1992). Slow inactivation differs from fast inactivation in that it is relatively insensitive to cytoplasmic manipulations that have profound effects on fast inactivation. For example, fast inactivation is readily abolished by intracellular enzymatic treatment (Rojas and Armstrong, 1971; Quandt, 1987) and by mutations in the cytoplasmic loop linking the third and fourth domains of the Na+ channel protein (Stühmer et al., 1989; Patton et al., 1992; West et al., 1992). These treatments do not disrupt slow inactivation (Rudy, 1978; Quandt, 1987; Valenzuela and Bennett, 1994; Cummins and Sigworth, 1996b ). The molecular nature of slow inactivation and the location of its gate are unknown.
We show here that both permeant and impermeant alkali metal cations in the extracellular solution affect the kinetics and steady-state levels of slow inactivation when compared with four different organic cations. These effects of alkali metal cations can be explained by their influence on two rate constants, one for entry into and one for recovery from a slow-inactivated state.
methods
The techniques used in this paper generally follow those described in the preceding paper (Townsend et al., 1997) with the following modifications.
Solutions
For whole cell recordings the solutions bathing the cells were exchanged with a large bore macropipet. For single channel recordings the bath Na+ concentration was changed by perfusing the entire recording chamber with the solution of interest. To avoid effects on the junction potential at the silver wire due to changes in Cl− concentration, we used a 3 M KCl agar bridge in the bath.
Data Analysis
Whole-cell and single-channel data were analyzed with a combination of pCLAMP programs, Microsoft Excel, Origin (Microcal, Northampton, MA), and our own Fortran and Basic programs. Unless otherwise specified, data are expressed as mean±SEM.
Steady-state inactivation curves were fit by the Boltzmann distribution:
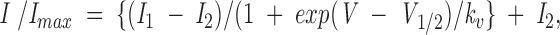 |
where I is the peak current for a test pulse to +60 mV, I max is the peak current at +60 mV measured from the most negative prepulse potential (−140 mV), I 1 and I 2 are the maximum and minimum values in the fit, V 1/2 is the midpoint of the curve, and k v is the slope factor.
Single-channel currents were idealized with TRANSIT (VanDongen, 1996) and further analyzed with our own software, including a program for estimating rate constants by maximum likelihood (Horn and Lange, 1983; Horn and Vandenberg, 1984). For the latter analytical method, asymptotic standard errors of the estimated rate constants were calculated from the inverse of the observed information matrix. The ability of the maximum likelihood program to invert the information matrix in all estimates is a good indication that the fits are unique and that the problem is identifiable. Because these are iterative fits, however, it is not possible to guarantee that any particular solution is the global maximum of the likelihood function, but different starting guesses always converged to the same answers in the cases we tested. The number of channels in a patch was assumed to be the maximum number simultaneously open at a depolarized voltage (Horn, 1991). Most of the data were analyzed for patches with only one channel. For two-channel patches the first latency distributions were corrected for the number of channels (Patlak and Horn, 1982). We also corrected the estimates for mean number of openings in a depolarization in two-channel patches, as described previously (Chahine et al., 1994). Because of considerable patch-to-patch variability, and variability between repeated samples from the same patch under the same conditions but at different times, the various estimated parameters were examined across patches and across external Na+ concentration ([Na+]o) using a two-way analysis of variance (ANOVA). The ANOVA model is an extension of a generalized linear model that accounts for both fixed effects ([Na+]o) and random effects (the individual patches) and involves the use of the MIXED algorithm from the SAS library (SAS Institute, Cary, NC).
Successive depolarizations (90 ms, presented every 2 s) in single channel patches tended to show a clustering of records with openings and those without (i.e., nulls). These data were analyzed by “runs analysis” as described previously (Horn et al., 1984). A run is defined as a sequence of depolarizations that results in either all nulls or records all with openings. The distribution of the number of runs can be approximated by an asymptotic distribution, forming a standardized random variable, Z, with a mean of zero and a variance of 1 (Wald and Wolfowitz, 1940). In the present case:
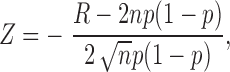 |
where R is the observed number of runs, n is the number of depolarizations, and p the probability that a channel will open at least once during a depolarization (Horn et al., 1984). The expected number of runs is 2np(1 − p), so that Z = 0 for a random ordering of null records, Z > 2 (P < 0.05) if the null records are significantly clustered, and Z < −2 (P < 0.05) for a tendency to alternate between null records and those with openings.
To test the effect of [Na+]o on the number of blank records in a run, we derived a likelihood ratio test based on the geometric distribution, as follows. Let X and Y be independent random variables representing the number of blanks in a run in either high or low [Na+]o, each variable having a geometric distribution with joint distribution:
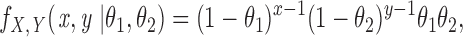 |
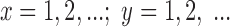 |
If there are m runs of X and n runs of Y, the maximum likelihood estimates of θi (i = 1, 2) are
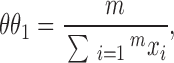 |
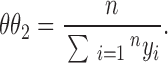 |
The null hypothesis (H 0) is that θ1 = θ2, and the alternative hypothesis (H 1) is that θ1 ≠ θ2, with estimates of θi as defined above. The maximum likelihood estimate of θi under H 0 is
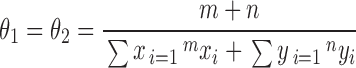 |
The natural logarithm of the maximum likelihood of hypothesis H j (j = 0,1) can be written as
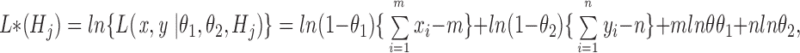 |
and 2{L*(H 1) − L*(H 0)} has an asymptotic χ2 distribution with 1 degree of freedom. We tested this hypothesis for 8 patches by adding the χ2 statistics of each patch. The resultant sum has an asymptotic χ2 distribution with 8 degrees of freedom.
results
[Na+]o Effects on Slow Inactivation of Macroscopic Currents of F1485Q Channels
To test whether slow inactivation of Na+ channels is influenced by [Na+]o, we examined the effects of [Na+]o on the kinetics of macroscopic Na+ current during prolonged depolarizations, using the mutant F1485Q of the human heart Na+ channel hH1a (Townsend et al., 1997). Whole-cell currents (Figs. 1–4) were obtained from transiently transfected tsA201 cells, and single channel currents (Figs. 5–7) were from outside-out patches of cRNA-injected Xenopus oocytes.
Figure 1.

Effects of [Na+]o on macroscopic F1485Q current inactivation. Na+ currents elicited by 1-s depolarizations to +60 mV (holding potential = −140 mV) obtained from cells sequentially bathed in either (A) 150, 10, and 150 mM [Na+]o, with NMG+ as a substitute for Na+, or (B) 150 Na+ o, 150 Rb+ o, and 150 mM Na+ o. Peak currents normalized to unity.
Figure 4.

Steady-state slow and fast inactivation in high and low [Na+]o. Steady-state slow inactivation (SΘ) curves for F1485Q (A) and WT hH1a (B). Transfected cells were held at holding potentials ranging from −160 to −30 mV in 10- or 20-mV increments. After 2 min at each holding potential, a 20-ms recovery pulse to −140 mV and a 9-ms test pulse to +60 mV were given (insets). The peak current measured at +60 mV is plotted as a fraction of the maximal current. (A) F1485Q SΘ curves. The data points are fit to the Boltzmann equation (solid lines) with midpoints at −79.0 ± 1.1 and −85.9 ± 1.7 mV and slope factors of 6.9 ± 0.4 and 6.0 ± 0.6 mV for 150 and 10 mM external Na+, respectively (n = 4 cells for 150 mM Na+ and n = 5 cells for 10 mM Na+). (B) WT SΘ curves. Best-fits to the Boltzmann equation have midpoints at −82.2 ± 2.8 and −102.0 ± 0.5 mV and slope factors of 25.4 ± 2.5 and 15.3 ± 0.6 for 150 and 0 mM Na+ o, respectively (n = 3 cells for each [Na+]o). (C and D) Steady-state fast inactivation (hΘ) induced by a 50-ms prepulse to the indicated voltage from a holding potential of −160 mV (see insets). Test pulse, +60 mV. The peak current at +60 mV is plotted as a fraction of the maximal current. (C) F1485Q hΘ curves. Data shown are from 3 cells sequentially bathed in 150, 10, and 150 mM Na+. The best-fit Boltzmann curves have midpoints at −73.8 ± 2.4, −78.0 ± 2.6, and −75.6 ± 3.0 mV and slope factors of 3.27 ± 0.16, 3.47 ± 0.35, and 3.53 ± 0.36 mV for 150, 10, and 150 mM Na+ o, respectively. (D) WT hΘ curves. Data from 3 cells transfected with WT hH1a and successively bathed in 150, 0, and 150 mM Na+. Inactivation curves are fitted to the Boltzmann equation with midpoints at −100.3 ± 0.9, −103.4 ± 0.6, and −103.5 ± 0.4 mV and slope factors of 8.0 ± 0.2, 8.8 ± 0.3, and 7.8 ± 0.2 for 150, 0, and 150 mM Na+ o, respectively. In each panel, the dashed lines are the Boltzmann curves for 10 (F1485Q) or 0 mM Na+ o (WT) multiplied by 0.67 (F1485Q) or 0.84 (WT), giving the expected reduction in P open for the decrease in [Na+]o (Townsend et al., 1997).
Figure 5.

Single-channel data analysis. (A) Ensemble averages obtained from a two-channel outside-out patch successively bathed in 150, 10, and 150 mM Na+. Currents were activated by 90-ms pulses to +60 mV from a holding potential of −140 mV with a frequency of 0.5 Hz (n = 200 depolarizations for each bath solution). (B) Effects of [Na+]o on open time distributions. The normalized square root of the number of events per bin (n 1/2) is plotted versus the logarithm of the open duration. The lines represent fits to single exponential distributions. Mean open times were 1.12 ± 0.03, 0.63 ± 0.01, and 0.93 ± 0.03 ms for 150, 10, and 150 mM Na+, respectively. Data from the two-channel patch shown in A. (C) Normalized first latency distributions, corrected for the number of channels, obtained for the same patch. (D) Cumulative distributions of the duration of the last (truncated) closing in a 90-ms depolarization for the same patch. (E and F) Cumulative distributions of the truncated closed time conditional on whether the following record contains openings (solid line) or not (dotted line) for a single-channel patch successively bathed in 10 (E) and 150 mM Na+ (F).
Figure 7.

Probability of a channel being in the open (A) or the slow-inactivated state (B) during a depolarization. (A) Ensemble average of bursts in 10 and 150 mM external Na+. The lines represent the open probability during a burst calculated from the two-channel patch of Fig. 5. The estimated rate constants for this patch, using the model in Fig. 6, were (150 mM Na+, first exposure): k 01 = 804 ± 18 s−1, k 10 = 1,915 ± 85 s−1, k 12 = 1,247 ± 89 s−1, k 21 = 154 ± 10 s−1, k 23 = 8.06 ± 1.53 s−1; (10 mM Na+): k 01 = 1,357 ± 33 s−1, k 10 = 1,865 ± 80 s−1, k 12 = 739 ± 63 s−1, k 21 = 102 ± 10 s−1, k 23 = 19.2 ± 2.6 s−1; (150 mM Na+): k 01 = 860 ± 15 s−1, k 10 = 2,361 ± 67 s−1, k 12 = 933 ± 46 s−1, k 21 = 131 ± 8 s−1, k 23 = 5.65 ± 2.08 s−1. The 4-state model predicts three time constants for the decay of burst open probability. These time constants were determined from the estimated rate constants by spectral expansion, and for this patch were (150 mM Na+): 0.27, 2.62, and 177.0 ms; (10 mM Na+): 0.27, 2.74, and 70.1 ms; and (150 mM Na+, first exposure): 0.25, 3.23, and 272.8 ms. (B) Probability for a channel to be in the absorbing inactivated state I3 (P(I)3) during a 90-ms pulse to +60 mV. P(I)3 was calculated from the above rate constants for the two-channel patch.
Fig. 1 A shows normalized whole-cell Na+ currents through F1485Q channels obtained during 1-s depolarizations to +60 mV from a transfected cell sequentially exposed to 150, 10, and 150 mM [Na+]o, using N-methyl- d-glucamine+ (NMG+) as a Na+ replacement. In both high and low [Na+]o, activation of Na+ current is very fast and is followed by a short phase (∼20–30 ms) of rapid inactivation. Thereafter the whole-cell Na+ currents continue to decay; however this rate of decay is clearly more rapid at the lower [Na+]o. The kinetics of the inactivation are complicated, requiring at least 2 exponentially decaying components. By contrast if extracellular Na+ is completely replaced by another impermeant cation, Rb+, there is no effect on the decay kinetics (Fig. 1 B).
Fig. 2 shows a similar experiment in which extracellular Na+ is replaced by K+, Cs+, Li+, or by the organic cations choline, tetramethylammonium, and Tris, all in the same cell. The decay kinetics for 1-s depolarizations to +60 mV are faster for the organic cations than for the alkali metal cations, consistent with the data in Fig. 1. These data suggest that external alkali metal cations can modulate a slow gating process, entry into a slow-inactivated state. To determine whether this is an effect on slow inactivation, rather than the residual fast inactivation of the F1485Q mutant, we also examined currents of the mutant IFM/QQQ in oocyte macropatches. This triple mutation more severely disrupts fast inactivation (Hartmann et al., 1994). The differential effect of Na+ and Cs+, by comparison with NMG+, was also observed for IFM/QQQ during 1-s depolarizations (data not shown). By contrast with the modulation of P open we reported in the previous paper, permeant and impermeant alkali metal cations are indistinguishable in terms of their effects on this slower kinetic process. We also tested the effect of substituting the permeant cation hydrazinium for extracellular Na+. In 11 out of 20 cells the current decay in response to a 1-s depolarization was reversibly slowed in hydrazinium. In the other 9 cells there was either no effect or the effects of hydrazinium were irreversible. We have no explanation for this variability, but it is possible that neutral hydrazine, a highly reactive compound, enters the cell and affects slow inactivation. Consistent with this idea is the fact that reversibility was improved for shorter exposures to hydrazinium. In the following experiments we used NMG+ as a substitute for extracellular Na+.
Figure 2.

Effects of external cations on the slow decay of F1485Q Na+ channels currents. Whole-cell currents were evoked as described in the legend of Fig. 1. Recordings from one cell sequentially bathed in 150 mM of the indicated monovalent cations. Normalized peak currents.
Entry into slow inactivation and recovery from slow inactivation were examined over longer periods of time by sequentially holding F1485Q-transfected cells at −70, −140, +40, and −140 mV for 5 min at each voltage (inset to Fig. 3 A). Brief pulses to +60 mV were given every 15 s to measure the fraction of noninactivated channels from the amplitudes of outward whole cell currents. To study slow inactivation in isolation from fast inactivation, the cells were hyperpolarized for a short time before the test pulse to allow recovery from fast inactivation. Recovery from fast inactivation is quite rapid for F1485Q channels expressed in tsA201 cells and is unaffected by changes in [Na+]o (data not shown). These channels recover from 90-ms prepulses to +60 mV with time constants of 2.4 ± 0.2 ms and 2.8 ± 0.4 ms in 150 and 10 mM Na+, respectively (−120 mV holding potential, n = 3). Thus, to ensure that channels fully recovered from fast inactivation, a 20-ms pulse to −140 mV was given to the cells immediately before the +60-mV test pulse. To avoid contamination by time-dependent shifts in the voltage dependence of inactivation in whole cell recordings (Wang et al., 1996), the effects of high and low [Na+]o were examined in different cells. Fig. 3 A shows peak currents at +60 mV for two cells bathed in either 10 or 150 mM Na+. At −70 mV the Na+ currents first decay quickly and then reach a steady-state level after about 2.5 min. This decay phase is voltage dependent as it is faster at +40 mV than at −70 mV (Fig. 3, A and C). In addition, the extent of the inactivation is greater at +40 mV (∼88% versus ∼75% at −70 mV for 10 mM Na+ o; Fig. 3, A and B). Recovery from slow inactivation at −140 mV after 5 min at either −70 or +40 mV is complete within 2 min and is moderately faster in 150 mM than in 10 mM Na+ o (Fig. 3, A and D). These results suggest that, in addition to modulating entry into a slow-inactivated state at depolarized voltages, external Na+ ions can also alter the rate at which channels leave that state at hyperpolarized voltages.
Figure 3.

Slow inactivation of F1485Q channels in 10 and 150 mM [Na+]o. (A) Changes in peak current in response to changes in holding potential for two F1485Q-transfected cells bathed in either 150 (closed squares) or 10 mM Na+ (open circles). The cells were held at −140 mV for at least 10 min before the holding potential was sequentially changed every 5 min to −70, −140, +40, and −140 mV. Every 15 s during the entire protocol, a 20-ms recovery pulse to −140 mV was applied followed by a 20-ms test pulse to +60 mV (inset). The normalized peak current at +60 mV is plotted. (B) Current levels after 5 min at either −70 or +40 mV expressed as percent of the maximal current at a −140 mV holding potential. (C) 10–90% decay times for entry into slow inactivation at −70 and +40 mV. The 10–90% time corresponds to the time it takes for the current to decay from 10 to 90% of the maximal decay measured after 5 min at either −70 or +40 mV. (D) 10–90% times for recovery at −140 mV after 5 min of inactivation at either −70 mV or +40 mV. Numbers in parentheses are number of cells.
The experimental protocols used in Fig. 3 could be used to measure the steady-state slow inactivation over a wide voltage range. However the duration of these experiments would be very long, compared to the slow shift of voltage-dependent gating seen with whole cell recording (Wang et al., 1996). Therefore we used a cumulative inactivation protocol in which channels were not allowed to recover from slow inactivation between inactivating prepulses (Cummins and Sigworth, 1996b ). Cells were held at voltages ranging from −140 to +30 mV in 10- or 20-mV increments. After 2 min at each voltage, a 20-ms recovery pulse to −140 mV was given followed by a 9-ms test pulse to +60 mV. Even this protocol lasted 20 min; therefore different cells were used for high and low [Na+]o. For each cell the peak current was largest for the most hyperpolarized holding potential, and data for more depolarized holding potentials are scaled to this value in Fig. 4. Fig. 4 A shows the cumulative slow inactivation (SΘ) curves obtained for 10 and 150 mM Na+ o. Consistent with the observed faster entry into slow inactivation and slower recovery from slow inactivation in 10 mM Na+ o (Fig. 3), the SΘ curve is significantly shifted (6.9 mV) in the hyperpolarizing direction in 10 mM Na+ o (P < 0.02, two-tailed t test). We also plot the SΘ curve expected for 10 mM [Na+]o (dashed line in Fig. 4 A) after consideration of the average reduction in P open caused by the decreased [Na+]o (Townsend et al., 1997). By contrast, steady-state fast inactivation induced by 50-ms conditioning pulses is not affected by [Na+]o (Fig. 4 C). Fig. 4 C also shows the corrected relationship for fast inactivation in 10 mM [Na+]o (dashed line) after consideration of the expected reduction in P open, as above. These experiments are consistent with external Na+ ions modulating channel gating by preventing F1485Q channels from entering a slow-inactivated state and speeding recovery from this state.
A [Na+]o-dependent shift of the cumulative SΘ curve is also observed for WT hH1a channels (Fig. 4 B). For these experiments we also used a 20-ms recovery pulse to −140 mV before each test pulse. The recovery time constants from fast inactivation for WT channels at −140 mV were 5.9 ± 0.8 ms (150 mM Na+; n = 4) and 6.7 ± 0.6 ms (0 mM Na+; n = 4). The −19.8-mV shift of the midpoint was statistically significant (P < 0.01, two-tailed t test). As for F1485Q channels, the steady-state fast inactivation of WT channels induced by 50-ms conditioning pulses is not affected by [Na+]o (Fig. 4 D). These data show that the effects of [Na+]o on slow inactivation are not unique to the F1485Q mutant. Furthermore, slow inactivation is more complete for the mutant, in support of the idea that fast-inactivated and slow-inactivated states are negatively coupled (see discussion).
Modulation of Single-channel Gating by [Na+]o
The reduced whole-cell currents and faster rate of slow inactivation observed in low [Na+]o should be manifested at the single-channel level as selective effects of [Na+]o on the probability of a channel opening and on the dwell time of a slow-inactivated state. To test this we conducted a detailed analysis of single-channel recordings obtained from outside-out oocyte patches exposed to high and low [Na+]o.
[Na+]o affects the probability of a channel opening after a depolarization. In 10 mM Na+ o, ∼45% of the depolarizations to +60 mV fail to activate single F1485Q channels in outside-out patches (45.3 ± 4.4%, n = 15 patches, −140 mV holding potential, 90-ms depolarizations presented at 0.5 Hz). By contrast, with 150 mM Na+ in the bath solution the proportion of blank (i.e., null) records is significantly lower (30.4 ± 5.2%, n = 10 patches, P < 0.05). This effect of [Na+]o on the percent of blank records was observed for all test potentials we examined, from +20 to +80 mV. Some of this effect is due to the influence of [Na+]o on P open. However, this phenomenon has rapid kinetics (Townsend et al., 1997). An effect on slow inactivation should, by contrast, be reflected by a clustering of records with and without openings (Horn et al., 1984). In fact, null records occur in clusters rather than at random in these single-channel experiments. This was especially clear in several patches bathed in 10 mM Na+ where as many as 30 records in a row were blanks. To evaluate the randomness of the occurrence of these null records, runs analysis was performed. A run is defined as a sequence of like events so that if null records are clustered, the number of runs will be smaller than expected for a random occurrence of a null record (see methods). Such analysis, when applied to F1485Q single-channel data, reveals that null records are significantly clustered regardless of [Na+]o, with Z = 5.64 ± 1.00 (150 Na+ o, n = 9) and 7.66 ± 0.6 (10 Na+ o, n = 15). The higher value of the Z statistic in low [Na+]o indicates an increased clustering of consecutive null records. Clustering is also observed for WT hH1a channels with Z = 2.2 ± 1.1 (n = 3) and 3.53 ± 1.61 (n = 3) for 150 and 10 mM Na+ o, respectively. This clustering pattern is indicative of channels slowly cycling in and out of a long-lived inactivated state. In most F1485Q single-channel patches studied, raising [Na+]o reduced the degree of clustering. Therefore external Na+ ions may modulate the number of activatable channels by changing the rates at which channels cycle in and out of a slow-inactivated state.
Fig. 5 A shows the normalized, ensemble-averaged open probability at +60 mV obtained from a two-channel outside-out patch sequentially bathed in 150, 10, and 150 mM Na+. In both high and low [Na+]o the activation of the channels is very fast and followed by a fast decay phase (<10 ms) to about 40–50% of the maximum current. Then, in 10 mM [Na+]o the open probability continues to decay slowly to about 10% of the maximum current while it remains relatively constant in 150 mM Na+ o (Fig. 5 A). The slower decay rate in high [Na+]o of the open probability after 10–20 ms shows that, in addition to decreasing the number of null records, high [Na+]o also affects channel gating during a depolarizing voltage pulse. High [Na+]o could sustain the open probability during a pulse by either increasing the amount of time a channel spends in the open state or by shortening the duration of channel closings or both. Fig. 5 B shows the open time distributions obtained from the same patch sequentially bathed in 150, 10, and 150 mM [Na+]o. The mean open times were moderately increased in high [Na+]o in this patch: 1.12 ± 0.03 ms, 0.63 ± 0.01 ms, and 0.93 ± 0.03 ms for 150, 10, and 150 mM Na+, respectively. However, across 13 patches channel open time was not significantly affected by Na+ o, with mean open times of 1.31 ± 0.12 for 150 mM [Na+]o and 1.23 ± 0.14 ms for 10 mM [Na+]o (P > 0.5, ANOVA). Similarly, closed times did not vary with [Na+]o, with mean closed times of 5.09 ± 0.80 and 4.83 ± 0.65 ms for 150 and 10 mM [Na+]o, respectively (P > 0.3, ANOVA).
If open and closed times are unaffected by [Na+]o, then the number of openings per 90-ms depolarization must vary with [Na+]o. The mean number of openings per depolarization (excluding blanks) was 10.1 ± 1.4 openings per depolarization in 10 mM Na+ (n = 11 patches); this number increased by 17% when [Na+]o was raised to 150 mM. Longer-duration “bursts” in high [Na+]o may result from either faster activation, inhibition of entry into a long-duration closed state, or both. To discriminate these possibilities, we examined the effect of [Na+]o on the distribution of first latencies and on the distribution of the duration of the last event of a 90-ms depolarization, if that event was a closing. As shown in Fig. 5 C for one patch, first latency distributions were not affected by [Na+]o (P > 0.1, Kolmogorov-Smirnov test). Median first latencies were 0.65 ± 0.19 and 0.53 ± 0.07 ms in 150 and 10 mM external Na+, respectively (P > 0.8, ANOVA; n = 13 patches). These data suggest that activation at +60 mV is not significantly altered by [Na+]o.
Because the number of openings in a depolarization increases in high [Na+]o, the duration of the last closing (if it terminates the 90-ms depolarization) must be decreased by high [Na+]o. This duration is not classified as a normal closed time, however, because it is truncated by the repolarization of the membrane. This truncated closed time was examined as a function of [Na+]o. Analysis of the cumulative distribution of the duration of the truncated closed time indeed reveals a significant difference between 10 and 150 mM Na+ o, with shorter durations in high [Na+]o (P < 0.05, Kolmogorov-Smirnov test, n = 6 out of 9 patches). Fig. 5 D shows an example of such a distribution where the duration of the truncated closing is reversibly and significantly shortened upon exposure to 150 mM Na+ o. The median durations of the truncated closing obtained for 12 patches were 33.1 ± 4.3 ms in 10 mM Na+ o and 25.3 ± 4.3 ms in 150 mM Na+ o (P < 0.02, ANOVA).
These results are consistent with high [Na+]o inhibiting the channels from entering a slow-inactivated state from which they cannot reopen during a 90-ms depolarization. The longer truncated closed intervals could, therefore, correspond to the entry into a slow-inactivated state that is responsible for runs of blank records. To test this we examined the duration of the truncated closed time conditional on whether the following record contained openings. If blank records reflect occupancy of a slow-inactivated state, longer truncated closed times should precede blank records. Fig. 5, E and F, show that longer closings indeed precede null records for both high and low [Na+]o. In 10 mM Na+ o, the median duration of the truncated closed time was 23.8 ± 3.3 ms if the following record had openings versus 54.5 ± 4.5 ms if it was a blank record (n = 10 patches). Similarly, the median duration of the truncated closed time was 19.0 ± 3.5 ms when preceding a record with openings and 45.3 ± 4.3 ms when preceding a blank one in 150 mM Na+ o (n = 10 patches).
These results provide evidence that a run of null records represents the residence of the channel in a slow-inactivated state accessed during a preceding depolarization. Consequently, the number of blank records in a row should give an estimate of the dwell time in the slow-inactivated state at the −140 mV holding potential.2 In 10 mM Na+ o, runs of blank records were on average 50% longer than in 150 mM Na+ o (4.80 ± 0.47 versus 3.15 ± 0.43, n = 8 patches). With a 2-s interval at −140 mV between 90-ms depolarizing pulses, these correspond to dwell times of roughly 9.6 s in 10 mM Na+ o versus 6.3 s in 150 mM Na+ o. Likelihood ratio analysis (see methods) of the distribution of the number of blanks in a run revealed a significant difference between 10 and 150 mM Na+ o (χ8 2 = 26; P < 0.01, n = 8 patches). Shorter runs of blank records in high [Na+]o therefore result from faster recovery from slow inactivation at −140 mV than occurs with low [Na+]o. This is in agreement with our whole-cell data (Fig. 3).
To further characterize the gating modifications associated with changes in [Na+]o, burst analysis of single-channel data was performed. We included in a burst all the events of a 90-ms record at +60 mV, beginning with the first opening. For a 2-channel patch the burst was defined as beginning the first time in a record that 2 channels were simultaneously open. This is justified because the kinetic model used to fit the data has one open state:
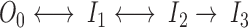 |
This model includes an open state, O0, and 3 inactivated states, I1, I2 and I3. Because the data were collected at +60 mV, the channels were maximally activated and transitions to or from a closed state in the activation pathway are extremely unlikely. Transitions between open and inactivated states are included because, for F1485Q channels, the repeated transitions between open and closed levels observed during a depolarization to large positive voltages are thought to arise from recurrent entry and recovery from fast-inactivated states destabilized by the mutation (Hartmann et al., 1994; Lawrence et al., 1996). To account for the long inactivated state from which channels cannot reopen (blank records), an absorbing inactivated state (I3) is included. I3 is identified with the slow-inactivated state in this model. Although this model is sequential, in that fast inactivation must precede slow inactivation, a nonsequential model produced equivalent results (see discussion). We estimated the rate constants for the above sequential model from the gating transitions of idealized single-channel data (Horn and Lange, 1983). Fig. 6 shows these estimates for 13 single-channel patches in which [Na+]o was changed. The model describes the data quite well, as shown by the open probability calculated from the estimated rate constants (Fig. 7 A). Fig. 7 represents analysis of the same patch shown in Fig. 5 during sequential exposure to 150, 10, and 150 mM Na+.
Figure 6.

Maximum likelihood estimates of the rate constants for the kinetic model. Data are means ± SEM from 13 patches. P values are derived by ANOVA from the natural logarithm of the rate constants.
Only one rate constant, k 23, is significantly altered by [Na+]o at +60 mV (P < 0.005, ANOVA). This rate constant, which corresponds to the rate of entry into the absorbing inactivated state I3, is about 35% larger in 10 mM Na+ o than in 150 mM Na+ o (13.11 ± 1.66 versus 9.74 ± 1.2 s−1). This change is consistent with the experiments described above which suggested that [Na+]o modulates entry into a slow-inactivated state. Moreover, this analysis suggests that [Na+]o is fairly selective in its effects, as a single rate constant is significantly altered by [Na+]o at a depolarized voltage. Fig. 7 B shows the probability of a channel being in the slow-inactivated state (PI3) as a function of time, as determined with the rate constants obtained for one patch. This shows that, in low [Na+]o, F1485Q channels are about twice more likely to enter this state at any time during a 90-ms depolarization than in high [Na+]o, and that this effect is reversible. Analysis of the average number of blanks in a run (see above) shows that the exit rate from this state at −140 mV is affected by [Na+]o. Because the exit rate is very slow, even at −140 mV (6 to 10 min−1), a channel is highly unlikely to recover from slow inactivation during a 90-ms depolarization (P < 0.02), and thus this rate is not included in our model.
discussion
Slow inactivation is a physiologically important (Cannon, 1996) but poorly understood process that is common to all Na+ channels. Defects in slow inactivation are thought to underlie certain hereditary muscle diseases, such as periodic paralyses (Ruff, 1994; Cummins and Sigworth, 1996b ). Slow inactivation is generally believed to be distinct from the fast inactivation that plays a key role in the termination of the action potential. For example, cytoplasmic application of proteolytic enzymes abolishes fast inactivation without disrupting slow inactivation (Rudy, 1978; Quandt, 1987; Valenzuela and Bennett, 1994). This effect of cytoplasmic enzymes may be due to the fact that fast inactivation depends critically on the integrity of the cytoplasmic linker between domains 3 and 4 (Stühmer et al., 1989; Patton et al., 1992; West et al., 1992). The location of the “gate” for slow inactivation is not known. However, mutations in cytoplasmic (Cummins and Sigworth, 1996b ), extracellular (Balser et al., 1996; Todt et al., 1997), and transmembrane (Fleig et al., 1994) regions affect slow inactivation.
Our results reveal another difference between slow and fast inactivation in that the kinetics and steady-state levels of slow inactivation are influenced by the concentration of extracellular alkali metal cations. Fast inactivation, by contrast, is rather insensitive to changes of [Na+]o (Armstrong and Bezanilla, 1974; Oxford and Yeh, 1985; Correa and Bezanilla, 1994; O'Leary et al., 1994; Bezanilla and Correa, 1995; Tang et al., 1996). Raising alkali metal cation concentration inhibits slow inactivation. Our single channel data indicate that at a depolarized voltage a single rate constant for entry into a slow-inactivated state is reduced at high [Na+]o, whereas the rate of leaving this state is increased at hyperpolarized voltages. Impermeant organic cations cannot substitute for the effects observed with both permeant and impermeant alkali metal cations; however, preliminary data with the permeant cation hydrazinium suggests that it also inhibits slow inactivation, perhaps more than the alkali metal cations (see results).
We have primarily studied the effects of [Na+]o on channels having the F1485Q mutation in the D3-D4 linker; this mutation strongly suppresses fast inactivation. Our results therefore raise the question of the relationship between slow and fast inactivation. Although channels may undergo slow inactivation from either closed, open, or fast-inactivated states (Ruff, 1996), the rate of slow inactivation may be dependent on the conformation of either activation or fast inactivation gates. Our results show that steady-state slow inactivation is more complete in the F1485Q mutant than in WT channels (Fig. 4); however, a dependence on extracellular cations is observed for both mutant and WT channels. Published results are inconclusive on the coupling between fast and slow inactivation, suggesting either that a channel capable of fast inactivation is more resistant (Rudy, 1978; Horn et al., 1984; Valenzuela and Bennett, 1994; Featherstone et al., 1996; Lawrence et al., 1996; Nuss et al., 1996; Hayward et al., 1997), less resistant (Balser et al., 1996; Cummins and Sigworth, 1996a ), or oblivious (Bezanilla et al., 1982; Cummins and Sigworth, 1996b ) to slow inactivation. Without strong evidence for a particular gating scheme we chose to analyze our data with a kinetic model in which slow inactivation is strictly linked to a fast inactivated state (Fig. 6). However an independent model is statistically indistinguishable (data not shown), indicating that the exact details of the model (i.e., the connectivity of the states) has little consequence on the conclusion that a single rate constant is affected by [Na+]o at a depolarized voltage. This may be due to the fact that the rate of slow inactivation is much smaller than other rates in these two gating models.
Blank records were not seen in a previous study of the F1485Q mutant of hH1a expressed in oocytes when the holding potential was as negative as we used (−140 mV) (Hartmann et al., 1994). There are, however, three notable differences between these two sets of experiments. Whereas we used excised patches, high internal [Na+], and depolarizations to +60 mV, the earlier study used cell-attached patches, the low intracellular [Na+] within the oocytes, and depolarizations to either −20 or 0 mV. We have, however, also observed blanks when pulsing to 0 mV in our experiments (data not shown). Therefore the appearance of blanks is a consequence of either internal [Na+] or the patch configuration. It is possible, for example, that the removal of cytoplasm from the inner surface of a patch by excision enhances slow inactivation, causing the appearance of blank records.
Mutation of residues contributing to the Na+ channel pore can have large effects on slow inactivation (Balser et al., 1996; Todt et al., 1997), suggesting a close relationship between the permeation pathway and the slow inactivation gate. Our results suggest that an alkali metal cation, perhaps bound in the external mouth of the channel, has two consequences for slow inactivation. It both reduces the entry rate into a slow-inactivated state at depolarized voltages, and increases the rate of leaving this state at hyperpolarized voltages. A similar dual effect of [K+]o has been observed for C-type inactivation in Shaker type K+ channels (Pardo et al., 1992; Lopez-Barneo et al., 1993; Baukrowitz and Yellen, 1995; Levy and Deutsch, 1996).
The effects of external cations on P open and slow inactivation have very different selectivities. The former effect has the selectivity of permeation, suggesting a binding site near the selectivity filter deep within the pore. This possibility is supported by the rather steep voltage dependence of P open in the absence of external permeant ions (Townsend et al., 1997). The lower selectivity of external cation effects on slow inactivation suggests a more superficial site in the external mouth of the channel. The relationship between these putative sites remains an open question.
In the Shaker subfamily of K+ channels, an inactivation mechanism called C-type inactivation seems to involve a cooperative movement of each channel subunit to constrict the outer mouth of the ion-conducting pore (Panyi et al., 1995; Ogielska et al., 1995; Liu et al., 1996), a conformational change that is apparently inhibited by the binding of a K+ ion in the pore. Moreover, C-type inactivation is inhibited by external, and enhanced by intracellular, pore blockers (Grissmer and Cahalan, 1989; Choi et al., 1991; Baukrowitz and Yellen, 1996). In many Shaker K+ channels C-type inactivation coexists with the typically faster N-type inactivation, which is caused by the occlusion of the inner mouth of the pore by a tethered inactivation particle (Hoshi et al., 1990; Zagotta et al., 1990; Demo and Yellen, 1991). An equivalent mechanism has been proposed for fast inactivation in Na+ channels (Armstrong and Bezanilla, 1977; West et al., 1992). Our data suggest the possibility that the molecular mechanism of slow inactivation in Na+ channels has some similarity with that of C-type inactivation in K+ channels. This similarity is testable by mutagenesis of pore residues that affect selectivity, and possibly Na+ binding, and by examining the effects on slow inactivation of both intracellular and extracellular pore blockers.
Acknowledgments
We thank Dr. John Lawrence for providing a copy of his manuscript prior to publication, and Dr. Carol Deutsch for extensive comments on the manuscript.
This work was supported by National Institutes of Health grant AR41691 (R. Horn).
Footnotes
Abbreviations used in this paper: ANOVA, analysis of variance; [Na+]o, external Na+ concentration; NMG, N-methyl-d-glucamine; P open, peak open probability; WT, wild type.
The rate of exit from this state may, however, be decreased by the 90-ms depolarizations every 2 s.
references
- Adelman WJ, Palti Y. The effects of external potassium and long duration voltage conditioning on the amplitude of sodium currents in the giant axon of the squid. J Gen Physiol. 1969;54:589–606. doi: 10.1085/jgp.54.5.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM, Bezanilla F. Charge movement associated with the opening and closing of the activation gates of the Na channels. J Gen Physiol. 1974;63:533–552. doi: 10.1085/jgp.63.5.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM, Bezanilla F. Inactivation of the sodium channel. II. Gating current experiments. J Gen Physiol. 1977;70:567–590. doi: 10.1085/jgp.70.5.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balser JR, Nuss HB, Chiamvimonvat N, Pérez-García MT, Marban E, Tomaselli GF. External pore-lining residue mediates slow inactivation in μ1 rat skeletal muscle sodium channels. J Physiol (Lond) 1996;494.2:431–442. doi: 10.1113/jphysiol.1996.sp021503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baukrowitz T, Yellen G. Modulation of K+ current by frequency and external [K+]: a tale of two inactivation mechanisms. Neuron. 1995;15:951–960. doi: 10.1016/0896-6273(95)90185-x. [DOI] [PubMed] [Google Scholar]
- Baukrowitz T, Yellen G. Use-dependent blockers and exit rate of the last ion from the multi-ion pore of a K+channel. Science (Wash DC) 1996;271:653–656. doi: 10.1126/science.271.5249.653. [DOI] [PubMed] [Google Scholar]
- Bezanilla F, Taylor RE, Fernandez JM. Distribution and kinetics of membrane dielectric polarization. I. Long-term inactivation of gating currents. J Gen Physiol. 1982;79:21–40. doi: 10.1085/jgp.79.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla, F., and A.M. Correa. 1995. Single-channel properties and gating of Na+ and K+ channels in the squid giant axon. In Cephalopod Neurobiology. N.J. Abbott, R. Williamson, and L. Maddock, editors. Oxford University Press, Oxford. 131–151.
- Cannon SC. Slow inactivation of sodium channels: more than just a laboratory curiosity. Biophys J. 1996;71:5–7. doi: 10.1016/S0006-3495(96)79203-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahine M, George AL, Jr, Zhou M, Ji S, Sun W, Barchi RL, Horn R. Sodium channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron. 1994;12:281–294. doi: 10.1016/0896-6273(94)90271-2. [DOI] [PubMed] [Google Scholar]
- Choi KL, Aldrich RW, Yellen G. Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-activated K+channels. Proc Natl Acad Sci USA. 1991;88:5092–5095. doi: 10.1073/pnas.88.12.5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa AM, Bezanilla F. Gating of the squid sodium channel at positive potentials. I. Macroscopic ionic and gating currents. Biophys J. 1994;66:1853–1863. doi: 10.1016/S0006-3495(94)80979-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins, T.R., and F.J. Sigworth. 1996a Comparison of slow inactivation in HYPP-T698M and F1304Q mutant rat skeletal muscle Na+ channels. Biophys. J. 70:A132.(Abstr.).
- Cummins TR, Sigworth FJ. Impaired slow inactivation in mutant sodium channels. Biophys J. 1996b;71:227–236. doi: 10.1016/S0006-3495(96)79219-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demo SD, Yellen G. The inactivation gate of the Shaker K+channel behaves like an open-channel blocker. Neuron. 1991;7:743–753. doi: 10.1016/0896-6273(91)90277-7. [DOI] [PubMed] [Google Scholar]
- Featherstone DE, Richmond JE, Ruben PC. Interaction between fast and slow inactivation in Skm1 sodium channels. Biophys J. 1996;71:3098–3109. doi: 10.1016/S0006-3495(96)79504-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleig A, Fitch JM, Goldin AL, Rayner MD, Starkus JG, Ruben PC. Point mutations in IIS4 alter activation and inactivation of rat brain IIA Na channels in Xenopusoocyte macropatches. Pflüg Archiv. 1994;427:406–413. doi: 10.1007/BF00374254. [DOI] [PubMed] [Google Scholar]
- Grissmer S, Cahalan M. TEA prevents inactivation while blocking open K+channels in human T lymphocytes. Biophys J. 1989;55:203–206. doi: 10.1016/S0006-3495(89)82793-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann HA, Tiedeman AA, Chen S-F, Brown AM, Kirsch GE. Effects of III-IV linker mutations on human heart Na+channel inactivation gating. Circ Res. 1994;75:114–122. doi: 10.1161/01.res.75.1.114. [DOI] [PubMed] [Google Scholar]
- Hayward LJ, Brown RH, Jr, Cannon SC. Slow inactivation differs among mutant Na channels associated with myotonia and periodic paralysis. Biophys J. 1997;72:1204–1219. doi: 10.1016/S0006-3495(97)78768-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn R. Estimating the number of channels in patch recordings. Biophys J. 1991;60:433–439. doi: 10.1016/S0006-3495(91)82069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn R, Lange K. Estimating kinetic constants from single channel data. Biophys J. 1983;43:207–223. doi: 10.1016/S0006-3495(83)84341-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn R, Vandenberg CA. Statistical properties of single sodium channels. J Gen Physiol. 1984;84:505–534. doi: 10.1085/jgp.84.4.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn R, Vandenberg CA, Lange K. Statistical analysis of single sodium channels. Effects of N-bromoacetamide. Biophys J. 1984;45:323–335. doi: 10.1016/S0006-3495(84)84158-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science (Wash DC) 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Lawrence JH, Orias DW, Balser JR, Nuss HB, Tomaselli GF, O'Rourke B, Marban E. Single-channel analysis of inactivation-defective rat skeletal muscle sodium channels containing the F1304Q mutation. Biophys J. 1996;71:1285–1294. doi: 10.1016/S0006-3495(96)79329-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DI, Deutsch C. Recovery from C-type inactivation is modulated by extracellular potassium. Biophys J. 1996;70:798–805. doi: 10.1016/S0006-3495(96)79619-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Jurman ME, Yellen G. Dynamic rearrangement of the outer mouth of a K+channel during gating. Neuron. 1996;16:859–867. doi: 10.1016/s0896-6273(00)80106-3. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J, Hoshi T, Heinemann SH, Aldrich RW. Effects of external cations and mutations in the pore region on C-type inactivation of Shakerchannels. Receptors and Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- Moorman JR, Kirsch GE, Brown AM, Joho RH. Changes in sodium channel gating produced by point mutations in a cytoplasmic linker. Science (Wash DC) 1990;250:688–691. doi: 10.1126/science.2173138. [DOI] [PubMed] [Google Scholar]
- Nuss HB, Balser JR, Orias DW, Lawrence JH, Tomaselli GF, Marban E. Coupling between fast and slow inactivation revealed by analysis of a point mutation (F1304Q) in μ1 rat skeletal muscle sodium channels. J Physiol (Lond) 1996;494.2:411–429. doi: 10.1113/jphysiol.1996.sp021502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary ME, Kallen RG, Horn R. Evidence for a direct interaction between tetra-alkylammonium cations and the inactivation gate of cardiac sodium channels. J Gen Physiol. 1994;104:523–539. doi: 10.1085/jgp.104.3.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogielska EM, Zagotta WN, Hoshi T, Heinemann SH, Haab J, Aldrich RW. Cooperative subunit interactions in C-type inactivation of K channels. Biophys J. 1995;69:2449–2457. doi: 10.1016/S0006-3495(95)80114-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxford GS, Yeh JZ. Interaction of monovalent cations with sodium channels in squid axon. I. Modification of physiological inactivation gating. J Gen Physiol. 1985;85:583–602. doi: 10.1085/jgp.85.4.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panyi G, Sheng Z, Tu L, Deutsch C. C-type inactivation of a voltage-gated K+channel occurs by a cooperative mechanism. Biophys J. 1995;69:896–903. doi: 10.1016/S0006-3495(95)79963-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo LA, Heinemann SH, Terlau H, Ludewig U, Lorra C, Pongs O, Stühmer W. Extracellular K+ specifically modulates a rat brain K+channel. Proc Natl Acad Sci USA. 1992;89:2466–2470. doi: 10.1073/pnas.89.6.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patlak J, Horn R. Effect of N-bromoacetamide on single sodium channel currents in excised membrane patches. J Gen Physiol. 1982;79:333–351. doi: 10.1085/jgp.79.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton DE, West JW, Catterall WA, Goldin AL. Amino acid residues required for fast Na+-channel inactivation: charge neutralizations and deletions in the III-IV linker. Proc Natl Acad Sci USA. 1992;89:10905–10909. doi: 10.1073/pnas.89.22.10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quandt FN. Burst kinetics of sodium channels which lack fast inactivation in mouse neuroblastoma cells. J Physiol (Lond) 1987;392:563–585. doi: 10.1113/jphysiol.1987.sp016797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas E, Armstrong CM. Sodium conductance activation without inactivation in pronase-perfused axons. Nature New Biology. 1971;229:177–178. doi: 10.1038/newbio229177a0. [DOI] [PubMed] [Google Scholar]
- Ruben PC, Starkus JG, Rayner MD. Steady-state availability of sodium channels. Interactions between activation and slow inactivation. Biophys J. 1992;61:941–955. doi: 10.1016/S0006-3495(92)81901-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy B. Slow inactivation of the sodium conductance in squid giant axons: pronase resistance. J Physiol (Lond) 1978;283:1–21. doi: 10.1113/jphysiol.1978.sp012485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff RL. Slow Na+channel inactivation must be disrupted to evoke prolonged depolarization-induced paralysis. Biophys J. 1994;66:542–545. doi: 10.1016/s0006-3495(94)80807-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff RL. Single-channel basis of slow inactivation of Na+channels in rat skeletal muscle. Am J Physiol. 1996;271:C971–C981. doi: 10.1152/ajpcell.1996.271.3.C971. [DOI] [PubMed] [Google Scholar]
- Ruff RL, Simoncini L, Stühmer W. Comparison between slow sodium channel inactivation in rat slow- and fast-twitch muscle. J Physiol (Lond) 1987;383:339–348. doi: 10.1113/jphysiol.1987.sp016412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoncini L, Stühmer W. Slow sodium channel inactivation in rat fast-twitch muscle. J Physiol (Lond) 1987;383:327–337. doi: 10.1113/jphysiol.1987.sp016411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stühmer W, Conti F, Suzuki H, Wang XD, Noda M, Yahagi N, Kubo H, Numa S. Structural parts involved in activation and inactivation of the sodium channel. Nature (Lond) 1989;339:597–603. doi: 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- Tang L, Kallen RG, Horn R. Role of an S4-S5 linker in sodium channel inactivation probed by mutagenesis and a peptide blocker. J Gen Physiol. 1996;108:89–104. doi: 10.1085/jgp.108.2.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todt, H., S. Dudley, and H. Fozzard. 1997. Ultra-slow inactivation in the skeletal muscle sodium channel is influenced by pore residues. Biophys. J. 72:A261. (Abstr.).
- Townsend C, Hartmann HA, Horn R. Anomalous effect of permeant ion concentration on peak open probability of cardiac Na+channels. J Gen Physiol. 1997;110:11–21. doi: 10.1085/jgp.110.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela C, Bennett PB., Jr Gating of cardiac Na+channels in excised membrane patches after modification by α-chymotrypsin. Biophys J. 1994;67:161–171. doi: 10.1016/S0006-3495(94)80465-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanDongen AM. A new algorithm for idealizing single ion channel data containing multiple unknown conductance levels. Biophys J. 1996;70:1303–1315. doi: 10.1016/S0006-3495(96)79687-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wald A, Wolfowitz J. On a test whether two samples are from the same population. Annals of Mathematical Statistics. 1940;11:147–162. [Google Scholar]
- Wang DW, George AL, Jr, Bennett PB. Comparison of heterologously expressed human cardiac and skeletal muscle sodium channels. Biophys J. 1996;70:238–245. doi: 10.1016/S0006-3495(96)79566-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na+-channel inactivation. Proc Natl Acad Sci USA. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science (Wash DC) 1990;250:568–571. doi: 10.1126/science.2122520. [DOI] [PubMed] [Google Scholar]