

Abstract
The ryanodine receptor (RyR)/Ca2+ release channel is an essential component of excitation–contraction coupling in striated muscle cells. To study the function and regulation of the Ca2+ release channel, we tested the effect of caffeine on the full-length and carboxyl-terminal portion of skeletal muscle RyR expressed in a Chinese hamster ovary (CHO) cell line. Caffeine induced openings of the full length RyR channels in a concentration-dependent manner, but it had no effect on the carboxyl-terminal RyR channels. CHO cells expressing the carboxyl-terminal RyR proteins displayed spontaneous changes of intracellular [Ca2+]. Unlike the native RyR channels in muscle cells, which display localized Ca2+ release events (i.e., “Ca2+ sparks” in cardiac muscle and “local release events” in skeletal muscle), CHO cells expressing the full length RyR proteins did not exhibit detectable spontaneous or caffeine-induced local Ca2+ release events. Our data suggest that the binding site for caffeine is likely to reside within the amino-terminal portion of RyR, and the localized Ca2+ release events observed in muscle cells may involve gating of a group of Ca2+ release channels and/or interaction of RyR with muscle-specific proteins.
Keywords: excitation–contraction coupling, calcium sparks, Chinese hamster ovary cells, Fura-2, green fluorescent protein
introduction
Ryanodine receptor (RyR)1 is an essential component of excitation–contraction (E-C) coupling in striated muscles, where it functions as a Ca2+ release channel in the sarcoplasmic reticulum (SR) membrane (Fleischer and Inui, 1989; McPherson and Campbell, 1993; Sutko and Airey, 1996). RyR is a single polypeptide of ∼560 kD, and exists in a homotetrameric structure with at least two functional domains: a carboxyl-terminal hydrophobic domain containing the conduction pore of the Ca2+ release channel (Takeshima et al., 1989; Zorzato et al., 1990; Bhat et al., 1997b ), and a large amino-terminal cytoplasmic domain referred to as the “foot structure” (Block et al., 1988; Lai et al., 1989; Wagenknecht et al., 1989; Franzini-Armstrong and Jorgensen, 1994). The activity of the skeletal muscle Ca2+ release channel is modulated by a wide spectrum of endogenous and exogenous ligands (Coronado et al., 1994; Meissner, 1994; Zucchi and Ronca-Testoni, 1997). Cytoplasmic Ca2+ activates the channel in nanomolar to micromolar concentrations, while in the micromolar to millimolar concentrations it inactivates the channel (Lai et al., 1988; Ma et al., 1988; Smith et al., 1988; Ma and Zhao, 1994). This dual effect of Ca2+ is thought to involve two independent high and low affinity Ca2+ binding sites on the RyR protein (Chu et al., 1993; Laver et al., 1995). Caffeine is one of the most widely used exogenous activators of the Ca2+ release channel (Zucchi and Ronca-Testoni, 1997). Rousseau et al. (1988) demonstrated a direct activation by caffeine of the isolated Ca2+ release channel reconstituted in planar lipid bilayer. Caffeine has been shown to activate Ca2+ release from SR by increasing the apparent affinity of the Ca2+ activation site for Ca2+ (Rousseau et al., 1988; Meissner et al., 1997). However, the site(s) on the Ca2+ release channel protein responsible for caffeine effect have not yet been identified.
Understanding the function and regulation of the Ca2+ release process is important in elucidating the mechanism(s) of E-C coupling. Recently, spontaneous local increases in the concentration of intracellular Ca2+, called “Ca2+ sparks,” have been observed in cardiac muscle (Cheng et al., 1993). The Ca2+ release process during normal cardiac E-C coupling has been suggested to result from the spatial and temporal summation of these elementary events of release (Cheng et al., 1993, 1996; Cannell et al., 1994, 1995; Lopez-Lopez et al., 1994, 1995). Studies in skeletal muscle have revealed these spontaneous Ca2+ release events to be smaller than cardiac sparks, suggesting that these events are controlled by different mechanisms in skeletal and cardiac muscles (Tsugorka et al., 1995; Klein et al., 1996; Blatter et al., 1997). It is not yet clear as to whether the “functional Ca2+ release units” underlying these events consist of a single or a cluster of Ca2+ release channels.
The RyRs of skeletal (Takeshima et al., 1989; Zorzato et al., 1990) and cardiac (Nakai et al., 1990; Otsu et al., 1990) muscle are coded by different genes, and share a high degree of sequence identity, especially in the carboxyl-terminal regions where the two proteins are well conserved in their amino acid sequence (Sorrentino and Volpe, 1993; Takeshima, 1993). This region of the protein contains several putative transmembrane segments (Takeshima et al., 1989; Zorzato et al., 1990), and the binding site(s) for Ca2+ and ryanodine (Callaway et al., 1994; Witcher et al., 1994). In recent studies, we have successfully used heterologous expression system to study the structure–function relationship of the Ca2+ release channel (Bhat et al., 1997a , 1997b ). Full length skeletal RyR expressed in Chinese hamster ovary (CHO) cells exhibits single channel properties similar to those of RyR from skeletal muscle SR. The carboxyl-terminal ∼20% of the RyR (RyR-C) was found to contain structures sufficient to form a functional Ca2+ release channel. The opening of the truncated RyR channel requires micromolar concentrations of Ca2+. However, unlike the full length RyR channel, the RyR-C channel fails to close at high cytoplasmic Ca2+ concentrations (Bhat et al., 1997b ). Moreover, the amino-terminal foot structure also appears to participate in both ion conduction and Ca2+-dependent regulation of the Ca2+ release channel (Bhat et al., 1997a , 1997b ).
In the present study, we have investigated the effect of caffeine on the Ca2+ release channel function of the full length and carboxyl-terminal portion of skeletal muscle RyR expressed in CHO cells using three different approaches: (a) single channel measurements using planar lipid bilayer reconstitution, (b) intracellular Ca2+ release measurements using Ca2+-sensitive dye Fura-2, and (c) Ca2+ imaging in single cells using laser scanning confocal microscopy. Caffeine was found to induce release of intracellular Ca2+ in cells expressing full length RyR in a concentration-dependent manner. This cooperative gating of the RyR channel is consistent with the Ca2+-induced Ca2+ release mechanism proposed for E-C coupling in striated muscles. However, caffeine did not show any effect on the function of the carboxyl-terminal portion of RyR, suggesting that the region(s) responsible for caffeine effect reside in the amino-terminal domain of the protein. Cells expressing the truncated RyR were found to have a higher resting intracellular Ca2+ compared with untransfected cells or those expressing full length RyR, which may be the result of spontaneous changes of intracellular Ca2+ observed in these cells. Furthermore, Ca2+ spark signals, which underlie localized Ca2+ release in muscle cells, were not present in CHO cells expressing either the full length or the carboxyl-terminal portion of RyR.
materials and methods
Cells and Expression System
The entire sequence of the rabbit skeletal muscle RyR cDNA (15.3 kb, RyR-wt) was cloned into the pRRS11 expression vector and the transcription occurs under the control of the SV40 promoter (Penner et al., 1989; Takeshima et al., 1989). A deletion mutant of pRRS11, pRyR-C, was generated through digestion with SalI (nt 546) and XhoI (nt 12018) restriction enzymes and religation. pRyR-C lacks the nucleotide sequence from 546 to 12018, which corresponds to a deletion of amino acids from 183 through 4006 (Bhat et al., 1997b ). CHO cells were grown at 37°C and 5% CO2 in Ham's F-12 medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. The expression plasmids were introduced into the cells (50– 60% confluent) using lipofectamine reagent (Life Technologies, Inc., Grand Island, NY). Stable transfectant cells were selected with G418 (0.5 mg/ml) 48 h after transfection. The level of RyR protein expression was tested using Western blot analysis.
Western Blot Analyses
Normal and transfected CHO cells were harvested and washed twice with ice cold PBS and lysed with ice cold modified RIPA buffer (150 mM NaCl, 50 mM Tris-Cl, pH 8.0, 1 mM EGTA, 1% Triton X-100, 0.1% sodium dodecyl sulfate [SDS], 1% sodium deoxycholate) in the presence of protease inhibitors (0.5 mM Pefabloc, 1 μM pepstatin, 1 μM leupeptin, 1 μg/ml aprotinin, and 1 mM benzamidine). The proteins in the whole cell lysate were mixed with the sample buffer (200 mM Tris-Cl, pH 6.7, 9% SDS, 6% β-mercaptoethanol, 15% glycerol, 0.01% bromophenol blue) and separated on a 3–12% linear SDS-PAGE gel after heating the samples at 85°C for 5 min. The proteins were then transferred to a polyvinylidene difluoride membrane and blotted with RR2 monoclonal antibody and horseradish peroxidase–linked secondary antibody using the electrochemiluminescence detection system (Amersham Corp., Arlington Heights, IL). The epitope for RR2 monoclonal antibody is known to reside in the carboxyl-terminal 656 amino acid region of the skeletal muscle RyR (Takeshima et al., 1993).
Expression and Detection of Fluorescence of Green Fluorescent Protein Fusion Proteins
The 15.2-kb HindIII-EcoRV fragment cDNA encoding the full length rabbit skeletal muscle RyR was cloned into pcDNA3 expression vector (Invitrogen Corp., San Diego, CA) to get pcDNA3-RyR. Similarly, a 4.2-kb BamHI-EcoRV fragment (nt 10982– 15230) encoding the carboxyl-terminal (amino acids 3661–5037) portion of RyR was cloned into pcDNA3 expression vector (Invi-trogen Corp.) to generate pcDNA3-RyR-C. The cDNA encoding the green fluorescent protein (GFP, 720 bp, 240 residues) was generated by PCR using pEGFP-N1 (Clontech, Palo Alto, CA) as template, with HindIII-KOZAK-6xHis sequence added to the 5′ terminal end and KpnI to the 3′ terminal end for ease of cloning. This sequence (HindIII-KOZAK-6xHis-GFP-KpnI) was then cloned in-frame to pcDNA3-RyR, by replacing the sequence (nt 1–866) that encodes the aminoterminal first 289 amino acids of RyR, to give pcDNA3 (GFP-RyR). A similar sequence (HindIII-KOZAK-6xHis-GFP-BamHI) was cloned into pcDNA3-RyR-C in tandem on the 3′ terminal end of RyR sequence to give pcDNA3 (GFP-RyR-C). Transfection of these plasmids into CHO cells was done as described above, using pEGFP-N1 to express GFP alone as positive control. Cells expressing GFP alone, GFP-RyR, and GFP-RyR-C fusion proteins were grown on coverslips and fixed with 4% paraformaldehyde and placed on a glass slide in inverted position for viewing. The cells are visualized with a Zeiss laser scanning confocal microscope using a 100× oil immersion objective (Carl Zeiss, Inc., Thornwood, NY). An FITC dichroic filter set with excitation at 450–490 nm and emission at 520 nm was used.
Measurement of Cytoplasmic [Ca2+]
All experiments were performed in saline solution containing 140 mM NaCl, 2.8 mM KCl, 1 mM CaCl2, 2 mM MgCl2, 12 mM glucose, 0.1% (wt/vol) bovine serum albumin, and 10 mM HEPES-NaOH, pH 7.2. Normal and transfected CHO cells were loaded with 2 μM Fura-2 AM (Molecular Probes Inc., Eugene, OR) for 45 min at room temperature. To remove nonhydrolyzed dye, cells were washed twice and resuspended at ∼106 cells/ml. Fluorescence (339 nm excitation/500 nm emission) was measured using cells maintained under continuous stirring in a cuvette at room temperature using previously described instrumentation (Dubyak and De Young, 1985). Ca2+-dependent fluorescence was calibrated using standard techniques as described (Grynkiewiz et al., 1985; Lam et al., 1993).
Confocal Imaging of Intracellular [Ca2+]
Single rat cardiac ventricular cells were obtained from 2-mo-old Sprague-Dawley rats by an enzymatic technique described in detail previously (Lopez-Lopez et al., 1995). Both cardiac and CHO cells were loaded with the Ca2+ indicator Fluo-3 by incubation for 30 min or longer in Tyrode's solution to which 10 μM Fluo-3 AM was added (Molecular Probes Inc.). Recordings of [Ca2+] were made in normal Tyrode's solution (composition [mM]: 140 NaCl, 10 dextrose, 10 Hepes, 4.0 KCl, 1 MgCl2, 1 CaCl2, pH adjusted to 7.3–7.4 with NaOH) at room temperature.
For ‘x-y' or ‘full-frame' imaging of calcium in the CHO cells, an MRC 600 confocal microscope (Bio-Rad Laboratories, Richmond, CA) was used. Fluo-3 fluorescence line-scan images were acquired with the ‘homebrew' confocal microscope (Parker et al., 1997) and were expressed as normalized increases in fluorescence compared with ‘resting' level (F/F0) (see Figs. 5–7), or, in some cases (see Fig. 8), simply in arbitrary units. Images of F/F0 were produced by dividing the fluorescence intensity (F) of each pixel in the original fluorescence image by its intensity at the beginning of the image (defined as F0), during a time when the cell was assumed to be in the ‘resting' state. All line-scan recordings of Fluo-3 fluorescence on single cells were obtained with the homebrew confocal microscope attached to the camera port of a Nikon Diaphot inverted microscope equipped with a 60× plan-apo oil-immersion objective (numerical aperture 1.4; Nikon Inc., Melville, NY), and the resolution was always 3 ms per scan line. As reported earlier, the spatial resolution of this instrument was near diffraction limited, at 300 nm laterally and 400 nm vertically (Parker et al., 1997). In some cases (see Fig. 5 B), the image was compressed (using neighborhood averaging) to display the fluorescence over a long period of time. Full frame fluorescence images of Fluo-3 fluorescence (see Fig. 8) were obtained with an MRC-600 confocal microscope (Bio-Rad Laboratories).
Figure 5.

Spatial and temporal patterns of caffeine-induced Ca2+ release in CHO cells expressing RyR. CHO cells expressing RyR-wt release Ca2+ upon exposure to caffeine. (A) Full frame confocal image of Fluo-3 fluorescence (Ca2+ activated) in CHO cells expressing the full length RyR: (a) control, (b) 0.5 s after addition of 10 mM caffeine, and (c) 5 min after washout of the caffeine. Increasing Ca2+-activated Fluo-3 fluorescence is indicated by the color bar on the right. Distance, in both x and y dimensions, is indicated by calibration bar indicating 10.0 μm. (B) Compressed line-scan image Fluo-3 fluorescence (proportional to [Ca2+]i) of a single CHO cell exposed to 10 mM caffeine. Calcium rises rapidly and cooperatively (see small initial “foot” in rise), and declines slowly.
Figure 7.
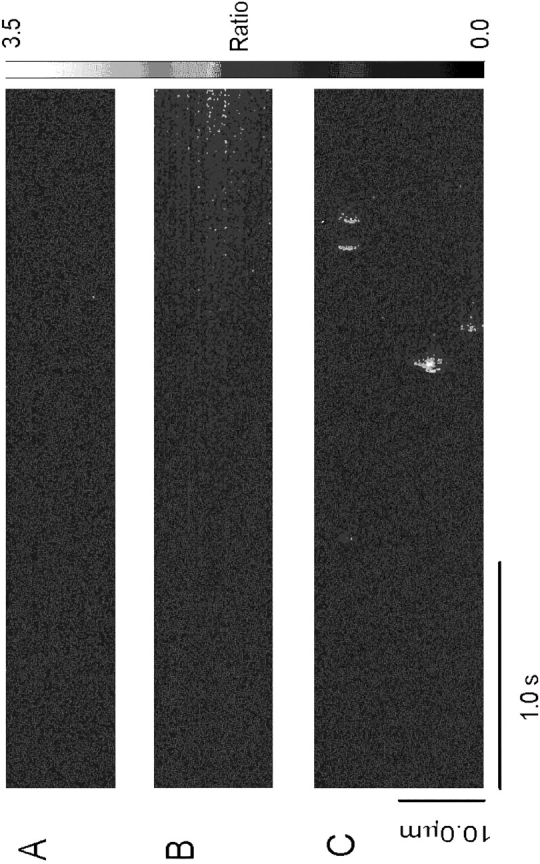
Lack of Ca2+ sparks in CHO cells expressing RyR-wt. (A) Line-scan image of fluorescence ratio (Fluo-3) in c1148 cells under control conditions. No local calcium transients or sparks are evident. (B) Line-scan image of c1148 cells during the initial rise in [Ca2+]i in the presence of 0.5 mM caffeine. [Ca2+] rises very slowly from resting levels, but no sparks are evident. (C) Typical spontaneous Ca2+ sparks in a cardiac myocyte. All line-scan images were obtained and presented at exactly the same spatial and temporal resolution, with the same confocal microscope.
Figure 8.

Spontaneous oscillation of intracellular Ca2+ in CHO cells expressing RyR-C. (A) Sequence of 16 full-frame images of Ca2+- activated Fluo-3 fluorescence taken at 20-s intervals. (B) Plot of Ca2+-activated Fluo-3 fluorescence as a function of time in the cell illustrated in A. The existence of spontaneous oscillations of [Ca2+]i may explain the apparent high resting [Ca2+]i in population measurements of [Ca2+]i in CHO cells expressing RyR-C.
The excitation of Fluo-3 was achieved using the 488-nm line from a 100 megawatt argon-ion laser (Omnichrome, Chino, CA), attenuated to 1–10%, beam expanded to overfill the back aperture of the objective, and deflected by a galvanometer-driven scan mirror (Cambridge Technology, Inc., Cambridge, MA) positioned at a conjugate telocentric plane formed by an eyepiece lens placed in the camera port. Fluorescence emission was de-scanned by the same mirror and wavelengths >510 nm were directed onto a confocal aperture placed in front of an avalanche photodiode. Computations and image analysis were performed on a Risc System/6000 workstation (IBM, Armonk, NY) with the software IDL (Research Systems, Inc., Boulder, CO).
Isolation of Microsomal Membrane Vesicles from CHO Cells
Microsomal membrane vesicles were isolated from transfected CHO cells as described (Bhat et al., 1997b ). The cells were homogenized on ice in hypotonic lysis buffer (10 mM HEPES, pH 7.4, 1 mM EDTA) containing protease inhibitors (0.5 mM pefabloc-SC, 1 μM pepstatin, 1 μM leupeptin, 1 μg/ml aprotinin, and 1 mM benzamidine) using nitrogen cavitation (300 psi for 15 min) with 10 strokes in tight-fitting Dounce homogenizer (Wheaton, Milville, NJ), followed by 15 strokes after addition of an equal volume of sucrose buffer (500 mM sucrose, 10 mM HEPES, pH 7.4, 1 mM EDTA). Microsome vesicles were collected by centrifugation of postnuclear supernatant (10,000 g, 15 min) at 100,000 g for 45 min at 4°C. The pellet was resuspended in a buffer containing 250 mM sucrose, 10 mM HEPES, pH 7.2. The membrane vesicles were stored at a protein concentration of 2–6 mg/ml at −75°C until use. Usually, 1–3 μl of microsomal membrane vesicles was used for reconstitution of Ca2+ release channels in the lipid bilayer system.
Reconstitution of Ca2+ Release Channels in Lipid Bilayer Membrane
Lipid bilayer membranes were formed across an aperture of ∼200 μm diameter using the Muller-Rudin method with a mixture of phosphatidylethanolamine:phosphatidylserine:cholesterol (6:6:1); the lipids were dissolved in decane at a concentration of 40 mg/ml. Incorporation of the Ca2+ release channel in bilayer was achieved by addition of membrane vesicles containing RyR-wt or RyR-C proteins to the cis solution, under a concentration gradient of 200 mM (cis)/50 mM (trans) cesium-gluconate. A symmetrical 200-mM Cs-gluconate was used as current carrier in the presence of 1 mM ATP. The pH in both cis and trans solution was maintained throughout the experiment at 7.4 with 10 mM HEPES-Tris. The free Ca2+ concentrations in both solutions were buffered with 1 mM EGTA, and measured using a Ca2+-sensitive electrode (Orion Research, Boston, MA). Orientation of the Ca2+ release channel in the lipid bilayer, usually in the cis-cytoplasmic trans-lumenal SR manner, was determined by the sensitivity of the channel to cytoplasmic Ca2+ (Bhat et al., 1997b ). Channels oriented in the opposite direction, which account for <10% of the experiments, were not used in the present study. To maintain stability of the bilayer membrane and the Ca2+ release channel activities, designed pulse protocols were used to measure currents through the single Ca2+ release channels. The bilayer membrane was kept at a holding potential of 0 mV, and pulsed to different test potentials of 0.5–1-s durations. Single channel currents were recorded with an Axopatch 200A patch clamp unit (Axon Instruments, Inc., Foster City, CA). Data acquisition and pulse generation were performed with a 486 computer and 1200 Digidata A/D-D/A convertor (Axon Instruments, Inc.). The currents were sampled at 0.05 ms/point and filtered at 1 kHz through an 8-pole Bessel filter. Single channel data analyses were performed with pClamp, TIPS, and custom programs.
results
Full Length and Carboxyl-Terminal RyRs are Expressed in Intracellular Membranes of CHO Cells
c1148 is a stable Chinese hamster ovary cell line that was transfected with pRRS11, which encodes the entire sequence of the rabbit skeletal muscle ryanodine receptor (Penner et al., 1989; Takekura et al., 1995). To study the function and regulation of the carboxyl-terminal RyR channel, we have generated another clone of CHO cells (ryr-c) that was stably transfected with a deletion mutant of RyR, pRyR-C. This mutant lacks 3,824 amino acids (amino acids 183–4006) from the cytoplasmic domain, but retains the carboxyl-terminal putative transmembrane domain of RyR, and has a predicted molecular weight of ∼130 kD (Bhat et al., 1997b ). The ryr-c cells expressed detectable amounts of the 130-kD RyR-C proteins (Fig. 1, lanes 3 and 6), recognized by a monoclonal antibody (RR2) whose epitope resides in the COOH-terminal region of skeletal muscle RyR (Takeshima et al., 1989). The c1148 cells expressed a high level of the full length RyR proteins (RyR-wt) (Fig. 1, lane 7), which was identical to the native RyRs from the rabbit skeletal muscle SR (Fig. 1, lane 1), in terms of molecular weight and immunoreactivity. No proteins were recognized by RR2 in untransfected CHO cells (Fig. 1, lane 2), or in those transfected with pcDNA3 alone, suggesting that CHO cells do not contain any detectable amount of endogenous skeletal isoform of RyR proteins.
Figure 1.

Heterologous expression of skeletal muscle RyR in CHO cells. Western blot analysis of RyR proteins expressed in Chinese hamster ovary cells. Total cell lysates were run on a 3–12% gradient SDS polyacrylamide gel, and blotted with monoclonal antibody (RR2) against the rabbit skeletal muscle RyR (Bhat et al., 1997). Lane 1, SR vesicles from rabbit skeletal muscle; lane 2, untransfected CHO cells; lanes 3 and 6, COOH-terminal RyR proteins (∼130 kD) stably expressed in CHO cells (ryr-c); lane 4, CHO cells transfected with GFP-RyR-C; lane 5, CHO cells transfected with GFP-RyR; lane 7, full length RyR (∼560 kD) stably expressed in CHO cells (c1148).
For subcellular localization of RyR proteins expressed in CHO cells, a fusion protein (GFP-RyR) was generated by replacing the amino-terminal first 289 amino acids of RyR-wt with those of the green fluorescent protein. In addition, a GFP-RyR-C construct was obtained by linking the GFP sequence in tandem with the COOH-terminal 1,377 amino acids of RyR (amino acids 3661–5037). For positive controls, CHO cells were transfected with pEGFP-N1 that encodes only GFP. As shown in Fig. 2, cells transfected with pEGFP-N1 exhibited a diffuse pattern of fluorescence, as expected for a soluble protein (Fig. 2 A), whereas cells expressing GFP-RyR and GFP-RyR-C fusion proteins exhibited a fluorescence signal only in certain subcellular areas, particularly in the perinuclear region (Fig. 2, B and C), indicating that the protein is probably localized to the endoplasmic reticulum (ER) membrane of CHO cells. Both GFP-RyR and GFP-RyR-C proteins were detected in these cells by the RR2 antibody in a Western blot (Fig. 1, lanes 5 and 4, respectively). These results suggest that the carboxyl-terminal portion of RyR contains the signal for retention of the RyR protein in the intracellular membranes.
Figure 2.

Localization of RyR in intracellular membranes of CHO cells. Confocal measurement of green fluorescence in CHO cells transfected with (A) pEGFP-N1, (B) pcDNA3 (GFP-RyR), and (C) pcDNA3 (GFP-RyR-C). Cells expressing GFP alone exhibit a diffuse pattern of fluorescence throughout the cell (A), whereas cells expressing the GFP-RyR and GFP-RyR-C fusion proteins show localized fluorescence signal near the perinuclear region of the cells (B and C). The panels from left to right show representative sections through the cells at 0.5-μm intervals. The cells have an average thickness of ∼10 μm.
Caffeine Enhances Activity of the Wild-Type, Not the COOH-Terminal RyR Ca2+ Release Channels
The Ca2+ release channel functions of the expressed RyR-wt and RyR-C were measured by reconstituting microsomal membrane vesicles from the c1148 and ryr-c cells into lipid bilayer membranes, using Cs-gluconate as current carrying ion in the recording solution. In previous studies, we showed that RyR-C forms a functional Ca2+ release channel that shared some of the single channel properties with RyR-wt, including activation by cytoplasmic Ca2+ and regulation by ryanodine (Bhat et al., 1997b ). But unlike the RyR-wt channel, which exhibited a linear current–voltage relationship and inactivated at millimolar Ca2+, the channels formed by RyR-C displayed significant inward rectification and failed to close at high cytoplasmic Ca2+ (Bhat et al., 1997b ).
Fig. 3 shows that openings of both RyR-wt and RyR-C channels were sensitive to activation by cytoplasmic Ca2+, as chelating the free [Ca2+]i from 220 to 0.08 μM resulted in decrease in open probability (Po) of the RyR-wt channel from Po = 0.121 ± 0.044 to 0.013 ± 0.002, and that of the RyR-C channel from Po = 0.201 ± 0.036 to 0.008 ± 0.003. It is interesting to note that the Po of RyR-C is higher than RyR-wt at free [Ca2+] of 220 μM, which could be due to the lack of Ca2+-dependent inactivation of the RyR-C channel (Bhat et al., 1997b ). The RyR-wt channel was sensitive to activation by caffeine, as addition of 10 mM caffeine to the cytoplasmic solution resulted in a significant increase of Po to 0.198 ± 0.063 at [Ca2+]i = 0.08 μM (Fig. 3 A). In contrast, the activity of the RyR-C channel did not change upon the addition of 10 mM caffeine (Fig. 3 B). Furthermore, the lack of effect of caffeine on the RyR-C channel was independent of [Ca2+]i in a concentration range from 0.08 to 220 μM (data not shown).
Figure 3.

Effect of caffeine on single RyR-wt and RyR-C channels. Selected single channel currents at +50 mV from a continuous experiment with the RyR-wt (A) and RyR-C (B) channels incorporated into the lipid bilayer membrane. Under control conditions (220 μM [Ca2+]i), the RyR-wt had an average Po of 0.121 ± 0.044 (n = 19), and the RyR-C channel had an average Po of 0.201 ± 0.036 (n = 12). The addition of 10 mM EGTA lowered the free [Ca2+] to 0.08 μM and reduced the Po of RyR-wt to 0.013 ± 0.002 (n = 5), and Po of RyR-C to 0.008 ± 0.003 (n = 4). Subsequent addition of 10 mM caffeine increased the Po of RyR-wt to 0.198 ± 0.063 (n = 4). Po of the RyR-C channel remained unchanged after the addition of 10 mM caffeine (Po = 0.008 ± 0.002, n = 4).
To further test the effect of caffeine on the Ca2+ release channels, we measured the change in cytoplasmic [Ca2+] in c1148 and ryr-c cells after stimulation with caffeine. The measurements were performed with a large number of cells (1–2 × 106) loaded with a fluorescent Ca2+ indicator, Fura-2, which provides a macroscopic assessment of the caffeine sensitivity of the RyR-wt and RyR-C channels. Untransfected CHO cells had negligible amounts of caffeine-induced Ca2+ release, but they maintained an intact intracellular Ca2+ pool, as addition of 500 nM thapsigargin resulted in a rise in [Ca2+]i due to inhibition of the Ca2+-ATPase in the ER (Fig. 4 A). With the c1148 cells, we observed a robust increase of cytoplasmic [Ca2+] from a resting level of 0.192 ± 0.038 μM to 1.222 ± 0.257 μM upon stimulation with 10 mM caffeine (Fig. 4 B). This rapid rise of [Ca2+]i in c1148 cells was followed by a slow decline phase, probably reflecting the exit of Ca2+ to the extracellular solution through the plasma membrane Ca2+ pump or Na/ Ca2+ exchanger. Different from the c1148 cells, the ryr-c cells did not respond to caffeine (Fig. 4 C), and the thapsigargin-sensitive ER Ca2+ pool appears to be smaller in the ryr-c cells than in the untransfected CHO cells (comparing Fig. 4, A and C), although the response to thapsigargin was variable (Table I). The ryr-c cells, however, had a resting [Ca2+]i = 0.422 ± 0.071 μM, which was significantly higher than that of the c1148 cells and the untransfected CHO cells ([Ca2+]i = 0.209 ± 0.012 μM). These results are summarized in Table I.
Figure 4.

Caffeine-induced release of Ca2+ in CHO cells. Changes in [Ca2+]i were measured in cells (106/ml) loaded with Fura-2 AM and maintained under continuous stirring in a cuvette. (A) Application of 10 mM caffeine (Caf) does not induce release of intracellular Ca2+ in untransfected CHO cells, whereas thapsigargin (Thg, 500 nM) causes a rise in [Ca2+], suggesting intact intracellular Ca2+ store in these cells. (B) In c1148 cells, an increase in [Ca2+]i from 0.192 ± 0.038 μM (n = 4) to 1.222 ± 0.257 μM (n = 3) is seen after stimulation with caffeine. (C) CHO cells expressing RyR-C do not respond to stimulation with caffeine. The resting intracellular [Ca2+] in these cells is 0.422 ± 0.071 μM (n = 7). All fluorescence traces are representative of at least three separate experiments. Gaps in the recordings are due to the opening of the compartment to make additions.
Table I.
Caffeine-induced Ca2+ Release in CHO Cells Expressing RyR-wt and RyR-C*
| Initial [Ca2+]i | [Ca2+]i after caffeine‡ | [Ca2+]i before Thg§ | [Ca2+]i after Thg§ | |||||
|---|---|---|---|---|---|---|---|---|
| nM | ||||||||
| Control | 170.2 | 170.2 | 170.2 | 285.1 | ||||
| 210.0 | 210.0 | 210.0 | 933.3 | |||||
| 191.2 | 191.2 | 191.2 | 665.6 | |||||
| 256.0 | 256.0 | 256.0 | 665.6 | |||||
| 200.2 | 200.2 | 200.2 | 1176 | |||||
| 227.9 | 227.9 | 227.9 | 512.0 | |||||
| Average ± SEM | 209.3 ± 12.2 | 209.3 ± 12.2 | 209.3 ± 12.2 | 706.3 ± 127.9 | ||||
| RyR-wt | 135.8 | 1808 | 487.2 | 693.7 | ||||
| 292.9 | 1456 | 386.9 | 476.0 | |||||
| 207.2 | 638.4 | 465.9 | 560.0 | |||||
| 131.5 | 987.0 | ND | ND | |||||
| Average ± SEM | 191.9 ± 37.9 | 1222.4 ± 257.2 | 446.7 ± 30.5 | 576.6 ± 63.4 | ||||
| RyR-C | 207.7 | 207.7 | 171.7 | 355.1 | ||||
| 476.0 | 476.0 | 206.7 | 5376 | |||||
| 310.9 | 310.9 | 336.0 | 1209 | |||||
| 600.3 | 600.3 | 600.3 | 806.4 | |||||
| 512.8 | 512.8 | 512.8 | 776.0 |
Each experiment was done with ∼106 cells.
10 mM caffeine was applied to the cell suspension under continuous stirring.
500 nM thapsigargin was applied to the cell suspension under continuous stirring.
Caffeine Exhibits Concentration-dependent Effect on RyR-wt Channels Expressed in CHO Cells
Confocal images of individual c1148 cells expressing the full length skeletal muscle RyR showed a rapid caffeine-induced Ca2+ release from the intracellular membranes (Fig. 5 A). The effect of caffeine is reversible, since [Ca2+]i returned completely to the resting level upon washout of caffeine (Fig. 5 A, c). Having established a positive caffeine response, we next obtained a series of line-scanning images of the same cell after reapplication of 10 mM caffeine (Fig. 5 B). These line scanning pictures show that the distribution of [Ca2+]i is inhomogeneous in response to caffeine. There appeared to be ‘hot spots' where the level of [Ca2+]i is higher than other places, as indicated by the arrows in Fig. 5 B. These hot spots may correspond to regions where the Ca2+ release channels are clustered together (Takekura et al., 1995). Also, the overall increase in [Ca2+]i is biphasic in that caffeine-induced transient rise in [Ca2+]i was followed by a slow decaying phase, consistent with the observation shown in Fig. 4 B. The rapid increase in [Ca2+]i may underline cooperative features of the Ca2+ release process.
To further examine the caffeine sensitivity of RyR-wt, we tested different concentrations of caffeine (ranging from 0.1 to 10 mM) in c1148 cells. Fig. 6 shows the concentration of intracellular Ca2+ measured at a given position from three c1148 cells exposed to 0.5, 2, and 10 mM caffeine. From the dose–response relationship, it is clear that caffeine exhibits a concentration-dependent Ca2+ release in these cells. The threshold for caffeine activation of the RyR-wt channels occurred at a concentration between 0.1 and 0.5 mM. The time course of increase in [Ca2+]i had a strong correlation with the concentration of caffeine—the lower the caffeine concentration, the slower the time course. It is evident that there is a plateau phase preceding the rise in [Ca2+]i, in particular at lower caffeine concentrations (0.5 and 2 mM). The cooperative nature of the response is clear in the sigmoidal onset of Ca2+ release, which is most evident at 2.0 and 10 mM caffeine (Fig. 6, B and C).
Figure 6.

Dose-dependent effect of caffeine on CHO cells expressing RyR-wt. Fluorescence ratio, derived from one selected pixel of line scan images such as those in Fig. 5 B, is plotted as a function of time. The pixel size was 0.2 μm. Results are from three different CHO cells. Doses of caffeine: (A) 0.5, (B) 2.0, and (C) 10 mM. The rate of rise in fluorescence ratio ([Ca2+]) is highly dose dependent, possibly indicating dose-dependent cooperativity of the opening of the Ca2+ release channels. The fact that the peak fluorescence ratio reached was greatest in 2.0 mM caffeine may be the result of variability in the amount of Ca2+ in the intracellular stores of different cells. The number of cells tested are n = 2 (0.1 mM), n = 5 (0.2 mM), n = 4 (0.5 mM), n = 4 (2 mM), n = 2 (5 mM), and n = 10 (10 mM).
RyR Channels Expressed in CHO Cells Do Not Exhibit “Localized Ca2+ Release” Typical of Muscle Cells
In cardiac muscle cells, local increases in intracellular Ca2+ have been observed that occur spontaneously (Cheng et al., 1993), as well as in response to activation of voltage-gated Ca2+ channels (Lopez-Lopez et al., 1994, 1995; Cannell et al., 1994, 1995). Elementary Ca2+ release events, similar to those in cardiac muscle, although smaller in size, have also been recorded in skeletal muscle cells (Tsugorka et al., 1995). It is not known if a single or a group of Ca2+ release channels acting in concert constitute the “Ca2+ release units” underlying the local Ca2+ transients in skeletal and cardiac muscles. We tested for the presence of spontaneous changes in intracellular Ca2+ in CHO cells expressing skeletal RyR (Fig. 7). Under resting conditions, no spontaneous local Ca2+ transients were evident in the line-scan images of c1148 cells (Fig. 7 A), although caffeine was capable of inducing Ca2+ release in these cells (Fig. 7 B). Furthermore, no local Ca2+ signals were observed in c1148 cells stimulated with various concentrations of caffeine (ranging from 0.1 to 10 mM, data not shown). Under identical spatial and temporal resolution, typical Ca2+ sparks could be recorded in rat cardiac ventricular myocytes (Fig. 7 C). Therefore, the lack of local Ca2+ transients in the CHO cells was not likely to be due to inability to resolve such events had they been present and similar to the calcium sparks typical of cardiac muscle.
CHO Cells Expressing RyR-C Exhibit Spontaneous Changes in [Ca2+]i
While CHO cells expressing the carboxyl-terminal portion of RyR did not respond to stimulation with caffeine (Figs. 3 B and 4 C), the intracellular [Ca2+] showed spontaneous changes in these cells, as shown in Fig. 8. This change in [Ca2+]i appears to occur due to release of Ca2+ from intracellular store since this response is restricted to an area of the cell surrounding the nucleus. During the period of observation (usually 10 min), these transient changes in [Ca2+]i were observed in ∼10% of the CHO cells expressing RyR-C. The higher resting [Ca2+]i observed in these cells (Fig. 4 C) may be attributed to these changes in [Ca2+]i. No evidence for such changes in intracellular Ca2+ was found in c1148 cells. These spontaneous changes in intracellular Ca2+ in cells expressing truncated, but not full length, RyR suggest the importance of the amino-terminal foot region of the protein in the regulation of Ca2+ release channel.
discussion
The present study examined the function and regulation of skeletal muscle Ca2+ release channel through heterologous expression of RyR in CHO cells. Using this approach, we have recently demonstrated that the carboxyl-terminal portion of RyR contains structures sufficient to form a functional Ca2+ release channel, but the amino-terminal portion of the protein also contributes to the ion conduction and regulation of the channel (Bhat et al., 1997a , 1997b ). In the present study, we show that the RyR protein is localized to the intracellular organelle, possibly ER, of CHO cells based upon the fluorescence pattern of GFP-RyR fusion proteins (Fig. 2). The sequence required for targeting of the RyR protein to the intracellular membrane is likely to reside in the carboxyl-terminal end of the protein since the COOH-terminal RyR protein is also localized to the perinuclear area of CHO cells. In addition, an earlier study by Takeshima et al. (1993) showed that a 75-kD protein containing the carboxyl-terminal 656 amino acids of the skeletal muscle RyR was expressed in the ER membranes of CHO cells.
Caffeine, an effective activator of Ca2+ release in both skeletal and cardiac muscles, was found to induce release of Ca2+ in cells expressing full length RyR in a dose-dependent manner. However, it failed to show any effect on cells expressing the carboxyl-terminal portion of RyR. The sigmoidal onset of Ca2+ release induced by caffeine in c1148 cells reveals the cooperative nature of the response (Fig. 6). There appears to be a threshold level of intracellular [Ca2+] required before a robust increase in [Ca2+]i can occur, especially at lower concentrations of caffeine. It is possible that Ca2+ released from one single or a cluster of RyRs triggers activation of adjacent Ca2+ release channels in these cells. The affinity of Ca2+ activation site for Ca2+ in RyR protein is increased in the presence of caffeine, causing a cooperative interaction between these sites. This is consistent with the Ca2+-induced Ca2+ release mechanism proposed for activation of Ca2+ release in both skeletal and cardiac muscles (Fabiato, 1983). The lack of effect of caffeine in activating Ca2+ release from cells expressing RyR-C suggests that the site(s) responsible for caffeine effect are lost in the truncated RyR protein. While the caffeine-binding site on the Ca2+-release channel protein has not been identified, our data suggest the following possibilities: first, the caffeine binding site resides within the amino-terminal foot region of the RyR protein; second, the binding site is formed by the ensemble of both the amino- and the carboxyl-terminal portion of RyR; and third, the spatial configuration of the binding site is disrupted in the absence of the amino-terminal portion of the protein. The lack of effect of caffeine in cells expressing RyR-C is not likely due to the higher resting [Ca2+]i in these cells, since caffeine was not able to influence the activity of single RyR-C channels in cytoplasmic [Ca2+] ranging from 0.08 to 220 μM.
In cells expressing the RyR-C protein, the resting intracellular [Ca2+] was found to be higher than that in control cells and in those expressing RyR-wt. Furthermore, spontaneous transient changes in intracellular [Ca2+] were recorded in cells expressing the truncated RyR, but not in those expressing the full length protein. The high level of resting [Ca2+]i and spontaneous changes in Ca2+ in these cells could be due to several possible factors. First, inability of the RyR-C channel to close at higher cytosolic [Ca2+]. Unlike the native RyR from skeletal muscle SR and the full length RyR expressed in CHO cells, RyR-C channels lack the apparent Ca2+-dependent inactivation property (Bhat et al., 1997b ). Recent studies by other investigators have demonstrated that dyspedic myotubes (skeletal muscle cells lacking native RyR), when transfected with cardiac subtype of RyR, exhibit spontaneous oscillation of intracellular Ca2+ (Nakai et al., 1997; Yamazawa et al., 1996, 1997). Thus, a sustained level of activation of the Ca2+ release channel may cause a higher intracellular Ca2+ concentration. It is interesting to note that the cardiac Ca2+ release channels, unlike the skeletal subtype, are less susceptible to inactivation by cytoplasmic Ca2+ (Chu et al., 1993; Laver et al., 1997). It remains to be seen if CHO cells expressing the cardiac RyR also exhibit any spontaneous changes in intracellular Ca2+. Second, abnormally large conductance states associated with the RyR-C protein. In a recent study, we have shown that RyR-C forms a large conductance channel in ∼20% of the single channel experiments, which seems to be less sensitive to Ca2+ regulation (see Bhat et al., 1997b ; Fig. 5). This large conductance channel may constitute a leak in the ER membranes, which may result in an elevated level of cytoplasmic [Ca2+]. Although the mechanism of this phenomenon is not understood, this may result from a defective or unstable oligomerization of the truncated RyR protein. Thus, the instability of the oligomeric structure together with a greater tendency of the truncated RyR channel to opening (see Fig. 3) may contribute to spontaneous increase in intracellular [Ca2+]i in cells expressing RyR-C protein. Third, the truncated RyR protein may have altered interaction with accessory proteins, such as FK506 binding protein (FKBP12), which is known to regulate the function of the Ca2+ release channel (Brillantes et al., 1994; Ma et al., 1995; Ahern et al., 1997). While the interaction of FKBP appears to be essential for setting normal gating properties of RyR, channels depleted of FKBP show a higher probability of channel opening, as well as an increased sensitivity to activation by cytoplasmic Ca2+ (Brillantes et al., 1994; Ahern et al., 1997). It is likely that the site of FKBP interaction with the Ca2+ release channel is missing in the truncated RyR protein (Wagenknecht et al., 1996).
In cells expressing full length or truncated RyR, no evidence was found for the presence of spontaneous local increase in intracellular Ca2+ such as described in cardiac muscle (i.e., sparks) (Cheng et al., 1993) and skeletal muscles (Tsugorka et al., 1995; Klein et al., 1996). In cardiac cells, Ca2+ sparks occur at about the rate of 1/100 μm/s per cell, or ∼100/s per cell (Cheng et al., 1993). In this study, at least 50 CHO cells expressing wild-type RyR were scanned over a distance of 25 μm (usually somewhat more than their diameter) for at least 25 s. This amount of scanning of cardiac cells would have produced 312.5 sparks. As shown in Fig. 7, had such sparks occurred in the CHO cells (Fig. 7, A and B), they would have been readily detectable since Ca2+ sparks were readily detected in cardiac cells when scanned at the same spatial and temporal resolution (Fig. 7 C). The release of Ca2+ from each RyR in the CHO cells seems to be too small to produce a resolvable local Ca2+ transient, perhaps similar to the situation in cardiac muscle, where small, unresolvable Ca2+ transients, termed “quarks” are thought to arise from Ca2+ released from a single RyR (Lipp and Niggli, 1996).
The absence of local Ca2+ transients associated with the expressed RyR, similar to those in muscle, may be attributed to the following possible factors. First, the absence of local cooperative opening of multiple channels in CHO cells, which is thought to underlie the origin of sparks. Although the channels exhibit Ca2+- induced Ca2+ release, as seen in response to stimulation with caffeine, the spatial distribution of protein may not mimic that seen in muscle cells. Second, the absence (in CHO cells) of muscle-specific proteins involved in E-C coupling. In skeletal muscle, the dihydropyridine receptor (DHPR) functions as a voltage sensor and is thought to be in close interaction with the RyR (Rios et al., 1991). Recent studies have suggested that RyR not only receives a signal from DHPR, but also transmits a retrograde signal back to regulate the activity of DHPR in skeletal muscle and neurons (Chavis et al., 1996; Nakai et al., 1996). The interaction between DHPR and RyR appears to be tissue specific since coexpression of these two proteins in CHO cells failed to form junctions between plasma and ER membranes such as typically exists between transverse tubule and SR membranes in muscle cells (Takekura et al., 1995). Thus, an intimate juxtaposition of RyR, DHPR, and/or other accessory proteins may be essential to form a “local response element” responsible for generation of detectable local Ca2+ release events (Yue, 1997; Suda et al., 1997). Alternatively, Ca2+ sparks require a close apposition among channels and other related proteins in a restricted diffusional space provided by the dyads and triads of cardiac and skeletal muscle. The absence of this structural arrangement in CHO cells may result in the lack of spontaneous or caffeine-induced Ca2+ sparks.
Acknowledgments
We thank Dr. George Dubyak for help with the Fura-2 measurement of cytoplasmic Ca2+ shown in Fig. 4, and Dr. Maryanne Pendergast for help with the confocal imaging of cells expressing GFP fusion proteins.
This work was supported by grants from National Institutes of Health (AG-15556 to J. Ma and HL-55280 to W.G. Wier), a Pilot Project from Howard Hughes Medical Institute, and an Established Investigatorship from the American Heart Association to J. Ma.
Footnotes
Abbreviations used in this paper: CHO, Chinese hamster ovary; DHPR, dihydropyridine receptor; E-C, excitation–contraction; ER, endoplasmic reticulum; GFP, green fluorescent protein; Po, open probability; RyR, ryanodine receptor; RyR-C, carboxyl-terminal ∼20% of the RyR; SDS, sodium dodecyl sulfate; SR, sarcoplasmic reticulum.
references
- Ahern GP, Junankar PR, Dulhunty AF. Subconductance states in single-channel activity of skeletal muscle ryano-dine receptors after removal of FKBP12. Biophys J. 1997;72:146–162. doi: 10.1016/S0006-3495(97)78654-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat MB, Ma J, Zhao JY, Hayek S, Freeman EC, Takeshima H. Deletion of amino acids 1641–2437 from the foot region of skeletal muscle ryanodine receptor alters the conduction properties of the Ca release channel. Biophys J. 1997a;73:1320–1328. doi: 10.1016/S0006-3495(97)78165-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat MB, Zhao JY, Takeshima H, Ma J. Functional calcium release channel formed by the carboxyl-terminal portion of ryanodine receptor. Biophys J. 1997b;73:1329–1336. doi: 10.1016/S0006-3495(97)78166-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatter LA, Huser J, Rios E. Sarcoplasmic reticulum Ca2+ release flux underlying Ca2+sparks in cardiac muscle. Proc Natl Acad Sci USA. 1997;94:4176–4181. doi: 10.1073/pnas.94.8.4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block BA, Imagawa T, Campbell KP, Franzini-Armstrong C. Structural evidence for a direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J Cell Biol. 1988;107:2587–2600. doi: 10.1083/jcb.107.6.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brillantes AB, Ondrias K, Scott A, Kobrinsky E, Ondriasova E, Moschella MC, Jayaraman T, Landers M, Ehrlich BE, Marks AR. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell. 1994;77:513–523. doi: 10.1016/0092-8674(94)90214-3. [DOI] [PubMed] [Google Scholar]
- Callaway C, Seryshev A, Wang JP, Slavik KJ, Needleman DH, Cantu C, Wu Y, Jayaraman T, Marks AR, Hamilton SL. Localization of the high and low affinity [3H]-ryanodine binding sites on the skeletal muscle Ca2+release channel. J Biol Chem. 1994;269:15876–15884. [PubMed] [Google Scholar]
- Cannell MB, Cheng H, Lederer WJ. Spatial non-uniformities in [Ca2+]iduring excitation–contraction coupling in cardiac myocytes. Biophys J. 1994;67:1942–1956. doi: 10.1016/S0006-3495(94)80677-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannell MB, Cheng H, Lederer WJ. The control of calcium release in heart muscle. Science. 1995;268:1045–1049. doi: 10.1126/science.7754384. [DOI] [PubMed] [Google Scholar]
- Chavis P, Fagni L, Lansman JB, Bockaert J. Functional coupling between ryanodine receptors and L-type calcium channels in neurons. Nature. 1996;382:719–722. doi: 10.1038/382719a0. [DOI] [PubMed] [Google Scholar]
- Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation–contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- Cheng H, Lederer MR, Lederer WJ, Cannell MB. Calcium sparks and [Ca2+] waves in cardiac myocytes. Am J Physiol. 1996;39:C148–C159. doi: 10.1152/ajpcell.1996.270.1.C148. [DOI] [PubMed] [Google Scholar]
- Chu A, Fill M, Stefani E, Entman ML. Cytoplasmic Ca2+ does not inhibit the cardiac muscle sarcoplasmic reticulum ryanodine receptor Ca2+ channel, although Ca2+-induced Ca2+release is observed in native vesicles. J Membr Biol. 1993;135:49–59. doi: 10.1007/BF00234651. [DOI] [PubMed] [Google Scholar]
- Coronado R, Morrissette J, Sukhareva M, Vaughan DM. Structure and function of ryanodine receptors. Am J Physiol. 1994;266:C1485–C1504. doi: 10.1152/ajpcell.1994.266.6.C1485. [DOI] [PubMed] [Google Scholar]
- Dubyak GR, De Young MB. Intracellular Ca2+mobilization activated by extracellular ATP in Ehrlich ascites tumor cells. J Biol Chem. 1985;260:10653–10661. [PubMed] [Google Scholar]
- Fabiato A. Calcium-induced calcium release from the cardiac sarcoplasmic reticulum. Am J Physiol. 1983;245:C1–C14. doi: 10.1152/ajpcell.1983.245.1.C1. [DOI] [PubMed] [Google Scholar]
- Fleischer S, Inui M. Biochemistry and biophysics of excitation–contraction coupling. Annu Rev Biophys Biophys Chem. 1989;18:333–364. doi: 10.1146/annurev.bb.18.060189.002001. [DOI] [PubMed] [Google Scholar]
- Franzini-Armstrong C, Jorgensen AO. Structure and development of E-C coupling units in skeletal muscle. Annu Rev Physiol. 1994;56:509–534. doi: 10.1146/annurev.ph.56.030194.002453. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Klein MG, Cheng H, Santana LF, Jiang Y-H, Lederer WJ, Schneider MF. Two mechanisms of quantized calcium release in skeletal muscle. Nature. 1996;379:455–458. doi: 10.1038/379455a0. [DOI] [PubMed] [Google Scholar]
- Lai FA, Erickson H, Rousseau E, Liu QY, Meissner G. Purification and reconstitution of the calcium release channel from skeletal muscle. Nature. 1988;331:315–319. doi: 10.1038/331315a0. [DOI] [PubMed] [Google Scholar]
- Lai FA, Misra M, Xu L, Smith HA, Meissner G. The ryanodine receptor-Ca2+release channel complex of skeletal muscle sarcoplasmic reticulum. Evidence for a cooperatively coupled, negatively charged homotetramer. J Biol Chem. 1989;264:16776–16785. [PubMed] [Google Scholar]
- Lam M, Dubyak G, Distelhorst CW. Effect of glucocorticoid treatment on intracellular calcium homeostasis in mouse Lymphoma cells. Mol Endocrinol. 1993;7:686–693. doi: 10.1210/mend.7.5.8316252. [DOI] [PubMed] [Google Scholar]
- Laver DR, Roden LD, Ahern GP, Eager KR, Junankar PR, Dulhunty AF. Cytoplasmic Ca2+inhibits the ryano-dine receptor from cardiac muscle. J Membr Biol. 1995;147:7–22. doi: 10.1007/BF00235394. [DOI] [PubMed] [Google Scholar]
- Lipp P, Niggli E. Submicroscopic calcium signals as fundamental events of EC-coupling in guinea pig cardiac myocytes. J Physiol (Camb) 1996;492:31–38. doi: 10.1113/jphysiol.1996.sp021286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Lopez JR, Shacklock PS, Balke CW, Wier WG. Local stochastic release of Ca2+in voltage-clamped rat heart cells: visualization with confocal microscopy. J Physiol (Camb) 1994;480:21–29. doi: 10.1113/jphysiol.1994.sp020337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Lopez JR, Shacklock PS, Balke CW, Wier WG. Local calcium transients triggered by single L-type calcium channel currents in cardiac cells. Science. 1995;268:1042–1045. doi: 10.1126/science.7754383. [DOI] [PubMed] [Google Scholar]
- Ma J, Bhat MB, Zhao JY. Rectification of skeletal muscle ryanodine receptor mediated by FK506 binding protein. Biophys J. 1995;69:2398–2404. doi: 10.1016/S0006-3495(95)80109-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Fill M, Knudson M, Campbell KP, Coronado R. Ryanodine receptor is a gap junction-type channel. Science. 1988;242:99–102. doi: 10.1126/science.2459777. [DOI] [PubMed] [Google Scholar]
- Ma J, Zhao JY. Highly cooperative and hysteretic response of the skeletal muscle ryanodine receptor to changes in proton concentrations. Biophys J. 1994;67:626–633. doi: 10.1016/S0006-3495(94)80522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson PS, Campbell KP. The ryanodine receptor/Ca2+release channel. J Biol Chem. 1993;268:13765–13768. [PubMed] [Google Scholar]
- Meissner G. Ryanodine receptor/Ca2+release channels and their regulation by endogenous effectors. Annu Rev Physiol. 1994;56:485–508. doi: 10.1146/annurev.ph.56.030194.002413. [DOI] [PubMed] [Google Scholar]
- Meissner G, Rios E, Tripathy A, Pasek DA. Regulation of skeletal muscle Ca2+ release channel (ryanodine receptor) by Ca2+and monovalent cations and anions. J Biol Chem. 1997;272:1628–1638. doi: 10.1074/jbc.272.3.1628. [DOI] [PubMed] [Google Scholar]
- Nakai J, Dirsksen RT, Nguyen HT, Pessah IN, Beam KG, Allen PD. Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature. 1996;380:72–75. doi: 10.1038/380072a0. [DOI] [PubMed] [Google Scholar]
- Nakai J, Imagawa T, Hakamata M, Shigekawa H, Takeshima H, Numa S. Primary structure and functional expression from cDNA of the cardiac ryanodine receptor/calcium release channel. FEBS Lett. 1990;271:169–177. doi: 10.1016/0014-5793(90)80399-4. [DOI] [PubMed] [Google Scholar]
- Nakai J, Ogura T, Protasi F, Franzini-Armstrong C, Allen PD, Beam KG. Functional nonequality of the cardiac and skeletal ryanodine receptors. Proc Natl Acad Sci USA. 1997;94:1019–1022. doi: 10.1073/pnas.94.3.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsu K, Willard HF, Khanna VK, Zorzato F, Green NM, MacLennan DH. Molecular cloning of cDNA encoding the Ca2+release channel (ryanodine receptor) of rabbit cardiac muscle sarcoplasmic reticulum. J Biol Chem. 1990;265:13472–13483. [PubMed] [Google Scholar]
- Parker I, Callamaras N, Wier WG. A high-resolution, confocal laser scanning microscope and flash photolysis system for physiological studies. Cell Calcium. 1997;21:441–452. doi: 10.1016/s0143-4160(97)90055-5. [DOI] [PubMed] [Google Scholar]
- Penner R, Neher E, Takeshima H, Nishimura S, Numa S. Functional expression of the calcium release channel from skeletal muscle ryanodine receptor cDNA. FEBS Lett. 1989;259:217–221. doi: 10.1016/0014-5793(89)81532-7. [DOI] [PubMed] [Google Scholar]
- Rios E, Ma J, Gonzalez A. The mechanical hypothesis of excitation–contraction (E-C) coupling in skeletal muscle. J Muscle Res Cell Motil. 1991;12:127–135. doi: 10.1007/BF01774031. [DOI] [PubMed] [Google Scholar]
- Rousseau E, Ladine J, Liu QY, Meissner G. Activation of the Ca2+release channel of skeletal muscle sarcoplasmic reticulum by caffeine and related compounds. Arch Biochem Biophys. 1988;267:75–86. doi: 10.1016/0003-9861(88)90010-0. [DOI] [PubMed] [Google Scholar]
- Smith JS, Imagawa T, Ma J, Fill M, Campbell KP, Coronado R. Purified ryanodine receptor from rabbit skeletal muscle is the calcium-release channel of sarcoplasmic reticulum. J Gen Physiol. 1988;92:1–26. doi: 10.1085/jgp.92.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrentino V, Volpe P. Ryanodine receptors: how many, where and why? . Trends Pharmacol Sci. 1993;14:98–103. doi: 10.1016/0165-6147(93)90072-r. [DOI] [PubMed] [Google Scholar]
- Suda N, Franzius D, Fleig A, Nishimura S, Bodding M, Hoth M, Takeshima H, Penner R. Ca2+-induced Ca2+release in chinese hamster ovary (CHO) cells co-expressing dihydropyridine and ryanodine receptors. J Gen Physiol. 1997;109:619–631. doi: 10.1085/jgp.109.5.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutko JL, Airey JA. Ryanodine receptor Ca2+ release channels: does diversity in form equal diversity in function? . Physiol Rev. 1996;76:1027–1071. doi: 10.1152/physrev.1996.76.4.1027. [DOI] [PubMed] [Google Scholar]
- Takekura H, Takeshima H, Nishimura S, Takahashi M, Tanabe T, Flockerzi V, Hofmann F, Franzini-Armstrong C. Co-expression in CHO cells of two muscle proteins involved in excitation-contraction coupling. J Muscle Res Cell Motility. 1995;16:465–480. doi: 10.1007/BF00126431. [DOI] [PubMed] [Google Scholar]
- Takeshima H. Primary structure and expression from cDNAs of the ryanodine receptor. Ann NY Acad Sci. 1993;707:165–177. doi: 10.1111/j.1749-6632.1993.tb38051.x. [DOI] [PubMed] [Google Scholar]
- Takeshima H, Nishimura S, Matsumoto T, Ishida H, Kangawa K, Minamino N, Matsuo H, Ueda M, Hanaoka M, Hirose T, Numa S. Primary structure and expression from complementary DNA of skeletal muscle ryanodine receptor. Nature. 1989;339:439–445. doi: 10.1038/339439a0. [DOI] [PubMed] [Google Scholar]
- Takeshima H, Nishimura S, Nishi M, Ikeda M, Sugimoto T. A brain-specific transcript from the 3′-terminal region of the skeletal muscle ryanodine receptor region. FEBS Lett. 1993;322:105–110. doi: 10.1016/0014-5793(93)81547-d. [DOI] [PubMed] [Google Scholar]
- Tsugorka A, Rios E, Blatter LA. Imaging elementary events of calcium release in skeletal muscle cells. Science. 1995;269:1723–1726. doi: 10.1126/science.7569901. [DOI] [PubMed] [Google Scholar]
- Wagenknecht T, Grassucci R, Berkowitz J, Wiederrecht GJ, Xin H-B, Fleischer S. Cryoelectron microscopy resolves FK506-binding protein sites on the skeletal muscle ryanodine receptor. Biophys J. 1996;70:1709–1715. doi: 10.1016/S0006-3495(96)79733-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenknecht T, Grassucci R, Frank J, Saito A, Inui M, Fleischer S. Three dimensional architecture of the calcium channel/foot structure of sarcoplasmic reticulum. Nature. 1989;338:167–170. doi: 10.1038/338167a0. [DOI] [PubMed] [Google Scholar]
- Witcher DR, McPherson PS, Kahl SD, Lewis T, Bentley P, Mullinix MJ, Windass JD, Campbell KP. Photoaffinity labeling of the ryanodine receptor/Ca2+release channel with an azido derivative of ryanodine. J Biol Chem. 1994;269:13076–13079. [PubMed] [Google Scholar]
- Yamazawa T, Takeshima H, Sakurai T, Endo M, Iino M. Subtype specificity of the ryanodine receptor for Ca2+signal amplification in excitation-contraction coupling. EMBO (Eur Mol Biol Organ) J. 1996;15:6172–6177. [PMC free article] [PubMed] [Google Scholar]
- Yamazawa T, Takeshima H, Shimuta M, Iino M. A region of the ryanodine receptor critical for excitation-contraction coupling in skeletal muscle. J Biol Chem. 1997;272:8161–8164. doi: 10.1074/jbc.272.13.8161. [DOI] [PubMed] [Google Scholar]
- Yue DT. Quenching the spark in the heart. Science. 1997;276:755–756. doi: 10.1126/science.276.5313.755. [DOI] [PubMed] [Google Scholar]
- Zorzato F, Fujii J, Otsu K, Green NM, Lai FA, Meissner G, MacLennan DH. Molecular cloning of cDNA encoding human and rabbit forms of the Ca2+release channel (ryanodine receptor) of skeletal muscle sarcoplasmic reticulum. J Biol Chem. 1990;265:2244–2256. [PubMed] [Google Scholar]
- Zucchi R, Ronca-Testoni S. The sarcoplasmic reticulum Ca2+channel/ryanodine receptor: modulation by endogenous effectors, drugs and disease states. Pharmacol Rev. 1997;49:1–51. [PubMed] [Google Scholar]