

Abstract
Modulation of L-type Ca2+ channels by tonic elevation of cytoplasmic Ca2+ was investigated in intact cells and inside-out patches from human umbilical vein smooth muscle. Ba2+ was used as charge carrier, and run down of Ca2+ channel activity in inside-out patches was prevented with calpastatin plus ATP. Increasing cytoplasmic Ca2+ in intact cells by elevation of extracellular Ca2+ in the presence of the ionophore A23187 inhibited the activity of L-type Ca2+ channels in cell-attached patches. Measurement of the actual level of intracellular free Ca2+ with fura-2 revealed a 50% inhibitory concentration (IC50) of 260 nM and a Hill coefficient close to 4 for Ca2+- dependent inhibition. Ca2+-induced inhibition of Ca2+ channel activity in intact cells was due to a reduction of channel open probability and availability. Ca2+-induced inhibition was not affected by the protein kinase inhibitor H-7 (10 μM) or the cytoskeleton disruptive agent cytochalasin B (20 μM), but prevented by cyclosporin A (1 μg/ ml), an inhibitor of protein phosphatase 2B (calcineurin). Elevation of Ca2+ at the cytoplasmic side of inside-out patches inhibited Ca2+ channels with an IC50 of 2 μM and a Hill coefficient close to unity. Direct Ca2+-dependent inhibition in cell-free patches was due to a reduction of open probability, whereas availability was barely affected. Application of purified protein phosphatase 2B (12 U/ml) to the cytoplasmic side of inside-out patches at a free Ca2+ concentration of 1 μM inhibited Ca2+ channel open probability and availability. Elevation of cytoplasmic Ca2+ in the presence of PP2B, suppressed channel activity in inside-out patches with an IC50 of ∼380 nM and a Hill coefficient of ∼3; i.e., characteristics reminiscent of the Ca2+ sensitivity of Ca2+ channels in intact cells. Our results suggest that L-type Ca2+ channels of smooth muscle are controlled by two Ca2+-dependent negative feedback mechanisms. These mechanisms are based on (a) a protein phosphatase 2B-mediated dephosphorylation process, and (b) the interaction of intracellular Ca2+ with a single membrane-associated site that may reside on the channel protein itself.
Keywords: L-type Ca2+ channels, gating, protein phosphatase 2B, vascular smooth muscle, patch clamp
introduction
The function of L-type Ca2+ channels is controlled by voltage as well as by the Ca2+ influx through the channel (Kass and Sanguinetti, 1984; Eckert and Chad, 1984; Lee et al., 1985). An inactivation process is induced by the charge carrier Ca2+ itself, providing a negative feedback mechanism that is considered crucial for Ca2+ homeostasis in a variety of cell types, including smooth muscle cells (Jmari et al., 1986; Granitkevich et al., 1987; Granitkevich and Isenberg, 1991). There is an ongoing debate as to whether Ca2+ induces channel inactivation via direct binding to proteins of the channel complex or via a more remote mechanism such as Ca2+-dependent dephosphorylation (Chad and Eckert, 1986; Armstrong, 1989) or disruption of the cells' cytoskeleton (Johnson and Byerly, 1993). Inhibition of L-type channels by cytosolic Ca2+ has been demonstrated in cell-free membranes, supporting the view of a rather direct inhibitory mechanism such as interaction of Ca2+ with the channel (Romanin et al., 1992; Haack and Rosenberg, 1994; Schmid et al., 1995). Recent evidence suggests that Ca2+-induced inactivation is a property of the pore-forming α1 subunit of the Ca2+ channel (Zong and Hofmann, 1996), and an EF-hand Ca2+ binding motif in the α1 subunit has recently been identified as a structure that is essential for Ca2+-dependent inactivation (De Leon et al., 1995). Nonetheless, different lines of investigations (Ohya et al., 1988; Hirano and Hiraoka, 1994; You et al., 1995) support the hypothesis of a more complex Ca2+-dependent control of L-type Ca2+ channels, involving Ca2+-dependent enzymatic mechanisms in addition to direct binding of Ca2+ to the channel. Both stimulatory (Hirano and Hiraoka, 1994) and inhibitory modulation (Ohya et al., 1988; Hirano and Hiraoka, 1994; You et al., 1995) of Ca2+ channels has been observed in response to increases in bulk cytoplasmic Ca2+ to levels lower than the concentrations reported for inhibition of L-type Ca2+ channels in cell-free membranes (Romanin et al., 1992; Haack and Rosenberg, 1994; Schmid et al., 1995). Candidate mechanisms for a remote control of L-type Ca2+ channels in cardiac and smooth muscle are Ca2+-dependent phosphorylation and dephosphorylation (Chad and Eckert, 1986; Armstrong, 1989; Hirano and Hiraoka, 1994), as well as Ca2+-dependent disruption of the cytoskeleton (Johnson and Byerly, 1993, 1994).
The present study was aimed at comparing Ca2+- dependent modulation of smooth muscle L-type Ca2+ channels in intact cells and cell-free patches and to test for a possible role of phosphorylation/dephosphorylation and modification of the cytoskeleton in intact cells. We present evidence for the control of L-type Ca2+ channels in vascular smooth muscle cells by two Ca2+-dependent mechanisms that comprise both dephosphorylation by protein phosphatase 2B and direct binding of Ca2+ to a membrane-associated site.
materials and methods
Cell Preparation
The media of human umbilical veins was enzymatically disaggregated to obtain single smooth muscle cells as described previously (Schuhmann and Groschner, 1994). In brief, endothelial cells were removed with dispase (type II; Boehringer Mannheim, Mannheim, Germany), and smooth muscle tissue was subsequently dissociated by filling the vessels with low Ca2+ (0.1 mM) Hanks buffer (Sera Lab Ltd., Sussex, UK), supplemented with 0.5 mg/ml collagenase (type II; Worthington Biochemical Corp., Freehold, NJ), 0.5 mg/ml trypsin inhibitor (Worthington Biochemical Corp.), and 1 mg/ml fatty acid–free bovine serum albumin. Vessels filled with collagenase containing Hanks solution were incubated at 37°C. After incubation for 10 min, the Hanks buffer contained single, mostly relaxed, elongated smooth muscle cells that were harvested by centrifugation (5 min, 250 g). The cells were resuspended and stored in a solution containing (mM): 110 K+ aspartate, 20 KCl, 2 MgCl2, 20 HEPES, 2 EGTA (pCa = 7, see below) at 4°C, and used for experimentation within 36 h.
Measurement of Single-channel Currents
Cell potentials were set to approximately zero by use of a high K+ low Cl− extracellular solution that contained (mM): 110 K+ aspartate, 20 KCl, 2 MgCl2, 20 HEPES, 2 EGTA, pH was adjusted to 7.4 with N -methyl-d-glucamine, and pCa was adjusted using a Ca2+-sensitive electrode. Patch pipettes were fabricated from borosilicate glass (Clark Electromedical Instruments, Pangbourne, UK), and had resistances of 5–10 MOhm. For recording of Ba2+ currents through single Ca2+ channels, the pipettes were filled with a solution containing (mM): 10 BaCl2, 100 NaCl, 30 TEA-Cl, and 15 HEPES, pH adjusted to 7.4. The dihydropyridine-Ca2+ channel activator S(−)-BayK 8644 (0.5 μM) was included in the pipette solution to facilitate stabilization of channel activity in inside-out patches. Run down of Ca2+ channel activity in inside-out experiments was prevented by addition of calpastatin (2 U/ml) plus 1 mM ATP/Na2 to the bath solution before patch excision (Romanin et al., 1992). In experiments recording Ca2+-activated K+ channels, the pipette solution contained (mM): 137 NaCl, 5 KCl, 2.5 CaCl2, 2 MgCl2, 10 HEPES, pH was adjusted to 7.4. All experiments were performed at room temperature. Exchange of bath solutions and administration of drugs was performed during constant flow perfusion.
Voltage clamp and current amplification was performed with a patch-clamp amplifier (EPC/7; List, Darmstadt, Germany). Current records were filtered at 1 kHz (−3 dB) and digitized at a rate of 5 kHz. Experiments were controlled using pClamp software (Axon Instruments, Foster City, CA). For idealization of current records, a custom-made level detection software was employed (Pastushenko and Schindler, 1997). Ca2+ channel activity was calculated as the mean number of open channels during depolarizing pulses. The dependency of channel activity on cytoplasmic free Ca2+ concentration was characterized in terms of 50% inhibitory concentration (IC50 values)1 and Hill coefficients by fitting the data with a four-parameter logistic function (De Lean et al., 1978). The gating properties of single channels were analyzed in terms of channel availability P s; i.e., the probability that a channel will open upon depolarization, as well as the open probability of available channels as outlined below.
Determination of Availability and Open Probability
Recently, a method was developed (Schmid et al., 1995; Baumgartner et al., 1997) to allow for an estimation of the number of channels, their availability P s and open probability P o from idealized channel traces with n < 9 channels based on the following assumptions: (a) the channels are identical and behave independently; (b) the total number of channels remains constant within the duration of the recording; and (c) the process regulating channels' gating within a sweep is distribution ergodic with respect to the number of sampling points in each conductance level. Hence, the probability of finding a sampling point in the k th conductance level can be described by a binomial distribution with the parameters n A and P o.
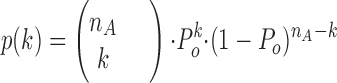 |
1 |
Eq. 1 and assumption c allow us to determine the probability for one sweep (Eq. 2) at given n A with the following variables defined as: n, maximum number of channels in a patch; n Ai, available channels in the i th sweep; t ik, number of sampling points in the k th conductance level in the i th sweep; T i, vector of t ik for the i th sweep; T, matrix of all t ik; T ges, number of sampling points in a sweep; P s, availability; P o, open probability; M, total number of sweeps; K i, set of conductance levels that occur in the i th sweep.
The elements of T i that describe the i th sweep obey a multinomial distribution (Weiß, 1987) and the probability of T i at given n Ai and P o is
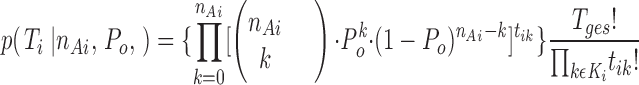 |
2 |
if n Ai is higher than or equal to the highest conductance level in T i. If n Ai is smaller than the highest conductance level, then
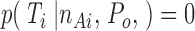 |
3 |
Because of assumptions a and b, the n Ai are binomially distributed, yielding the probability for T i at given n, P S, and P o to be
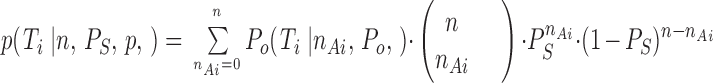 |
4 |
leading to the probability for T comprising all sweeps
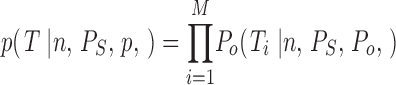 |
5 |
Using Eq. 5, a maximum likelihood estimator for n, P S, and P o was constructed by maximizing the probability for T.
Measurement of Intracellular Concentrations of Ca2+ and H+
Cytoplasmic free Ca2+([Ca2+]i) and intracellular pH (pHi) were determined using the fluorescent Ca2+ and pH indicators fura-2 and BCECF ((2′,7′)-bis(carboxyethyl)-(5,6)-carboxyfluorescein), as described by Wakabayashi and Groschner (1996). In brief, cells were suspended in 5 ml of physiological solution containing (mM): 137 NaCl, 5 KCl, 2.5 CaCl2, 2 MgCl2, 10 HEPES, pH 7.4, and loaded with fura-2 or BCECF for 60 min at 37°C. After loading, the cells were once washed and resuspended in high K+ low Cl− extracellular solution (see above). Fluorescence measurements were carried out using a dual wavelength spectrophotofluorometer (F2000; Hitachi Ltd., Tokyo, Japan). The cell suspension (2 ml) was stirred and maintained at room temperature. For [Ca2+]i measurement, excitation wavelengths were 340 and 380 nm, and emission was collected at 510 nm. For pHi measurement, excitation wavelengths were 506 and 455 nm, and emission was collected at 530 nm. Using the ratios (R) of fluorescence intensity (F), F340/F380 and F506/F455, the fractional changes in [Ca2+]i and pHi were determined, respectively. Fluorescence after sequential addition of 0.1% Triton X-100 and EGTA to the cell suspension provided the respective maximum fluorescence ratio (Rmax) and minimum fluorescence ratio (Rmin). [Ca2+]i was calculated as described (Wakabayashi and Groschner, 1996). Calibration of pHi measurements were performed using nigericin (7 μM)-containing high K+ solution at various extracellular pHi values (pHo).
Assay of p-Nitrophenyl Phosphate Phosphatase Activity
The activity of protein phosphatase 2B was measured employing p-nitrophenyl phosphate (pNPP) as substrate as described (Takai and Mieskes, 1991). 1 U phosphatase activity was the amount that catalyzed dephosphorylation of 1 mmol pNPP/min.
Statistics
Averaged data are given as mean ± SEM from the indicated number of experiments. Statistical analysis was performed using Student's t test for unpaired values. Differences were considered statistically significant at P < 0.05.
Materials
PP2B was obtained from Upstate Biotechnology Inc. (Lake Placid, NY), collagenase, type CLS II, and soybean trypsin inhibitor were obtained from Worthington Biochemical Corp., dispase type II was from Boehringer Mannheim, fatty acid–free bovine serum albumin was from Behring (Marburg, Germany), Hanks balanced salt solution was from Sera Lab Ltd., H-7, cytochalasin B, and cyclosporin A were from Research Biochemicals, Inc. (Natic, MA), and calpastatin and all other chemicals were from Sigma Chemical Co. (Deisenhofen, Germany). Calpastatin was dialyzed overnight against bath solutions (high K+ low Cl− solutions, see above).
results
Ca2+-dependent Inhibition of L-Type Ca2+ Channels in Intact Cells
Ca2+-dependent modulation of L-type Ca2+ channels in intact cells was studied by raising intracelluar Ca2+ of the cells via elevation of extracellular Ca2+ in the presence of the Ca2+ ionophore A23187 (1 μM). A typical experiment is illustrated in Fig. 1. The cell was initially bathed in a solution containing ∼10 nM free Ca2+ (pCa 8). A23187 by itself did not affect channel activity in the cell-attached patch under these conditions. Extracellular Ca2+ was increased in the presence of A23187 to 10 μM, and subsequently to 100 μM. Channel activity was barely effected at 10 μM extracellular Ca2+, but clearly suppressed when Ca2+ of the bath solution was raised to 100 μM, and activity recovered partially during a following period of reduction of extracellular Ca2+. The actual level of average cytoplasmic free Ca2+ ([Ca2+]i) obtained during elevation of extracellular Ca2+ was measured in parallel experiments using the Ca2+-sensitive fluorescent dye fura-2. As shown in Fig. 2 A, [Ca2+]i increased barely upon application of the Ca2+ ionophore at 10 nM free extracellular Ca2+; i.e., an extracellular pCa (pCao) of 8. As expected from the ability of A23187 to deplete intracellular Ca2+ stores, a small and transient rise in [Ca2+]i was observed occasionally. During elevation of extracellular Ca2+ to 10 μM, [Ca2+]i increased rapidly from a resting level of ∼50 nM to 216 ± 19 nM (n = 7). Upon further elevation of extracellular Ca2+ to 100 μM, [Ca2+]i increased to a level of 326 ± 14 nM (∼pCai 6.5, n = 7). These values of [Ca2+]i did not change significantly within a period of 2–4 min after elevation of extracellular Ca2+. To obtain additional information on the actual levels of [Ca2+]i at the cytoplasmic face of the plasma membrane of single cells, we measured the activity of large conductance Ca2+-activated (“maxi”) K+ channels, which are known to exhibit a typical Ca2+ dependence in the low micromolar range. Fig. 2 B shows a representative recording of maxi-K+ channel activity under conditions corresponding to those of the Ca2+ channel recordings illustrated in Fig. 1. Fig. 2 B (left) illustrates K+ channel activity in a cell-attached patch recorded at pCao 8 (top) and pCao 4 (bottom) in the presence of 1 μM A23187. It is clearly evident that channel activity increased only slightly when extracellular Ca2+ was raised to 100 μM (pCao 4). Fig. 2 B (right) illustrates the Ca2+ sensitivity of this K+ channel in the inside-out configuration. Channel activity is shown after excision of the patch into the bath solution of pCa 4. At the cytoplasmic side, this Ca2+ concentration caused full activation of the K+ channels. Reduction of the free Ca2+ concentration at the cytoplasmic side to 300 nM (pCai 6.5) diminished channel activity to a level comparable with that recorded in the cell-attached configuration at elevated extracellular Ca2+ (pCao 4, Fig. 2 B, left). In all of three experiments, the activity of maxi-K+ channels recorded at pCai 6.5 in inside-out patches was equal to or even somewhat higher than the activity observed in intact cells during a challenge with elevated Ca2+ (pCao 4) in the presence of A23187. Thus, the measurement of maxi-K+ channel activity in single cells was consistent with our determination of Cai by measurement of fura-2 fluorescence, confirming that Ca2+-dependent inhibition of L-type Ca2+ channels in intact cells indeed occurred at a level of [Ca2+]i as low as ∼300 nM. Fig. 3 A shows the concentration dependence obtained for Ca2+-induced inhibition of L-type Ca2+ channels in intact cells using the [Ca2+]i values determined with fura-2. The IC50 value was 260 nM and a Hill coefficient (nH) of ∼4 (3.9) was calculated.
Figure 1.

Elevation of extracellular Ca2+ in the presence of A23187 inhibits the activity of L-type Ca2+ channels in cell-attached patches. Time course of Ca2+ channel activity recorded in a cell- attached patch given as the mean number of open channels during individual depolarizing voltage pulses (holding potential −70 mV, test potential −20 mV, duration 500 ms, rate 0.66 Hz). Administration of 1 μM A23187 and changes in the extracellular Ca2+ concentration, given as pCao values, are indicated. (bottom) Individual current responses to depolarizing voltage pulses at the times indicated. Records were filtered at 500 Hz and digitized at 2 kHz. Closed state is indicated by arrows.
Figure 2.

Determination of intracellular Ca2+ levels during elevation of extracellular Ca2+ in the presence of A23187. (A) Time course of intracellular free Ca2+ ([Ca2+]i) during elevation of extracellular Ca2+ in the presence of 1 μM A23187 measured by fura-2 fluorescence. Administration of 1 μM A23187 and changes in the extracellular Ca2+ concentration, given as pCao values, are indicated. Mean values ± SEM (n = 7) of [Ca2+]i determined for the period of 2–4 min after elevation of extracellular Ca2+ to the respective value, are shown for pCao 5 and 4. (B, left) Activity of Ca2+-activated K+ channels in a cell-attached patch (test potential = 0 mV) during elevation of extracellular Ca2+, given as pCao values, in the presence of 1 μM A23187. (right) Channel activity in the same patch after excision (inside-out configuration) at pCa 4 (100 μM) and pCa 6.5 (300 nM). Records were filtered at 500 Hz and digitized at 2 kHz. Unitary current levels are indicated by dashed lines.
Figure 3.

Concentration dependence of Ca2+-induced inhibition of Ca2+ channel activity in cell-attached patches. Mean values ± SEM of channel activity obtained from two to four experiments. Data were fitted by a four-parameter logistic function, and the derived Hill coefficient (nH) is given.
Since Ca2+ channel activity in smooth muscle is determined by intracellular pH (Klöckner and Isenberg, 1994), we first tested whether changes in intracellular pH might account for the inhibitory effects observed in intact cells. Using BCECF as a pH indicator, we found that pHi did not change significantly upon administration of A23187 and subsequent elevation of extracelluar Ca2+. A pHi of 7.41 ± 0.16 (n = 9) in controls and 7.40 ± 0.11 (n = 4) in the presence of A23187 plus 100 μM Ca2+ was measured.
We have recently reported on a mechanism of Ca2+ channel inhibition that involves Ca2+-phospholipid- dependent protein kinases (Schuhmann and Groschner, 1994); thus, we tested whether the protein kinase inhibitor H-7 is able to blunt Ca2+-dependent inhibition of channels in intact cells. H-7 (10 μM) did not affect the inhibitory modulation in intact cells (n = 4). Similarly, cytochalasin B, a cytoskeletal disrupter, failed to suppress Ca2+-dependent inhibition of L-type channels (n = 3).
Cyclosporin A Prevents Ca2+-dependent Inhibition of Ca2+ Channels in Intact Cells
A role of Ca2+-dependent dephosphorylation has previously been implicated in Ca2+-induced negative feedback mechanisms (Armstrong, 1989). Thus, we tested cyclosporin A as an inhibitor of the Ca2+-dependent protein phosphatase 2B (calcineurin). Elevation of extracellular Ca2+ from 10 nM to 100 μM (pCao 4) in the presence of 1 μM A23187 resulted in an increase in intracellular free Ca2+ to ∼300 nM (pCai 6.5, Fig. 2) and suppressed channel activity substantially within 1 min (Fig. 4 A). However, when cells were pretreated with cyclosporin A (1 μg/ml), channel activity remained constant even during prolonged elevation of intracellular Ca2+ (Fig. 4 B; n = 3). Removal of cyclosporin A resulted in the expected inhibition of Ca2+ channel activity, which recovered upon reduction of extracellular Ca2+ to pCa 8 (Fig. 4 B). Cyclosporin A by itself did not affect channel activity significantly under basal conditions (n = 3). Experiments with fura-2 confirmed that elevation of [Ca2+]i induced by extracellular Ca2+ in the presence of A23187 remained unchanged by cyclosporin A (n = 3). Thus, cyclosporin A specifically antagonized Ca2+-induced suppression of channel activity. These results demonstrate that cyclosporin A interferes with Ca2+-dependent negative feedback control of Ca2+ channels in intact smooth muscle cells, and indicate a role of protein phosphatase 2B, the classical target of cyclosporin A.
Figure 4.

Cyclosporin A prevents Ca2+-induced inhibition of Ca2+ channels in intact cells. Time courses of Ca2+ channel activity recorded in cell-attached patches. A23187 (1 μM) is present throughout the experiments. Changes in the extra- and intracellular Ca2+ concentration are given as pCa values (pCao and pCai). (A) Control experiment. (B) Experiment in the presence of cyclosporin A (1 μg/ml; 10 min). Channel activity is given as the mean number of open channels during individual depolarizing voltage pulses (holding potential −70 mV, test potential −20 mV, duration 500 ms, rate 0.66 Hz). (bottom) Individual current responses to depolarizing voltage pulses at the times indicated. Records were filtered at 500 Hz and digitized at 2 kHz. Closed state is indicated by arrows.
In an attempt to further characterize the cyclosporin A–sensitive effect of cytoplasmic Ca2+, we evaluated the effects of Ca2+ on open probability (Po) and availability (Ps); i.e., the probability for a channel to open once during a depolarization. Using a recently developed procedure that allows for determination of these parameters from multichannel records (Schmid et al., 1995), we found that Ca2+-induced suppression of channel activity in intact cells was associated with a reduction of Po and in addition a marked reduction of Ps. These effects were clearly antagonized by cyclosporin A (Fig. 5).
Figure 5.

Cyclosporin A prevents Ca2+-induced inhibition of open probability (Po) and availability (Ps) of channels in intact cells. Po and Ps of channels are shown under control conditions (pCai 8.5) and at elevated cytoplasmic Ca2+ (pCai 6.5) in the absence and presence of cyclosporin A (1 μg/ml). Mean values ± SEM (n = 4) are given. *Significant difference versus control values.
Inhibition of L-Type Ca2+ Channels in Cell-free Patches by Ca2+ and Protein Phosphatase 2B
Ca2+ channel activity in excised, inside-out patches remained stable over more than 30 min when the cytoplasmic side was exposed to a solution containing calpastatin (2 U/ml) and ATP (1 mM) (Romanin et al., 1992; Schmid et al., 1995). This experimental protocol allowed for investigation of the sensitivity of smooth muscle L-type Ca2+ channels to cytoplasmic Ca2+ as well as to dephosphorylation by protein phosphatase 2B. As a first step, we studied channel inhibition by Ca2+ itself. As illustrated in Fig. 6, Ca2+ channels recorded in excised patches were apparently not affected when the Ca2+ concentration of the solution facing the cytoplasmic side was increased to 300 nM (pCai 6.5), but significantly inhibited at 10 μM free Ca2+ (pCai 5). This effect was rapidly reversible upon subsequent reduction of the free Ca2+ concentration to 10 nM (pCai 8). Fig. 7 depicts the concentration dependence obtained for Ca2+-induced inhibition of L-type Ca2+ channels in excised patches. Inhibition was characterized by an IC50 value of 2 μM and a Hill coefficient (nH) close to unity (1.1).
Figure 6.

Ca2+-induced inhibition of Ca2+ channels in cell-free patches. Time course of Ca2+ channel activity in the cell-attached and inside-out configuration, given as the mean number of open channels during individual depolarizing voltage pulses (holding potential −70 mV, test potential −20 mV, duration 500 ms, rate 0.66 Hz). Patch excision and subsequent changes in the Ca2+ concentration at the cytoplasmic side (pCai) are indicated. (bottom) Individual current responses to depolarizing voltage pulses at the times indicated. Records were filtered at 500 Hz and digitized at 2 kHz. Closed state is indicated by arrows.
Figure 7.

Concentration dependence of Ca2+-induced inhibition of Ca2+ channel activity in excised inside-out patches. Mean values ± SEM of channel activity obtained from two to four experiments. Data were fitted by a four parameter logistic function, and the derived Hill coefficient (nH) is given.
Since our experiments with cyclosporin A indicated a role of PP2B in Ca2+-dependent downregulation of Ca2+ channels in intact cells, the next step was to test whether purified PP2B is able to suppress Ca2+ channel activity in excised patches. The effects of PP2B were studied in the presence of 1 μM calmodulin. Fig. 8 illustrates that administration of purified PP2B (1 μg/ ml) to the cyctoplasmic side of Ca2+ channels in inside-out patches failed to affect channel activity at pCa 8, but inhibited channels at pCa 6 within 1–2 min to a level below 10% of control (n = 3). Administration of 1 μM calmodulin alone at pCa 6 did not affect channel activity (n = 3). The concentration dependence of Ca2+-induced channel inhibition in the presence of PP2B (1 μg/ml) is illustrated in Fig. 9. Elevation of cytoplasmic Ca2+ in the presence of PP2B inhibited Ca2+ channel activity in inside-out patches with an IC50 of 379 nM and a Hill coefficient (nH) of ∼3 (3.1). Consistent with the idea of PP2B effects being mediated by protein dephosphorylation, PP2B phosphatase activity was enhanced by Ca2+ in the same range of concentrations, with half-maximal stimulation at 430 nM free Ca2+ and a Hill coefficient of 4.5 (n = 4). To test whether downregulation of Ca2+ channels by PP2B is due to an impairment of Ca2+ channel stabilization by calpastatin, we preincubated calpastatin for 10 min with PP2B (1 μg/ml) plus calmodulin (1 μM) at pCa 6. This preincubation did not affect the ability of calpastatin to stabilize Ca2+ channel activity in excised patches (n = 2).
Figure 8.

PP2B-induced inhibition of Ca2+ channels in cell-free patches. Time courses of Ca2+ channel activity recorded in inside-out patches at a pCai of 8 (A) and 6 (B). Addition of PP2B (1 μg/ ml) to the cytoplasmic side is indicated. Channel activity is given as the mean number of open channels during individual depolarizing voltage pulses (holding potential −70 mV, test potential −20 mV, duration 500 ms, rate 0.66 Hz). (B, bottom) Individual current responses to depolarizing voltage pulses in the absence and presence of PP2B (12 U/ml). Records were filtered at 500 Hz and digitized at 2 kHz. Closed state is indicated by arrows.
Figure 9.

Concentration dependence of Ca2+-induced inhibition of Ca2+ channel activity in excised inside-out patches and Ca2+-induced stimulation of PP2B phosphatase activity. Mean values ± SEM of channel activity (○; n = 3–4) and of PP2B phosphatase activity (•; n = 5) measured in parallel with pNPP as substrate. Data were fitted by four-parameter logistic functions and the derived Hill coefficient (nH) of Ca2+-dependent inhibition of channel activity is given.
To further characterize Ca2+- and PP2B-mediated modulation of channels in excised patches, we analyzed channel function in terms of Po and Ps. As shown in Fig. 10, elevation of cytoplasmic Ca2+ to 1 μM (pCai 6) barely reduced Po and Ps in the absence of PP2B. Further elevation of cytoplasmic Ca2+ to 10 μM (pCai 5) inhibited channel activity mainly by suppression of Po. Thus, direct inhibition of Ca2+ channels in excised patches by cytoplasmic Ca2+ concentrations >1 μM was different from Ca2+-dependent inhibition in intact cells, which was associated with a reduction of both Po and Ps. The Ca2+-dependent effect of PP2B in excised patches was characterized by a substantial reduction of Po and Ps. Thus, channel modulation by PP2B mimicked Ca2+-dependent inhibition in intact smooth muscle cells. These results demonstrate two types of Ca2+- dependent downregulation of L-type Ca2+ channels in smooth muscle. Elevation of intracellular Ca2+ by itself inhibits specifically Po of the channel, whereas in the presence of calmodulin plus PP2B, elevation of Ca2+ induces suppression of both Po and Ps.
Figure 10.

PP2B inhibits open probability (Po) and availability (P s) of Ca2+ channels in cell-free patches. Po and Ps of channels in inside-out patches are shown at different cytoplasmic Ca2+ (given as pCa) in the absence or presence of PP2B (12 U/ml). Mean values ± SEM (n = 3–4) are given. *Significant difference versus control values.
discussion
The present study provides evidence for the existence of two distinct mechanisms by which intracellular Ca2+ governs L-type Ca2+ channel function in smooth muscle. These Ca2+-mediated mechanisms exhibit different concentration dependencies and are based on different changes in single-channel properties. Our results suggest that intracellular Ca2+ inhibits smooth muscle L-type Ca2+ channels by induction of a dephosphorylation process mediated by PP2B and by direct binding to a membrane-associated regulatory site.
Ca2+ Sensitivity of Smooth Muscle L-Type Ca2+ Channels in Intact Cells and Cell-free Patches
In the present study, Ca2+ channel function was measured with the charge carrier Ba2+, which fails to mimic the negative feedback inhibition mediated by Ca2+. Consequently, the use of Ba2+ as charge carrier allows us to determine the dependency of channel inhibition on steady state Ca2+ concentrations at the cytoplasmic side. It is of note that this approach does not allow us to evaluate the effects of Ca2+ permeation through the pore on channel function. Nonetheless, this approach allows for a detailed characterization of regulatory Ca2+ interaction sites at the cytoplasmic face of the membrane and of Ca2+-dependent intracellular regulatory mechanisms. We demonstrate that L-type Ca2+ channels exhibit different Ca2+ sensitivities in intact cells and cell-free patches. The observed Ca2+ sensitivity of smooth muscle channels in inside-out patches (IC50 = 2 μM) is in accordance with reports on the Ca2+ sensitivity of cardiac L-type channels in excised patches (Romanin et al., 1992) and in planar lipid bilayers (Haack and Rosenberg, 1994). In a previous study on the Ca2+ sensitivity of L-type channels in excised patches of rat mesenteric artery, inhibition of Ca2+ channels required millimolar concentrations of cytoplasmic Ca2+, suggesting a lower Ca2+ sensitivity of the channel (Huang et al., 1989). The reason for this discrepancy is unclear.
In the present study, a Hill coefficient close to 1 was calculated for the inhibition observed in cell-free patches, indicating interaction of Ca2+ with a single target site. This finding is in line with the idea that inhibition in excised patches is due to a direct interaction of Ca2+ with a binding site on the α1 subunit of the channel, which is a single EF-hand motif present both in cardiac and in smooth muscle channels (De Leon et al., 1995). In intact cells, accurate determination of the actual intracellular concentration of Ca2+, in particular at the cell membrane, is difficult. We have therefore employed two independent methods to estimate the level of intracellular free Ca2+; i.e., fura-2 fluorescence and open probability of Ca2+-activated K+ channels. Both methods yielded consistent results. When intracellular Ca2+ was elevated by exposure of the cells to A23187 plus 100 μM extracellular Ca2+, an intracellular Ca2+ concentration of ∼300 nM was obtained and Ca2+ channel activity was significantly suppressed. This result is in agreement with a previous study demonstrating that whole-cell Ba2+ currents in smooth muscle cells are inhibited when the cells' cytoplasmic Ca2+ concentration is elevated in the high nanomolar range (Ohya et al., 1988). Similarly, in cardiac muscle, a mechanism of Ca2+-dependent inhibition of L-type channels was detected that apparently required a rather modest rise in intracellular Ca2+; i.e., to levels below 1 μM (Ohya et al., 1988). These reports on inhibition of L-type channels in intact cells at submicromolar concentrations of cytoplasmic Ca2+ are in clear contrast to the observation that Ca2+-dependent inhibition of Ca2+ channels in excised patches was found to require concentrations above 1 μM (Romanin et al., 1992; Schmid et al., 1995). Thus, in intact cells, Ca2+-dependent inhibition occurred at about one order of magnitude lower concentrations with a substantially steeper (nH ≅ 4) concentration dependence than in excised patches. These data suggest that elevation of bulk cytoplasmic Ca2+ up to the low micromolar range is able to suppress Ca2+ entry through L-type Ca2+ channels in intact smooth muscle cells via a rather complex cellular mechanism, while local elevation of Ca2+ at the cytoplasmic face of the channel above 1 μM induces inactivation via interaction with a single target site.
Role of Protein Phosphatase Type 2B
In an attempt to identify the Ca2+-dependent mechanism that suppresses Ca2+ channel activity in intact cells, we tested the involvement of putative indirect pathways. Up to now, several Ca2+-dependent signaling pathways have been implicated in the control of Ca2+ channel function. Our measurements of intracellular pH demonstrated that pH remained constant during the observed Ca2+-dependent suppression of channel activity.
L-type channel function is known to be governed via Ca2+-dependent protein kinase activity (Schuhmann and Groschner, 1994) as well as by Ca2+-dependent changes in the cells cytoskeleton ( Johnson and Byerly, 1994). Thus, we tested the kinase inhibitor H-7 and the cytoskeleton destabilizer cytochalasin B for its ability to interfere with Ca2+-dependent channel inhibition. Neither agent affected Ca2+-induced inhibition of L-type Ca2+ channel activity, arguing against a role of kinases and/or the cells cytoskeleton.
Since it has been proposed previously that functionality of Ca2+ channels requires basal phosphorylation of channel proteins, and that Ca2+-dependent dephosphorylation represents an important mechanism of downregulation of cellular Ca2+ channel activity (Chad and Eckert, 1986; Armstrong, 1989), we tested cyclosporin A, an inhibitor of the Ca2+-dependent phosphatase type 2B (calcineurin). Cyclosporin A by itself did not affect basal channel function, indicating that PP2B-dependent dephosphorylation is not involved in the control of Ca2+ channel function at basal, resting conditions. This is not surprising since PP2B is not expected to be active at the low level of intracellular free Ca2+ (50 nM) measured in our cell preparation under control conditions (Klee et al., 1979). Nonetheless, Ca2+-dependent inhibition of channel activity was completely occluded in cells pretreated with a relatively low concentration (1 μg/ml) of cylosporin A. Based on the known potency and specificity of cyclosporin A as an inhibitor of PP2B, this result suggests that PP2B may be involved in Ca2+-mediated negative feedback control of L-type Ca2+ channels in smooth muscle. Consequently, we tested whether PP2B is able to inhibit Ca2+ channel activity and to promote Ca2+-dependent inhibition of smooth muscle Ca2+ channels. Addition of purified PP2B in the presence of 1 μM calmodulin and 1 μM free Ca2+ resulted in a profound suppression of channel activity. Addition of calmodulin (1 μM) to the cytoplasmic side of excised patches failed to inhibit channel activity at 1 μM free Ca2+ in the absence of exogenous PP2B, arguing against the presence of endogenous PP2B in the patches of membrane. In the presence of PP2B, Ca2+-dependent inhibition of Ca2+ channel activity in excised patches exhibited a concentration dependence corresponding to that of Ca2+-dependent stimulation of PP2B phosphatase activity, indicating that the effects of PP2B are due to downregulation of Ca2+ channels by a Ca2+-dependent dephosphorylation process. The characteristics of this concentration dependence (i.e., IC50 value and Hill coefficient) resembled those observed for Ca2+-dependent inhibition of Ca2+ channels in intact smooth muscle cells, supporting the idea that PP2B is involved in Ca2+-dependent control of L-type Ca2+ channel function in smooth muscle.
Modulation of Single Channel Properties by Cytoplasmic Ca2+ and PP2B
Analysis of the effects of cytoplasmic Ca2+ on single channel properties revealed a striking difference between Ca2+-dependent modulation of channels in intact cells and in cell-free patches. In contrast to the inhibition of channels in inside-out patches, which was mainly due to a reduction in Po, inhibition of Ca2+ channels in intact cells was based on a reduction of both Po and Ps. The observation of a Ca2+-induced low Po gating mode in cell-free membranes is consistent with the recently proposed model of direct binding of Ca2+ ions to an EF-hand motif of the channels' α1 subunit, which results in typically low Po gating (“mode Ca”) of the channel (Imredy and Yue, 1994; De Leon et al., 1995). Nonetheless, a specific effect of cytoplasmic Ca2+ on channels in intact smooth muscle cells was reduction of Ps. This reduction of Ps was not induced by elevation of Ca2+ at the cytoplasmic side of cell-free patches, but mimicked when Ca2+ was elevated in the low micromolar range in the presence of PP2B. These results of the present study demonstrate for the first time Ca2+- induced suppression of the availability of Ca2+ channels in intact smooth muscle cells and in cell-free membranes exposed to PP2B. We have recently reported on the downregulation of smooth muscle L-type Ca2+ channels by PP2A-induced dephosphorylation (Groschner et al., 1996). In contrast to channel modulation by PP2B, PP2A did not suppress channel availability but affected specifically fast gating properties (Po) of the channels by suppression of long lasting channel openings (mode 2 gating). Interestingly, the reduction of Po observed in the presence of PP2B was not based on suppression of mode 2 gating, as evident from two experiments that allowed analysis of open time distributions (our unpublished observations). Taken together, these data strongly support the idea of multiple regulatory phosphorylation sites that are dephosphorylated in a rather selective manner by specific phosphatases and control different kinetic properties of the channel.
Physiological and Pharmacological Implications
Fine adjustment of Ca2+ entry according to the actual Ca2+ concentration in the cytosol is an important cellular mechanism. The results of the present study suggest that in vascular smooth muscle, this negative feedback control involves at least two sensors for Ca2+ within the cell. One of these sensors appears to be located close to the Ca2+ entry pathway, while another more remote sensor detects changes in the bulk cytosolic Ca2+ concentration. Albeit the present study does not provide information as to how Ca2+ affects channel function by its permeation through the channel pore, it is clearly demonstrated that the cytoplasmic Ca2+ controls channel function through two distinct intracellular mechanisms. The existence of multiple feedback mechanisms is not unexpected since cellular Ca2+ homeostasis is based on a variety of mechanisms, including Ca2+ transport across the plasma membrane and across the membrane of intracellular Ca2+ stores. It is already well established that Ca2+ signaling within a cell involves Ca2+ gradients (van Breemen et al., 1995). According to the present results, elevation of intracellular Ca2+ within the smooth muscle cell causes a reduction of Ca2+ entry due to PP2B-mediated dephosphorylation that may take place even at low or moderate rates of Ca2+ entry. On the other hand, the rate of Ca2+ entry through the L-type channel is unequivocally subject to a tight, local feedback mechanism. This local mechanism is likely to serve rapid, short term modulation of Ca2+ entry. Nonetheless, both the local and the remote mechanism may be of particular importance for smooth muscle Ca2+ homeostasis. Suppression of one of these mechanisms may produce severe disturbances in smooth muscle Ca2+ homeostasis. In the present study, we demonstrate that cyclosporin A, which is widely used as an immunosuppressant drug, effectively inhibits the remote Ca2+-dependent control of L-type Ca2+ channels in human vascular smooth muscle. The interference of cyclosporin A with the negative feedback control of Ca2+ entry may contribute significantly to the severe vascular side effects (Sturrock et al., 1992) (i.e., promotion of vasoconstriction and hypertension) observed during cyclosporin A treatment.
In summary, our results demonstrate that in smooth muscle cells, the cytoplasmic Ca2+ concentration governs the function of L-type Ca2+ channels via two distinct negative feedback systems involving both Ca2+- dependent dephosphorylation and direct Ca2+ binding.
Acknowledgments
We thank Mrs. R. Schmidt for excellent technical assistance.
This work was supported by the Fonds zur Förderung der Wissenschaftlichen Forschung (S6605, S6606, S6607, and F708-SFB Biomembranes) and the Austrian National Bank (6121).
Footnotes
Abbreviations used in this paper: IC50, 50% inhibitory concentration; pNPP, p -nitrophenyl phosphate.
references
- Armstrong DL. Calcium-channel regulation by calcineurin, a Ca2+-activated phosphatase in mammalian brain. TINS (Trends Neurosci) 1989;12:117–122. doi: 10.1016/0166-2236(89)90168-9. [DOI] [PubMed] [Google Scholar]
- Baumgartner W, Hohentanner K, Höfer GF, Groschner K, Romanin C. Estimating the number of channels in patch-clamp recordings: application to kinetic a of multichannel data from voltage operated channels. Biophys J. 1997;72:1143–1152. doi: 10.1016/S0006-3495(97)78763-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chad JE, Eckert R. An enzymatic mechanism for calcium current inactivation in dialyzed Helix neurons. J Physiol (Camb) 1986;378:31–51. doi: 10.1113/jphysiol.1986.sp016206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lean A, Rodbard D. Simultaneous analysis of families of sigmoidal curves: application to bioassay, radioligand assay, and physiological dose-response curves. Am J Physiol. 1978;235:E97–E102. doi: 10.1152/ajpendo.1978.235.2.E97. [DOI] [PubMed] [Google Scholar]
- De Leon M, Wang Y, Jones L, Perez-Reyes E, Wie X, Soong TW, Snutch TP, Yue DT. Essential Ca2+-binding motif for Ca2+-sensitive inactivation of L-type Ca2+channels. Science (Wash DC) 1995;270:1502–1506. doi: 10.1126/science.270.5241.1502. [DOI] [PubMed] [Google Scholar]
- Eckert R, Chad JE. Inactivation of Ca2+channels. Prog Biophys Mol Bio. 1984;44:215–267. doi: 10.1016/0079-6107(84)90009-9. [DOI] [PubMed] [Google Scholar]
- Granitkevich VY, Shuba MF, Smirnov SV. Calcium- dependent inactivation of potential-dependent calcium inward current in an isolated guinea pig smooth muscle cell. J Physiol (Camb) 1987;392:431–449. doi: 10.1113/jphysiol.1987.sp016789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granitkevich VY, Isenberg G. Depolarisation-mediated intracellular calcium transients in isolated smooth muscle cells of guinea pig urinary bladder. J Physiol (Camb) 1991;435:187–205. doi: 10.1113/jphysiol.1991.sp018505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groschner K, Schuhmann K, Mieskes G, Baumgartner W, Romanin C. A type 2A phosphatase-sensitive phosphorylation site controls modal gating of L-type Ca2+channels in human vascular smooth muscle. Biochem J. 1996;318:513–517. doi: 10.1042/bj3180513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haack JA, Rosenberg RL. Calcium-dependent inactivation of L-type calcium channels in planar lipid bilayers. Biophys J. 1994;66:1051–1060. doi: 10.1016/S0006-3495(94)80886-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano Y, Hiraoka M. Dual modulation of unitary L-type Ca2+ channel currents by [Ca2+]iin fura-2-loaded guinea-pig ventricular myocytes. J Physiol (Camb) 1994;480:449–463. doi: 10.1113/jphysiol.1994.sp020374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Quayle JM, Worley JH, Standen NB, Nelson MJ. External cadmium and internal calcium block of calcium channels in smooth muscle cells from rabit mesenteric artery. Biophys J. 1989;56:1023–1028. doi: 10.1016/S0006-3495(89)82747-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imredy JP, Yue DT. Mechanism of Ca2+-sensitive inactivation of L-type Ca2+channels. Neuron. 1994;12:1301–1318. doi: 10.1016/0896-6273(94)90446-4. [DOI] [PubMed] [Google Scholar]
- Jmari K, Mironneau C, Mironneau T. Inactivation of calcium channel current in rat uterine smooth muscle: evidence for calcium and voltage mediated mechanism. J Physiol (Camb) 1986;380:111–126. doi: 10.1113/jphysiol.1986.sp016275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BD, Byerly L. A cytoskeletal mechanism for Ca2+ channel metabolic dependence and inactivation by intracellular Ca2+ . Neuron. 1993;10:797–804. doi: 10.1016/0896-6273(93)90196-x. [DOI] [PubMed] [Google Scholar]
- Johnson BD, Byerly L. Ca2+ channel Ca2+-dependent inactivation in a mammalian central neuron involves the cytoskeleton. Pflügers Arch. 1994;429:14–21. doi: 10.1007/BF02584025. [DOI] [PubMed] [Google Scholar]
- Kass RS, Sanguinetti MC. Inactivation of calcium channel current in the calf cardiac Purkinje fibre: evidence for voltage- and calcium-mediated mechanism. J Gen Physiol. 1984;84:705–726. doi: 10.1085/jgp.84.5.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klee CB, Crouch TH, Krinks MH. Calcineurin: a calcium- and calmodulin-binding protein of the nervous system. Proc Natl Acad Sci USA. 1979;76:6270–6273. doi: 10.1073/pnas.76.12.6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KS, Marban E, Tsien RW. Inactivation of calcium channels: joint dependence on membrane potential and intracellular calcium. J Physiol (Camb) 1985;364:395–411. doi: 10.1113/jphysiol.1985.sp015752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klöckner U, Isenberg G. Intracellular pH modulates the availability of vascular L-type Ca2+channels. J Gen Physiol. 1994;103:647–663. doi: 10.1085/jgp.103.4.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohya Y, Kitamura K, Kuriyama H. Regulation of calcium current by intracellular calcium in smooth muscle cells of rabbit portal vein. Circ Res. 1988;62:375–383. doi: 10.1161/01.res.62.2.375. [DOI] [PubMed] [Google Scholar]
- Pastushenko VP, Schindler H. Level detection in ion channel records via idealization by statistical filtering and likelihood optimization. Philos Trans R Soc Lond B Biol Sci. 1997;352:39–51. doi: 10.1098/rstb.1997.0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanin C, Karlsson JO, Schindler H. Activity of cardiac L-type Ca2+channels is sensitive to cytoplasmatic calcium. Pflügers Arch. 1992;421:516–518. doi: 10.1007/BF00370266. [DOI] [PubMed] [Google Scholar]
- Schmid R, Seydl K, Baumgartner W, Groschner K, Romanin C. Trypsin increases availability and open probability of cardiac L-type Ca2+ channels without affecting inactivation induced by Ca2+ . Biophys J. 1995;69:1847–1857. doi: 10.1016/S0006-3495(95)80055-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuhmann K, Groschner K. Protein kinase-C mediates dual modulation of L-type Ca2+channels in human vascular smooth muscle. FEBS Lett. 1994;341:208–212. doi: 10.1016/0014-5793(94)80458-3. [DOI] [PubMed] [Google Scholar]
- Sturrock NDC, Lang CC, Struthers AD. Cyclosporine-induced nephrotoxicity and hypertension. Br J Hosp Med. 1992;48:434. [PubMed] [Google Scholar]
- Takai A, Mieskes G. Inhibtory effect of okadaic acid on p -nitrophenyl phosphate phosphatase activity of protein phosphatases. Biochem J. 1991;275:233–239. doi: 10.1042/bj2750233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Breemen C, Chen Q, Laher I. Superficial buffer barrier function of smooth muscle sarcoplasmic reticulum. TIPS (Trends Pharmacol Sci) 1995;16:98–105. doi: 10.1016/s0165-6147(00)88990-7. [DOI] [PubMed] [Google Scholar]
- Wakabayashi I, Groschner K. Evidence for a direct inhibitory effect of an extracellular H+ on store depletion-activated Ca2+entry in vascular endothelial cells. Biochem Biophys Res Commun. 1996;221:762–767. doi: 10.1006/bbrc.1996.0670. [DOI] [PubMed] [Google Scholar]
- Weiß, P. 1987. Stochastische Modelle für Anwender. B.G. Teubner, Stuttgart, Germany. 53–54.
- You Y, Pelzer DJ, Pelzer S. Trypsin and forskolin decrease the sensitivity of L-type calcium current to inhibition by cytoplasmic free calcium in guinea pig heart muscle cells. Biophys J. 1995;69:1838–1846. doi: 10.1016/S0006-3495(95)80054-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong XG, Hofmann F. Ca2+-dependent inactivation of class-C L-type Ca2+channel is a property of the α1 subunit. FEBS Lett. 1996;378:121–125. doi: 10.1016/0014-5793(95)01434-9. [DOI] [PubMed] [Google Scholar]