

Abstract
We investigated the molecular basis for Ca-dependent inactivation of the cardiac L-type Ca channel. Transfection of HEK293 cells with the wild-type α1C or its 3′ deletion mutant (α1C−3′del) produced channels that exhibited prominent Ca-dependent inactivation. To identify structural regions of α1C involved in this process, we analyzed chimeric α1 subunits in which one of the major intracellular domains of α1C was replaced by the corresponding region from the skeletal muscle α1S subunit (which lacks Ca-dependent inactivation). Replacing the NH2 terminus or the III–IV loop of α1C with its counterpart from α1S had no appreciable effect on Ca channel inactivation. In contrast, replacing the I–II loop of α1C with the corresponding region from α1S dramatically slowed the inactivation of Ba currents while preserving Ca-dependent inactivation. A similar but less pronounced result was obtained with a II–III loop chimera. These results suggest that the I–II and II–III loops of α1C may participate in the mechanism of Ca-dependent inactivation. Replacing the final 80% of the COOH terminus of α1C with the corresponding region from α1S completely eliminated Ca-dependent inactivation without affecting inactivation of Ba currents. Significantly, Ca-dependent inactivation was restored to this chimera by deleting a nonconserved, 211–amino acid segment from the end of the COOH terminus. These results suggest that the distal COOH terminus of α1S can block Ca-dependent inactivation, possibly by interacting with other proteins or other regions of the Ca channel. Our findings suggest that structural determinants of Ca-dependent inactivation are distributed among several major cytoplasmic domains of α1C.
Keywords: α1S, skeletal muscle, L-type Ca channel, chimeric proteins, heart
INTRODUCTION
L-type Ca channels perform essential roles in the cardiovascular system, where they trigger excitation–contraction coupling and contribute to pacemaker and action potentials (Boyett et al., 1996). Inactivation of L-type channels is induced by membrane depolarization and elevations in intracellular [Ca], although these two types of Ca channel inactivation appear to proceed via distinct and independent mechanisms (Hadley and Lederer, 1991; Obejero-Paz et al., 1991; Shirokov et al., 1993). Ca-dependent inactivation is a prominent feature of cardiac L-type Ca channels that has important implications for the function of these channels in cardiovascular physiology.
Voltage-gated Ca channels are heteromultimeric complexes composed of pore-forming α1 and accessory α2δ and β subunits (Hofmann et al., 1994). Transfection of mammalian cell lines or Xenopus oocytes with the cardiac α1 subunit (α1C) by itself produces voltage-gated Ca channels that exhibit Ca-dependent inactivation (Neely et al., 1994; Perez-Garcia et al., 1995; Zong and Hofmann, 1996), suggesting that this type of inactivation is an intrinsic property of the α1C subunit. Because Ca-dependent inactivation is induced by a rise in intracellular Ca concentration (Haack and Rosenberg, 1994), it is reasonable to postulate that cytoplasmic domains of α1C participate in its molecular mechanism. The five major putative cytoplasmic domains of α1C include the NH2 and COOH termini and three linkers (the I–II, II–III, and III–IV loops) that connect the four major transmembrane domains (Mikami et al., 1989).
Two previous studies have provided evidence that one or more of these cytoplasmic domains play important roles in Ca-dependent inactivation. Thus, Ca- dependent inactivation is abolished by simultaneous replacement of all five of the major cytoplasmic domains of α1C with the corresponding regions from the skeletal muscle α1S subunit (Zong et al., 1994). Ca-dependent inactivation is also eliminated by replacing all or a portion of the COOH terminus of α1C with the corresponding region of the neuronal α1E subunit (de Leon et al., 1995). Because α1S and α1E both appear to exhibit only voltage-dependent inactivation (Donaldson and Beam, 1983; Beam and Knudson, 1988; de Leon et al., 1995), these results imply that the structural determinants of Ca-dependent inactivation are encoded within the cytoplasmic domains of α1C. The goal of the present study was to test this hypothesis. Toward this end, we have studied a series of chimeric α1 subunits in which the major cytoplasmic domains of α1C were individually replaced by their counterpart from α1S. Our results suggest that the cytoplasmic COOH terminus, and I–II and II–III loops are all involved in the molecular mechanism of Ca-dependent inactivation. In addition, our findings indicate that Ca-dependent inactivation can be prevented by the distal COOH terminus from the skeletal muscle α1S. Some of these results have appeared previously in abstract form (Adams and Tanabe, 1996).
MATERIALS AND METHODS
Cell Culture and Transfection
Human embryonic kidney cells were obtained from the American Type Culture Collection (CRL 1573; Rockville, MD) and propagated using standard techniques. The culture medium contained 90% DMEM (11995-065; GIBCO BRL, Gaithersburg, MD), 10% heat-inactivated horse serum (26050-13; GIBCO BRL) and 50 μg/ml gentamicin (15710-015; GIBCO BRL). Every 2–3 d, these cells were briefly trypsinized and replated onto the maintenance culture at a fourfold lower density. At the time of replating, additional 35-mm culture dishes (3001; Becton Dickinson & Co., Franklin Lakes, NJ) were seeded with ∼103 cells/dish. Approximately 16 h later, the CaPO4 precipitation technique (Cell Phect Kit; Pharmacia LKB Biotechnology Inc., Piscataway, NJ) was used to transfect the seeded cells with a combination (at 1 μg of each plasmid cDNA per dish) of expression plasmids encoding the rabbit cardiac α1C (or chimeras constructed between α1C and the rabbit α1S) and the rabbit skeletal muscle α2δa and β1a subunits. The transfection mixture also included an expression plasmid (EBO-pCD-Leu2; 59565; American Type Culture Collection) encoding the human CD8 protein at a fivefold lower concentration (0.2 μg per dish). 1–3 d later, paramagnetic beads (4.5 μm diameter) coated with anti–CD8 antibody (Dynal, Inc., Great Neck, NY) were added to each dish. Cells expressing CD8 protein on the surface membrane were visually identified by virtue of being decorated with the beads (Jurman et al., 1994) and were selected for electrophysiological analysis.
Molecular Biology
The amino acid compositions and construction of the expression plasmids encoding the 3′ deletion mutant of α1C (α1C−3′del) and the chimeric α1 subunits CSk1, CSk2, CSk3, and CSk4 have been previously described (Tanabe et al., 1990b ; Zong et al., 1994). The cDNAs encoding the chimeras CSk5 and CSk8 are composed of the following restriction fragments (the origin of the fragments is given in parentheses). pCSk5: 4.2-kb pair HindIII-AatII (pCARD1; see Mikami et al., 1989), 0.88-kb pair AatII-BglII (pCARD1), and 0.55-kb pair BglII-HindIII (pC6Δ1; see Beam et al., 1992). pCSk8: 3.8-kb pair HindIII-BspHI (pCARD1), 0.24-kb pair BspHI-BstXI (pCAC6; see Tanabe et al., 1988), 0.19-kb pair BstXI-AatII (pCARD1), and 2.9-kb pair AatII-HindIII (pCARD1). The expression plasmids pCSk5 and pCSk8, carrying the cDNAs encoding the individual chimeric Ca channels, were constructed by inserting the corresponding cDNAs into the HindIII site of the plasmid pKCRH2 (Mishina et al., 1984).
The amino acid compositions of CSk5 and CSk8 are as follows (C and Sk, cardiac and skeletal muscle Ca channel, respectively; numbers in parentheses, amino-acid numbers [Tanabe et al., 1987; Mikami et al., 1989]; the junctional sequences common to the two Ca channels are represented by amino acid numbers of the cardiac Ca channel). CSk5: C (1–1634) and Sk (1510–1662). CSk8: C (1–1204), Sk (1074–1129), and C (1261–2171).
Electrophysiology
Patch pipettes were fabricated from 100-μl borosilicate micropipettes (53432-921; VWR Scientific, West Chester, PA) and filled with a solution containing (mM): 155 CsCl, 10 Cs2EGTA, 4 MgATP, 0.38 Tris-GTP, and 10 HEPES, with pH adjusted to 7.4 using CsOH. Aliquots of this solution were stored at −80°C and kept on ice after thawing. The internal solution was filtered (0.22 μm) immediately before use. Filled pipettes had DC resistances of 1–2 MΩ. Pipette tips were coated with paraffin to reduce capacitance, and then fire polished. Residual pipette capacitance was compensated in the cell-attached configuration, using the negative capacitance circuit of the Axopatch 200A amplifier (Axon Instruments, Inc., Foster City, CA). In earlier experiments, the external solution contained (mM): 145 tetraethylammonium (TEA)1 chloride, 40 CaCl2 or BaCl2, and 10 HEPES, with pH adjusted to 7.4 using TEAOH. In later experiments, an external solution containing (mM) 140 NaCl, 2 KCl, 40 CaCl2 or BaCl2, and 10 HEPES (pH 7.4 with NaOH or HCl) was used because the cells appeared to remain healthy for longer periods in this solution. Because the voltage dependence of ionic currents recorded in the NaCl-based solution was shifted by −10 mV relative to currents recorded in the TEACl-based solution, data obtained using the two different external solutions have been analyzed separately. Experiments were performed at room temperature (20– 23°C). Voltages reported in this paper have not been corrected for liquid junction potentials.
Ca or Ba currents were recorded using the whole-cell patch-clamp technique (Hamill et al., 1981). After establishment of the whole-cell configuration, electronic compensation was used to minimize the access resistance and the time required to charge the cell capacitance. The DC resistance of the whole-cell configuration typically exceeded 1 GΩ, and leakage currents were usually <50 pA at the steady holding potential of −80 mV. Linear cell capacitance was monitored throughout each experiment and was calculated from the integral of charges required to clamp the membrane from −80 to −70 mV. The compensated series resistance (RS) was calculated from the time constant for decay of the capacity transient and the linear cell capacitance. Depolarizing test pulses were delivered at 5-s intervals. Linear membrane capacitance and leakage currents were subtracted from test currents using the −P/6 method, and all analyzed and reported data were obtained from corrected currents. Currents were filtered at 0.5–10 kHz using the built-in Bessel filter (4-pole low pass) of the Axopatch 200A patch-clamp amplifier, and were sampled at 1–50 kHz using a Digidata 1200 analogue-to-digital board installed in a Gateway 486-66V personal computer. The pCLAMP software programs Clampex and Clampfit (version 6.0) were used for data acquisition and analysis, respectively. Figures were made using Origin (version 4.1).
RESULTS
Fig. 1 shows whole-cell Ca or Ba currents recorded from a human embryonic kidney cell expressing the wild-type α1C subunits. As demonstrated by previous studies (Perez-Reyes et al., 1994; Zong et al., 1994; de Leon et al., 1995; Perez-Garcia et al., 1995; Ferreira et al., 1997), the heterologously expressed cardiac α1C exhibits prominent Ca-dependent inactivation, as evidenced by the faster inactivation of Ca than Ba currents. The inactivation rate of currents mediated by the subunit combination expressed here (α1C, α2δa, and β1a) closely approximates the inactivation rate of natively expressed cardiac channels (see Imredy and Yue, 1994), and is considerably faster than the inactivation rate of currents mediated by expression of α1C and β2a in the absence of α2δa (Perez-Reyes et al., 1994; de Leon et al., 1995; Perez-Garcia et al., 1995; Ferreira et al., 1997).
Figure 1.

Ca-dependent inactivation of wild-type α1C Ca channels coexpressed in HEK293 cells with skeletal muscle α2δa and β1a subunits. (A) Representative whole-cell Ca currents mediated by α1C. The illustrated currents were evoked by depolarizations from −10 to +90 mV. The compensated series resistance (RS) was 4.2 MΩ. Linear cell capacitance (C) = 41 pF. File 96506016. (B) Representative Ba currents mediated by α1C. Test pulses −20 to +90 mV. Same cell as in A. RS = 2.1 MΩ. File 96506018. (C) Average I–V relations for Ca and Ba currents mediated by α1C. Each plotted point represents the mean (±SEM) of 13 (Ca) and 6 (Ba) different cells. (D) Time constants for inactivation of Ca or Ba currents mediated by α1C. Currents were evoked by 250-ms test pulses and were fit with either a single or double exponential function. For currents requiring two exponentials, only the fastest component was included in the analysis. Each plotted point represents the mean (±SEM) of 2–14 (Ca) and 3–7 (Ba) different cells. Experiments summarized in this figure were done using the TEACl-based external solution.
To quantify the time course of inactivation, the decaying phase of Ca or Ba currents was fit with a single or double exponential function. Most Ca currents required two exponentials for a good fit, whereas Ba currents evoked by relatively short test pulses (250 ms) could usually be well fit by a single exponential. However, some Ba currents displayed two distinct components of inactivation. Fig. 1 D plots the time constants for inactivation of α1C currents as a function of test potential. For Ca currents, the time constants for inactivation had a U-shaped dependence on test potential, whereas time constants for inactivation of Ba currents decreased progressively with increasing test potential. These results are consistent with the expectation that α1C undergoes Ca- but not Ba-dependent inactivation. However, in some cells the availability of Ba currents (measured using a double-pulse protocol) displayed a weak U-shaped dependence on test potential (data not shown), consistent with the idea that Ba can also trigger ion-dependent inactivation, although less effectively than Ca.
To investigate the structural basis for Ca-dependent inactivation, we expressed a series of chimeric α1 subunits in which one of the major intracellular domains of α1C was replaced by the corresponding region from the skeletal muscle α1S subunit. The composition of these chimeras is represented diagrammatically in Fig. 2.
Figure 2.
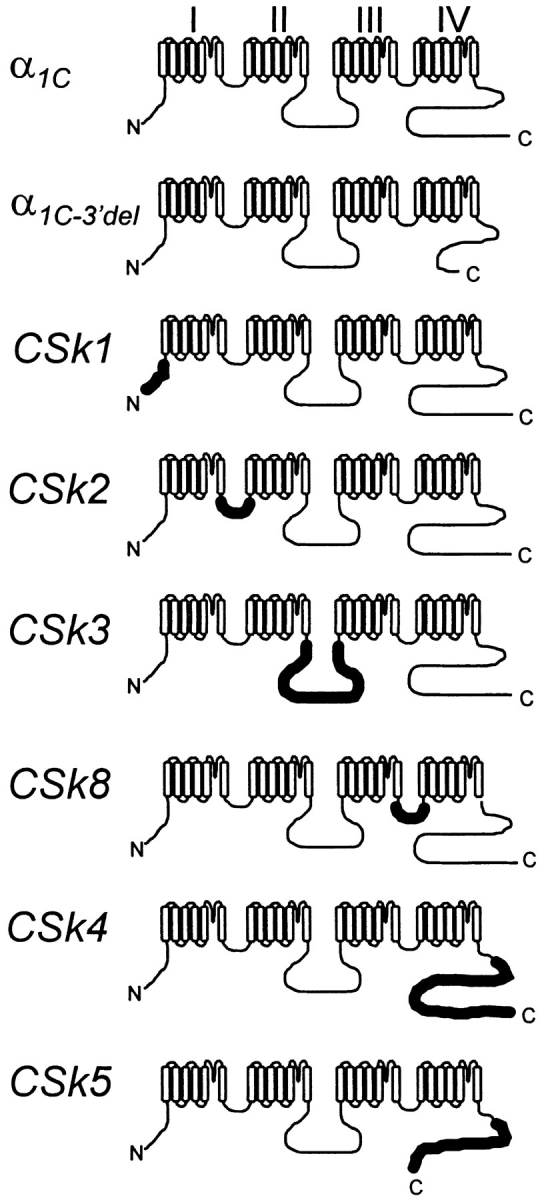
Schematic representations of the expressed α1 subunits. Thinner lines denote regions having amino acid sequence corresponding to the rabbit cardiac α1C subunit (Mikami et al., 1989); thicker lines denote regions corresponding to the rabbit skeletal muscle α1S subunit (Tanabe et al., 1987). The amino acid composition of α1C−3′del and construction of its cDNA has been previously described; the final five amino acid residues from the COOH terminus of α1C (GVSSL) are retained in α1C−3′del (Zong et al., 1994). The amino acid composition and cDNA construction of CSk1, CSk2, CSk3, and CSk4 are described in Tanabe et al. (1990b). The compositions of CSk5 and CSk8 and the constructions of their cDNAs are described in materials and methods.
Previous studies have shown that deletion of the distal COOH terminus of α1C or α1S produces a fully functional Ca channel (Beam et al., 1992; Zong et al., 1994; de Leon et al., 1995). Fig. 3 A confirms this result for the deletion mutant α1C−3′del in which amino acids 1813–2166 have been removed from the COOH terminus of α1C. As shown in Table I, inactivation of α1C−3′del proceeded at the same rate as the wild-type α1C, confirming that deletion of the distal COOH terminus of α1C does not appreciably alter the process of Ca-dependent inactivation.
Figure 3.

Ca-dependent inactivation is normal in α1C−3′del, CSk1, and CSk8. (A) Representative Ca (top) and Ba (bottom) currents mediated by α1C−3′del, a deletion mutant of α1C lacking amino acid residues 1813–2166 from the COOH terminus. Test pulses from 0 to +40 mV (Ca) and −10 to +60 mV (Ba). Files 95929002, 95929006. C = 42 pF. RS = 3.8 MΩ. (B) Representative Ca and Ba currents mediated by CSk1, a chimera in which the NH2 terminus of α1C was replaced by the corresponding region from α1S. Test pulses from 0 to +40 mV (Ca) and 0 to +60 mV (Ba). Files 95D01016, 95D01018. C = 21 pF. RS = 6.3 MΩ. (C) Representative Ca and Ba currents mediated by chimera CSk8, in which the III–IV loop of α1C was replaced by the corresponding region from α1S. Test pulses from +30 to +50 mV (Ca) and +20 to +40 mV (Ba). Files 96118045, 96118053. C = 15 pF. RS = 4.9 MΩ. Experiments summarized in this figure were done using the TEACl-based external solution.
Table I.
Data from HEK293 Cells Cotransfected with Expression Plasmids Encoding α1, α2δa, and β1a Ca Channel Subunits
| α1 construct | Ca current density | Fast tau ICa | Fast tau IBa | External solution | ||||
|---|---|---|---|---|---|---|---|---|
| pA/pF | ms | ms | ||||||
| α1C | −56 ± 9 (17) | 17 ± 2 (14) | 88 ± 8 (7) | TEAC1 | ||||
| α1C | −17 ± 3 (7) | 38 ± 6 (7) | — | TEAC1 | ||||
| α1C−3′del | −68 ± 21 (7) | 20 ± 4 (8) | 93 ± 21 (6) | TEAC1 | ||||
| CSk1 | −28 ± 5 (12) | 20 ± 1 (11) | 57 ± 11 (6) | TEAC1 | ||||
| CSk2 | −28 ± 11 (8) | 35 ± 4 (9) | 295 ± 38 (8) | TEAC1 | ||||
| CSk2 | −20 ± 7 (5) | 51 ± 8 (5) | 467 ± 107 (4) | NaCl | ||||
| CSk3 | −23 ± 6 (9) | 31 ± 5 (9) | 129 ± 13 (4) | TEAC1 | ||||
| CSk3 | −45 ± 9 (6) | 38 ± 5 (6) | 106 ± 22 (3) | NaCl | ||||
| CSk4 | −16 ± 3 (10) | 70 ± 4 (5) | 75 ± 5 (5) | TEAC1 | ||||
| CSk4 | −13 ± 5 (4) | 82 ± 15 (4) | 80 ± 10 (4) | NaCl | ||||
| CSk5 | −51 ± 12 (8) | 32 ± 4 (10) | 97 ± 9 (8) | TEAC1 | ||||
| CSk5 | −17 ± 6 (5) | 76 ± 7 (5) | 111 ± 6 (7) | NaCl | ||||
| CSk8 | −51 ± 12 (8) | 23 ± 4 (8) | 110 ± 10 (7) | TEAC1 |
The external solution was based upon TEAC1 or NaCl (see materials and methods for compositions). Time constants for inactivation were derived by fitting maximal Ca or Ba currents (i.e., at the peak of the I–V relationship) with one or two exponential functions. Only the fast time constants (fast tau) were analyzed. Currents were evoked by test pulses of 250 or 500 ms, or 1.25 or 5 s, as dictated by the inactivation rate. Mean ± SEM, with the number of cells (n) in parentheses.
We next examined the potential role of the NH2 terminus in Ca-dependent inactivation. The NH2 terminus of α1C is 103 amino acids longer than that of α1S (Mikami et al., 1989) and contains four consensus sites for potential phosphorylation by PKC, whereas the NH2 terminus of α1S lacks predicted PKC sites. In chimera CSk1, the NH2 terminus of α1C has been replaced by its counterpart from α1S (Fig. 2). Currents mediated by CSk1 closely resembled those produced by α1C and α1C−3′del (Fig. 3 B). Thus, Ca currents inactivated faster than Ba currents, and time constants for inactivation of Ca currents were indistinguishable between CSk1 and α1C (Table I). However, inactivation of Ba currents was slightly faster for CSk1 than for α1C. Overall, these results suggest that Ca-dependent inactivation was not significantly altered by replacing the NH2 terminus of α1C with the corresponding region from α1S.
The Ca channel α1 subunit is highly homologous to the α subunit of voltage-gated Na channels, and the cytoplasmic linker between transmembrane domains III and IV (the III–IV loop) of Na channels is a critical structural determinant of fast inactivation (Vassilev et al., 1988; Stühmer et al., 1989). To examine the possibility that the homologous III–IV loop of Ca channels is involved in Ca-dependent inactivation, we constructed chimera CSk8 in which the III–IV loop and the first half of IVS1 of α1C were replaced by the corresponding region from α1S (Fig. 2). As shown in Fig. 3 C, Ca currents mediated by CSk8 inactivated faster than Ba currents and in general closely resembled currents produced by expression of α1C−3′del or CSk1. Furthermore, the time constants for inactivation of currents mediated by CSk8 were comparable to those obtained for α1C, α1C−3′del, and CSk1 (Table I), suggesting that Ca-dependent inactivation is not altered in CSk8.
In both α1C and α1S, the I–II loop contains a number of negatively charged aspartate and glutamate residues that could potentially form a Ca-coordination site or sites. There are 25 negatively charged residues within the I–II loop of α1C and 19 such residues within the I–II loop of α1S. To examine the possibility that the extra acidic residues within the I–II loop of α1C might play a functional role in Ca-dependent inactivation, we expressed chimera CSk2 in which the I–II loop and most of the IIS1 segment of α1C were replaced by the corresponding region from α1S (Fig. 2).
Fig. 4 A presents Ca and Ba currents mediated by CSk2. For comparison, currents recorded under identical conditions from cells expressing α1C are shown in Fig. 4 B. Ca currents produced by CSk2 inactivated significantly faster than Ba currents, indicating the presence of Ca-dependent inactivation. Furthermore, when relatively long test pulses (1.25 s) were used, both Ca and Ba currents exhibited two distinct components of inactivation. As recently shown by Ferreira et al. (1997) for α1C, the fast component of Ba current inactivation likely represents an ion-dependent process because it parallels Ba influx, whereas the slow component of inactivation likely represents a voltage-dependent process because it parallels the immobilization of gating charge. The presence of two components for inactivation of currents mediated by CSk2 is consistent with the thesis of Ferreira et al. (1997) that Ba as well as Ca can trigger inactivation. In the present work, we have analyzed only the faster time constants for inactivation of Ca and Ba currents.
Figure 4.

CSk2 retains Ca-dependent inactivation, but Ba current inactivation is dramatically slowed. (A) Representative Ca and Ba currents mediated by CSk2. Test pulses from −10 to +60 mV (Ca) and −10 to +50 mV (Ba). Files 97428010, 97428014. C = 17 pF. RS = 2.2 MΩ. (B) Representative Ca and Ba currents mediated by α1C. Test pulses from −10 to +40 mV (Ca) and −10 to +50 mV (Ba). Files 97502005, 97502012. C = 31 pF. RS = 2.3 MΩ. (C) I–V relations for CSk2 currents. Plotted points represent the mean (±SEM) of 4–10 different cells. (D) Voltage dependence of fast time constants for inactivation of CSk2. Plotted points represent mean (±SEM) of four to five different cells. Experiments summarized in this figure were done using the NaCl-based external solution.
The fast time constants for inactivation of Ca currents mediated by CSk2 exhibited a U-shaped dependence on test potential, whereas those for Ba currents progressively decreased with increasing test potential (Fig. 4 D). The time constants for inactivation of Ca currents were similar for CSk2 and α1C currents of comparable densities (Table I). These results demonstrate that chimera CSk2 undergoes Ca-dependent inactivation. In marked contrast, the inactivation of Ba currents mediated by CSk2 was dramatically slowed (Fig. 4 A), with the fast time constants for inactivation being approximately threefold larger than for α1C (Table I). If the fast phase of Ba current inactivation primarily reflects an ion-dependent process as proposed by Ferreira et al. (1997), then the slower inactivation of CSk2 may indicate that Ba is less effective in triggering ion-dependent inactivation when the I–II loop has skeletal muscle as opposed to cardiac sequence. Such an interpretation would imply that the I–II loop of α1C has a functional role in Ca-dependent inactivation if Ba- and Ca-dependent inactivation are equivalent. It is also possible that the process of voltage-dependent inactivation is affected somewhat by replacement of the I–II loop region.
We have previously demonstrated that the II–III loop of α1S performs a critical function in skeletal muscle– type excitation–contraction coupling (Tanabe et al., 1990b ), perhaps by interacting directly with the ryanodine receptor. The II–III loops of α1C and α1S also contain numerous (35 and 27, respectively) negatively charged aspartate or glutamate residues that could potentially be involved in Ca coordination (Fujita et al., 1993). In addition, the II–III loop of α1S contains a consensus site for phosphorylation by PKA (Tanabe et al., 1987), whereas the II–III loop of α1C lacks predicted PKA sites (Mikami et al., 1989). To test whether the II– III loop of α1C performs a unique function in Ca-dependent inactivation, we expressed chimera CSk3 in which the II–III loop of α1C was replaced by its counterpart from α1S (Fig. 2). CSk3 undergoes Ca-dependent inactivation because Ca currents inactivated much faster than Ba currents (Fig. 5). Furthermore, both Ca and Ba currents exhibited two distinct components of inactivation. The fast time constants for inactivation of Ca currents exhibited a U-shaped dependence on test potential; in contrast, such a relationship was not apparent for Ba currents (Fig. 5 D). Similar to the results obtained for CSk2, the fast component of Ba current inactivation was slightly slower for CSk3 than for α1C (Table I), raising the possibility that the II–III loop may perform a functional role in Ca- or voltage-dependent inactivation.
Figure 5.

CSk3 exhibits Ca-dependent inactivation. (A) Representative Ca currents mediated by CSk3. Test pulses from −10 to +40 mV. File 97512062. C = 41 pF. RS = 4.0 MΩ. (B) Representative Ba currents mediated by CSk3. Test pulses from −10 to +40 mV. File 97512067. Same cell as in A. (C) I–V relations for CSk3 currents. Plotted points represent the mean (±SEM) of three to eight different cells. (D) Voltage dependence of fast time constants for inactivation of CSk3. Plotted points represent mean (±SEM) of three to six different cells. Experiments summarized in this figure were done using the NaCl-based external solution.
Several previous studies have identified the COOH terminus of α1C as an important structural determinant of Ca-dependent inactivation (de Leon et al., 1995; Soldatov et al., 1997; Zhou et al., 1997). To obtain further information regarding this issue, we expressed chimera CSk4 in which the distal 80% of the COOH terminus of α1C was replaced by the corresponding region from α1S (Fig. 2). In contrast to the other chimeras examined, Ca and Ba currents mediated by CSk4 inactivated at equivalent rates (Fig. 6, A and B). Both Ca and Ba currrents exhibited two distinct components of inactivation. However, there was no difference between the fast time constants for inactivation of Ca currents and those for Ba currents over a wide range of test potentials and time constants for inactivation of Ca currents did not have a U-shaped dependence upon test potential (Fig. 6 D). These results demonstrate that CSk4 lacks Ca- dependent inactivation. As shown in Table I, the fast time constants for inactivation of Ca currents were three- to fourfold larger for CSk4 than for α1C. This slow inactivation of Ca currents cannot be explained by low expression of CSk4, because inactivation was also slow compared with low density currents mediated by α1C (Table I). Interestingly, the fast time constants for inactivation of Ba currents were not different between CSk4 and α1C, suggesting that the fast component of Ba current inactivation is unaltered in CSk4.
Figure 6.

CSk4 lacks Ca-dependent inactivation. (A) Representative Ca currents mediated by CSk4, in which the distal 80% of the COOH terminus of α1C has been replaced by the corresponding region from α1S. File 97501003. C = 26 pF. RS = 3.6 MΩ. (B) Representative Ba currents mediated by CSk4. File 97501008. Same cell as in A. For the illustrated currents, inactivation appears to be slightly slower for Ba than for Ca currents, but this is only apparent: the Ba currents are larger and therefore less contaminated by outward currents. (C) I–V relations for CSk4 currents. Each point represents the mean (±SEM) of four different cells. (D) Voltage dependence of fast time constants for inactivation of CSk4 currents. Each point represents the mean (±SEM) of four different cells. Experiments summarized in this figure were done using the NaCl-based external solution.
The results obtained with α1C-3′del (Fig. 3) confirm that a large segment of the distal COOH terminus is not required for Ca-dependent inactivation (Zong et al., 1994; de Leon et al., 1995). In contrast, the results obtained with CSk4 (Fig. 6) suggest that the COOH terminus of α1S can somehow prevent Ca-dependent inactivation. A comparison of the COOH termini of α1C and α1S (Fig. 7) reveals substantial conservation of their sequences for ∼200 amino acids after the end of the last predicted transmembrane segment (IVS6). Beyond this point, however, the COOH termini of α1C and α1S diverge significantly. To test whether the distal, nonconserved portion of the COOH terminus from α1S could be responsible for the inability of CSk4 to undergo Ca-dependent inactivation, we constructed chimera CSk5 (Fig. 2). This construct is a truncation mutant of CSk4 in which the final 211 amino acids of the COOH terminus have been deleted, effectively removing the majority of the sequence that is not conserved between α1C and α1S (Fig. 7).
Figure 7.
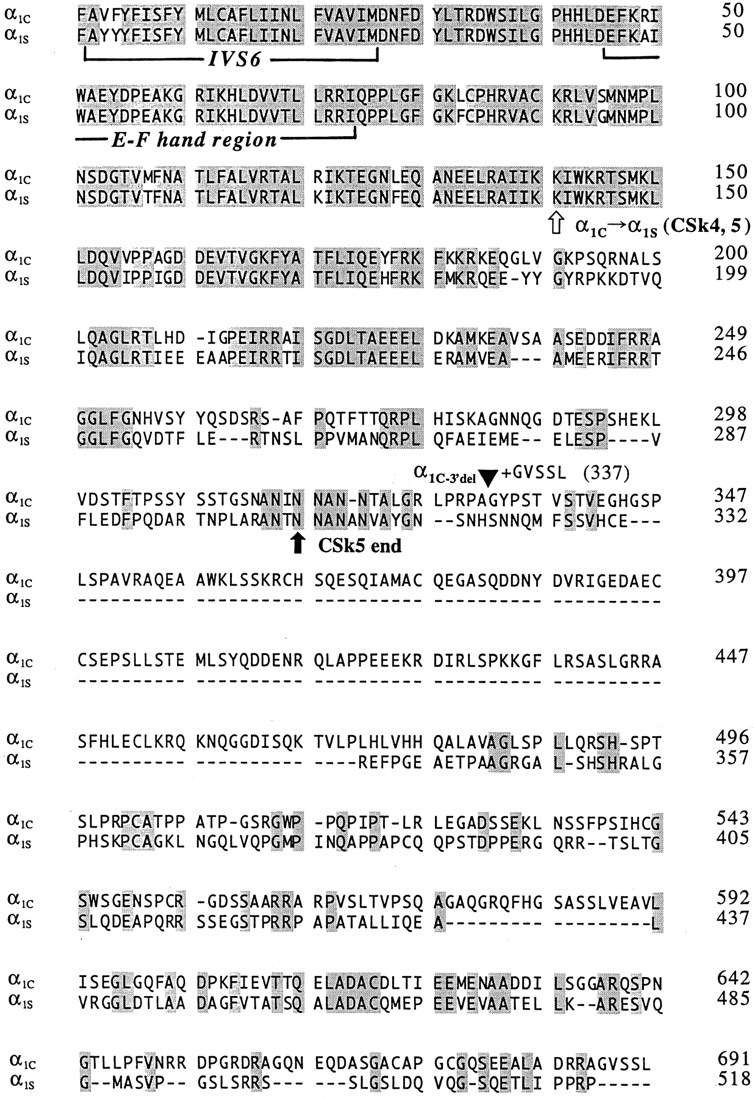
Sequence alignment of the COOH-terminal regions of the rabbit cardiac α1C (Mikami et al., 1989) and rabbit skeletal muscle α1S subunits (Tanabe et al., 1987). The sequences begin just before the last predicted transmembrane segment (S6) of domain IV (first bracket). The putative EF hand motif is indicated (second bracket), as is the junctional site within CSk4 and CSk5 where the sequence changes from α1C to α1S (white arrow). The termination of mutant α1C−3′del is indicated by a filled triangle; this construct retains the final five residues (GVSSL) of the wild-type COOH terminus. The termination point of chimera CSk5 is indicated (black arrow). Shaded amino acids are identical between α1C and α1S; dashes indicate gaps in the alignment.
CSk5 undergoes Ca-dependent inactivation, as evidenced by the significantly faster inactivation of Ca than of Ba currents (Fig. 8, A and B; Table I). Two distinct components of inactivation were present in both Ca and Ba currents mediated by CSk5. The fast time constants for inactivation of Ca currents exhibited a U-shaped voltage dependence, whereas those for Ba currents did not (Fig. 8 D). At test potentials of +10, +20, and +30 mV, the fast time constants for inactivation of Ca currents were significantly smaller than for Ba currents (Fig. 8 D). The fast component of Ca current inactivation was slower for CSk5 than for α1C, suggesting that Ca-dependent inactivation proceeds at a slower rate than in α1C. As was found for CSk4, the fast time constant for inactivation of Ba currents was not different between CSk5 and α1C (Table I).
Figure 8.

Ca-dependent inactivation is restored in CSk5. (A) Representative Ca currents mediated by CSk5, which is identical to CSk4 except that the nonconserved region (211 amino acids) has been deleted from the end of the COOH terminus. File 97522014. C = 43 pF. RS = 3.0 MΩ. (B) Representative Ba currents mediated by CSk5. File 97522021. Same cell as in A. (C) I–V relations for CSk5 currents. Each plotted point represents the mean (±SEM) of eight different cells. (D) Voltage dependence of fast time constants for inactivation of CSk5 currents. Each plotted point represents the mean (±SEM) of five to six different cells. Experiments summarized in this figure were done using the NaCl-based external solution.
DISCUSSION
The goal of the present study was to gain new insights into the molecular mechanism of Ca-dependent inactivation by identifying structural regions of α1C involved in this phenomenon. Toward this end, we compared inactivation of Ca and Ba currents mediated by chimeras constructed between the cardiac α1C, which exhibits prominent Ca-dependent inactivation, and the skeletal muscle α1S, which lacks this property (Donaldson and Beam, 1983; Beam and Knudson, 1988).
The protein sequence homology between α1C and α1S is ∼66%, with the majority of the amino acid differences occurring within the major cytoplasmic domains (Mikami et al., 1989). For example, the NH2 terminus of α1C contains 154 amino acids, whereas that of α1S contains only 50 amino acids. At the other extreme, the III–IV loops of α1C and α1S are highly conserved, both being 53 amino acids in length with only 7 amino acid differences. Our results with chimeras CSk1 and CSk8 suggest that neither the NH2 terminus nor the III–IV loop of α1C performs an essential function in Ca-dependent inactivation, because Ca and Ba currents mediated by these constructs are nearly identical to those mediated by α1C (Fig. 3). However, because the III–IV loop is so highly conserved between α1C and α1S, this region may function interchangeably in Ca-dependent inactivation.
We have also found that inactivation of Ba current is dramatically slowed for chimera CSk2, in which the I–II loop of α1C is replaced by the corresponding region from α1S (Fig. 4; Table I). This result is consistent with the relative inactivation rates of α1C and α1S, when these two different L-type Ca channels are expressed in dysgenic myotubes (Tanabe et al., 1990a ). Our results with CSk2 are also consistent with the finding of Page et al. (1997) that inactivation was slowed by replacing the entire I–II loop of the relatively fast inactivating α1E with the I–II loop from the more slowly inactivating α1B. It is also interesting to compare our results for CSk2 with those of Zhang et al. (1994), who identified transmembrane segment IS6 and its immediately flanking regions as important determinants of voltage-dependent inactivation. A comparison of the I–II loops of α1C and α1S reveals that most amino acid differences occur within the COOH-terminal half, whereas the NH2-terminal half of the I–II loop is comparatively well conserved (Mikami et al., 1989). The slowed inactivation of CSk2 may thus indicate an important role for the COOH-terminal portion of the I–II loop in Ca channel inactivation. However, because the NH2-terminal portion of the I–II loop contains an interaction site for the Ca channel β subunit (Pragnell et al., 1994), and different β subunit isoforms can modulate the rate of Ca channel inactivation (Hullin et al., 1992), it is also possible that altered interactions between CSk2 and the β subunit are partially responsible for its slower inactivation. No consensus sites for phosphorylation by PKA or PKC are present within the I–II loop of either α1C or α1S; thus, it seems unlikely that differential phosphorylation could account for the slower inactivation of CSk2.
Ca-dependent inactivation is usually defined as the faster inactivation of Ca than Ba currents and by a U-shaped voltage dependence of the time constants for Ca current inactivation. Inactivation of Ba currents is usually assumed to proceed through a voltage-dependent process. However, Ferreira et al. (1997) have recently demonstrated that Ba can trigger the ion-dependent inactivation of α1C. They found that Ba currents inactivate with two distinct components, and that the rate and extent of the fast component parallels Ba influx, whereas the rate and extent of the slow component parallels immobilization of gating charge (Ferreira et al., 1997). If the fast component of Ba current inactivation measured in our experiments reflects an ion-dependent process, then this process is significantly slowed in chimera CSk2, and to a lesser extent in chimera CSk3. In this view, our results with CSk2 suggest that the I–II loop of α1C may be an important structural determinant of ion- rather than voltage-dependent inactivation. Such inactivation could be triggered (physiologically) by Ca or (experimentally) by Ba binding to the I–II and II–III loops of α1C but not to the homologous regions of α1S. If this interpretation is correct, then the I–II and II–III loops of α1C are structural determinants of Ca-dependent inactivation.
We have demonstrated that CSk4, a chimera in which the COOH terminus of α1C has been replaced by the corresponding region from α1S, lacks Ca-dependent inactivation. This result is not an artifact stemming from low channel expression because the current density in cells expressing CSk4 was not significantly different from that in cells expressing CSk2 or CSk3 (Table I), which both displayed prominent Ca-dependent inactivation. Furthermore, Ca-dependent inactivation was absent even from relatively high density CSk4 currents (not shown), whereas it was present in relatively low density α1C, CSk1, CSk2, or CSk3 currents (e.g., Figs. 3 and 4). The lack of Ca-dependent inactivation by CSk4 may explain why this property is not exhibited by the skeletal muscle L-type Ca channel. In this regard, it would be interesting to know whether the property of Ca-dependent inactivation was gained or retained by α1C during the course of Ca channel evolution. A recent report that the neuronal α1D (an L-type Ca channel) also exhibits Ca-dependent inactivation (Hans et al., 1997) suggests that this property has been retained by α1C and α1D and lost by α1S.
The mechanism of Ca-dependent inactivation is not known, but it has been proposed that a putative EF hand motif located within the proximal COOH terminus of α1C functions as the essential Ca-binding site responsible for triggering Ca-dependent inactivation (de Leon et al., 1995). However, recent evidence from other laboratories suggests that the putative EF hand motif is not important in the mechanism of Ca-dependent inactivation. Thus, transfer of the putative EF hand from α1C into α1E fails to confer Ca-dependent inactivation and, conversely, transfer of the EF hand from α1E into α1C fails to disrupt it (Zhou et al., 1997). Additionally, Ca-dependent inactivation is not abolished by point mutations within α1C that eliminate the Ca-coordination site from the putative EF hand motif but leave the remainder of the COOH terminus intact (Zhou et al., 1997). These results strongly suggest that the exact site or sites of Ca binding remain to be identified.
Because CSk5 undergoes Ca-dependent inactivation (Fig. 8), it is reasonable to suppose that it contains one or more Ca binding sites. It follows that CSk4 contains the same site or sites, because it encompasses the entire sequence of CSk5 (Fig. 7). However, CSk4 lacks Ca-dependent inactivation (Fig. 6), which leads to the conclusion that Ca binding to the α1 subunit is only a prerequisite for Ca-dependent inactivation and is by itself insufficient. Presumably, Ca-dependent inactivation requires both Ca binding and a subsequent conformational shift of the channel protein(s).
Perhaps the most significant result of the present study is that Ca-dependent inactivation was restored in chimera CSk5 by deletion of the nonconserved, distal region of the COOH terminus present in CSk4 (Figs. 2 and 8). The COOH terminus of CSk5 is similar in length and composition to that of α1C−3′del (Fig. 7). The behavior of α1C−3′del clearly demonstrates that the most distal ∼350 amino acids of the COOH terminus of α1C are not required for Ca channel inactivation (Fig. 3 A; Zong et al., 1994; de Leon et al., 1995). In contrast, Ca-dependent inactivation is conferred upon the α1E backbone by replacing a 134–amino acid segment immediately downstream from the putative EF hand region with the homologous 142–amino acid segment from α1C (Zhou et al., 1997). Furthermore, Ca-dependent inactivation is profoundly influenced by splice variations within the proximal COOH terminus of α1C immediately downstream from the putative EF hand region (Soldatov et al., 1997). Our results with CSk5 suggests that the proximal COOH termini of α1C and α1S (which are mostly conserved) can function interchangeably in Ca-dependent inactivation. When considered altogether, our results and those of other studies indicate that the proximal COOH terminus downstream from the putative EF hand region is an important structural determinant of Ca-dependent inactivation. However, de Leon et al. (1995) showed that Ca-dependent inactivation was only partially conferred upon the neuronal α1E subunit by replacing its entire COOH terminus with 217 amino acids from the corresponding region of α1C. Thus, while the proximal COOH terminus appears to be important for Ca-dependent inactivation, the participation of additional channel regions may also be required.
Our findings that CSk4 lacks Ca-dependent inactivation, whereas this property is restored in CSk5, suggests that the distal COOH terminus of α1S (which is not well conserved between α1C and α1S) can somehow block Ca-dependent inactivation. The mechanism by which this block occurs is, at present, purely speculative. However, because ion channels appear to associate with many other proteins in vivo (Sheng and Kim, 1996), it seems plausible that CSk4 might be tethered through its distal COOH terminus to other proteins (such as ryanodine receptors, the cytoskeleton, kinases, phosphatases, or other ion channels), and that such interactions might prevent the conformational shift underlying Ca-dependent inactivation.
Acknowledgments
We thank two anonymous reviewers for constructive criticisms. We also thank C. Adams and Drs. N. Artemyev, K. Melliti, and U. Meza for helpful comments on the manuscript.
Footnotes
B. Adams was supported by a Grant-In-Aid from the American Health Association (Iowa Affiliate), a research grant from the Muscular Dystrophy Association, and grant NS-34422 from the National Institutes of Health. T. Tanabe was supported by the Ministry of Education, Science and Culture of Japan and Howard Hughes Medical Institute.
Dr. Tanabe's present address is Department of Pharmacology, Tokyo Medical and Dental University, School of Medicine, Tokyo 113, Japan
Abbreviations used in this paper: RS, compensated series resistance; TEA, tetraethylammonium.
REFERENCES
- Adams B, Tanabe T. Calcium-dependent inactivation of heterologously expressed cardiac L-type calcium channel. Biophys J. 1996;70:238a. . (Abstr.) [Google Scholar]
- Beam KG, Knudson CM. Calcium currents in embryonic and neonatal mammalian skeletal muscle. J Gen Physiol. 1988;91:781–798. doi: 10.1085/jgp.91.6.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beam KG, Adams BA, Niidome T, Numa S, Tanabe T. Function of a truncated dihydropyridine receptor as both voltage sensor and calcium channel. Nature (Lond) 1992;360:169–171. doi: 10.1038/360169a0. [DOI] [PubMed] [Google Scholar]
- Boyett MR, Harrison SM, Janvier NC, McMorn SO, Owen JM, Shui Z. A list of vertebrate cardiac ionic currents: nomenclature, properties, function and cloned equivalents. Cardiovasc Res. 1996;32:455–481. [PubMed] [Google Scholar]
- de Leon M, Wang Y, Jones L, Perez-Reyes E, Wei XY, Soong TW, Snutch TP, Yue DT. Essential Ca2+-binding motif for Ca2+-sensitive inactivation of L-type Ca2+channels. Science (Wash DC) 1995;270:1502–1506. doi: 10.1126/science.270.5241.1502. [DOI] [PubMed] [Google Scholar]
- Donaldson PL, Beam KG. Calcium currents in a fast-twitch skeletal muscle of the rat. J Gen Physiol. 1983;83:449–468. doi: 10.1085/jgp.82.4.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira G, Yi J, Rios E, Shirokov R. Ion-dependent inactivation of barium current through L-type calcium channels. J Gen Physiol. 1997;109:449–461. doi: 10.1085/jgp.109.4.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y, Mynlieff M, Dirksen RT, Kim M-S, Niidome T, Nakai J, Friedrich T, Iwabe N, Miyata T, Furuichi T, et al. Primary structure and functional expression of the ω-conotoxin-sensitive N-type calcium channel from rabbit brain. Neuron. 1993;10:585–598. doi: 10.1016/0896-6273(93)90162-k. [DOI] [PubMed] [Google Scholar]
- Haack JA, Rosenberg RL. Calcium-dependent inactivation of L-type calcium channels in planar lipid bilayers. Biophys J. 1994;66:1051–1060. doi: 10.1016/S0006-3495(94)80886-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadley RW, Lederer WJ. Ca2+ and voltage inactivate Ca2+channels in guinea-pig ventricular myocytes through independent mechanisms. J Physiol (Oxf) 1991;444:257–268. doi: 10.1113/jphysiol.1991.sp018876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflüg Arch Eur J Physiol. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hans M, Urrutia A, Brust P, Nesterova A, Sacaan AI, Harpold M, Stauderman K. Biophysical and pharmacological properties of human neuronal α1Dα2bδβ3a Ca2+channels stably expressed in HEK293 cells. Biophys J. 1997;72:A146. . (Abstr.) [Google Scholar]
- Hofmann F, Biel M, Flockerzi V. Molecular basis for Ca2+channel diversity. Annu Rev Neurosci. 1994;17:399–418. doi: 10.1146/annurev.ne.17.030194.002151. [DOI] [PubMed] [Google Scholar]
- Hullin R, Singer-Lahat D, Freichel M, Biel M, Dascal N, Hofmann F, Flockerzi V. Calcium channel β subunit heterogeneity: functional expression of cloned cDNA from heart, aorta and brain. EMBO (Eur Mol Biol Organ) J. 1992;11:885–890. doi: 10.1002/j.1460-2075.1992.tb05126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imredy JP, Yue DT. Mechanism of Ca2+-sensitive inactivation of L-type Ca2+channels. Neuron. 1994;12:1301–1318. doi: 10.1016/0896-6273(94)90446-4. [DOI] [PubMed] [Google Scholar]
- Jurman ME, Boland LM, Liu Y, Yellen G. Visual identification of individual transfected cells for electrophysiology using antibody-coated beads. Biotechniques. 1994;17:876–881. [PubMed] [Google Scholar]
- Mikami A, Imoto K, Tanabe T, Niidome T, Mori Y, Takeshima H, Narumiya S, Numa S. Primary structure and functional expression of the cardiac dihydropyridine-sensitive calcium channel. Nature (Lond) 1989;340:230–233. doi: 10.1038/340230a0. [DOI] [PubMed] [Google Scholar]
- Mishina M, Kurosaki T, Tobimatsu T, Morimoto Y, Noda M, Yamamoto T, Terao M, Lindstrom J, Takahashi T, Kuno M, Numa S. Expression of functional acetylcholine receptor from cloned cDNAs. Nature (Lond) 1984;307:604–608. doi: 10.1038/307604a0. [DOI] [PubMed] [Google Scholar]
- Neely A, Olcese R, Wei X, Birnbaumer L, Stefani E. Ca2+-dependent inactivation of a cloned cardiac Ca2+ channel α1 subunit (α1C) expressed in Xenopusoocytes. Biophys J. 1994;66:1895–1903. doi: 10.1016/S0006-3495(94)80983-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obejero-Paz CA, Jones SW, Scarpa A. Calcium currents in the A7r5 smooth muscle-derived cell line: increase in current and selective removal of voltage-dependent inactivation by intracellular trypsin. J Gen Physiol. 1991;98:1127–1140. doi: 10.1085/jgp.98.6.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page KM, Stephens GJ, Berrow NS, Dolphin AC. The intracellular loop between domains I and II of the B-type calcium channel confers aspects of G-protein sensitivity to the E-type calcium channel. J Neurosci. 1997;17:1330–1338. doi: 10.1523/JNEUROSCI.17-04-01330.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Garcia MT, Kamp TJ, Marbán E. Functional properties of cardiac L-type calcium channels transiently expressed in HEK293 cells. Role of α1and β subunits. J Gen Physiol. 1995;105:289–306. doi: 10.1085/jgp.105.2.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Reyes E, Yuan W, Wei X, Bers DM. Regulation of the cloned L-type cardiac calcium channel by cyclic-AMP- dependent protein kinase. FEBS Lett. 1994;342:119–123. doi: 10.1016/0014-5793(94)80484-2. [DOI] [PubMed] [Google Scholar]
- Pragnell M, De Waard M, Mori Y, Tanabe T, Snutch TP, Campbell KP. Calcium channel β-subunit binds to a conserved motif in the I–II cytoplasmic linker of the α1-subunit. Nature (Lond) 1994;368:67–70. doi: 10.1038/368067a0. [DOI] [PubMed] [Google Scholar]
- Sheng M, Kim E. Ion channel associated proteins. Curr Opin Neurobiol. 1996;6:602–608. doi: 10.1016/s0959-4388(96)80091-2. [DOI] [PubMed] [Google Scholar]
- Shirokov R, Levis R, Shirokova N, Rios E. Ca2+-dependent inactivation of cardiac L-type Ca2+channels does not affect their voltage sensor. J Gen Physiol. 1993;102:1005–1030. doi: 10.1085/jgp.102.6.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldatov NM, Zühlke RD, Bouron A, Reuter H. Molecular structures involved in L-type calcium channel inactivation. Role of the carboxyl-terminal region encoded by exons 40– 42 in α1C subunit in the kinetics and Ca2+dependence of inactivation. J Biol Chem. 1997;272:3560–3566. doi: 10.1074/jbc.272.6.3560. [DOI] [PubMed] [Google Scholar]
- Stühmer W, Conti F, Suzuki H, Wang X, Noda M, Yahagi N, Kubo H, Numa S. Structural parts involved in activation and inactivation of the sodium channel. Nature (Lond) 1989;339:597–603. doi: 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- Tanabe T, Mikami A, Numa S, Beam KG. Cardiac-type excitation–contraction coupling in dysgenic skeletal muscle injected with cardiac dihydropyridine receptor cDNA. Nature (Lond) 1990a;344:451–453. doi: 10.1038/344451a0. [DOI] [PubMed] [Google Scholar]
- Tanabe T, Beam KG, Adams BA, Niidome T, Numa S. Regions of the skeletal muscle dihydropyridine receptor critical for excitation-contraction coupling. Nature (Lond) 1990b;346:567–569. doi: 10.1038/346567a0. [DOI] [PubMed] [Google Scholar]
- Tanabe T, Takeshima H, Mikami A, Flockerzi V, Takahashi H, Kangawa K, Kojima M, Matsuo H, Hirose T, Numa S. Primary structure of the receptor for calcium channel blockers from skeletal muscle. Nature (Lond) 1987;328:313–318. doi: 10.1038/328313a0. [DOI] [PubMed] [Google Scholar]
- Tanabe T, Beam KG, Powell JA, Numa S. Restoration of excitation-contraction coupling and slow calcium current in dysgenic muscle by dihydropyridine receptor complementary DNA. Nature (Lond) 1988;336:134–139. doi: 10.1038/336134a0. [DOI] [PubMed] [Google Scholar]
- Vassilev P, Scheuer T, Catterall WA. Identification of an intracellular peptide segment involved in sodium channel inactivation. Science (Wash DC) 1988;241:1658–1661. doi: 10.1126/science.241.4873.1658. [DOI] [PubMed] [Google Scholar]
- Zhang J-F, Ellinor PT, Aldrich RW, Tsien RW. Molecular determinants of voltage-dependent inactivation in calcium channels. Nature (Lond) 1994;372:97–100. doi: 10.1038/372097a0. [DOI] [PubMed] [Google Scholar]
- Zhou JM, Olcese R, Qin N, Noceti F, Birnbaumer L, Stefani E. Feedback inhibition of Ca2+ channels by Ca2+ depends on a short sequence of the C terminus that does not include the Ca2+-binding function of a motif with similarity to Ca2+-binding domains. Proc Natl Acad Sci USA. 1997;94:2301–2305. doi: 10.1073/pnas.94.6.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong S, Zhou J, Tanabe T. Molecular determinants of calcium-dependent inactivation in cardiac L-type calcium channels. Biochem Biophys Res Commun. 1994;201:1117–1123. doi: 10.1006/bbrc.1994.1821. [DOI] [PubMed] [Google Scholar]
- Zong XG, Hofmann F. Ca2+-dependent inactivation of the class C L-type Ca2+channel is a property of the α1 subunit. FEBS Lett. 1996;378:121–125. doi: 10.1016/0014-5793(95)01434-9. [DOI] [PubMed] [Google Scholar]