

Abstract
Protein kinase C inhibits inactivation gating of Kv3.4 K+ channels, and at least two NH2-terminal serines (S15 and S21) appeared involved in this interaction (Covarrubias et al. 1994. Neuron. 13:1403–1412). Here we have investigated the molecular mechanism of this regulatory process. Site-directed mutagenesis (serine → alanine) revealed two additional sites at S8 and S9. The mutation S9A inhibited the action of PKC by ∼85%, whereas S8A, S15A, and S21A exhibited smaller reductions (41, 35, and 50%, respectively). In spite of the relatively large effects of individual S → A mutations, simultaneous mutation of the four sites was necessary to completely abolish inhibition of inactivation by PKC. Accordingly, a peptide corresponding to the inactivation domain of Kv3.4 was phosphorylated by specific PKC isoforms, but the mutant peptide (S[8,9,15,21]A) was not. Substitutions of negatively charged aspartate (D) for serine at positions 8, 9, 15, and 21 closely mimicked the effect of phosphorylation on channel inactivation. S → D mutations slowed the rate of inactivation and accelerated the rate of recovery from inactivation. Thus, the negative charge of the phosphoserines is an important incentive to inhibit inactivation. Consistent with this interpretation, the effects of S8D and S8E (E = Glu) were very similar, yet S8N (N = Asn) had little effect on the onset of inactivation but accelerated the recovery from inactivation. Interestingly, the effects of single S → D mutations were unequal and the effects of combined mutations were greater than expected assuming a simple additive effect of the free energies that the single mutations contribute to impair inactivation. These observations demonstrate that the inactivation particle of Kv3.4 does not behave as a point charge and suggest that the NH2-terminal phosphoserines interact in a cooperative manner to disrupt inactivation. Inspection of the tertiary structure of the inactivation domain of Kv3.4 revealed the topography of the phosphorylation sites and possible interactions that can explain the action of PKC on inactivation gating.
Keywords: potassium channels, protein phosphorylation, inactivation gating, site-directed mutagenesis, structure–function relation
introduction
A-type K+ channels activate, open, and inactivate in response to membrane depolarization. In excitable tissues, inactivation of these channels helps to control the frequency of repetitive spike firing, to shape the action potential (Hille, 1992) and to regulate the amplitude of back-propagating action potentials (Hoffman et al., 1997). There are two well-characterized mechanisms of inactivation of voltage-gated K+ channels: N- and C-type. The N-type mechanism is determined by an amphipathic region at the NH2 terminus of the channel protein, which acts as an internal blocking particle that occludes the inner mouth of the pore (Hoshi et al., 1990; Demo and Yellen, 1991; Ruppersberg et al., 1991; Murrell-Lagnado and Aldrich, 1993a , 1993b ; Baukrowitz and Yellen, 1995; Gomez-Lagunas and Armstrong, 1995). The C-type mechanism is determined by residues that contribute to the K+-selective pore and is thought to involve cooperative intersubunit interactions that alter the structure of the outer mouth of the channel (Hoshi et al., 1991; Lopez-Barneo et al., 1993; Baukrowitz and Yellen, 1995; Ogielska et al., 1995; Panyi et al., 1995; Starkus et al., 1997).
Inactivation of voltage-gated K+ channels may be modulated by various physiological factors (e.g., protein kinases, auxiliary subunits, redox potential, and external K+). Phosphorylation of these channels by protein kinases is of importance because this post-translational modification appears to be associated with physiological processes that underlie synaptic plasticity (Levitan, 1994; Kandel et al., 1995; Jonas and Kaczmarek, 1996). Several reports have shown that protein kinases regulate N-type inactivation of A-type K+ channels by phosphorylating specific amino acids (Covarrubias et al., 1994; Drain et al., 1994; Roeper et al., 1997). However, the molecular mechanisms of action are not yet known. PKA-dependent phosphorylation appears to be necessary for rapid N-type inactivation of Drosophila ShakerD K+ channels (Drain et al., 1994). Two serines (S507 and S508) located at the cytoplasmic COOH-terminal domain of ShakerD are involved in this process. Thus, it was proposed that PKA indirectly modulates N-type inactivation by phosphorylation of regulatory sites at the COOH terminus of ShakerD. CaMK II (Ca++/calmodulin-dependent protein kinase) slows inactivation of Kv1.4 by phosphorylation of S123 (Roeper et al., 1997). This residue is located downstream of the inactivation domain at the NH2 terminus. Roeper et al. (1997) postulated that phosphorylation of S123 may slow inactivation either by reducing the flexibility of the “chain” that links the NH2-terminal inactivation particle with the core of the channel, or by interfering with the binding of the inactivation particle to its receptor at the inner mouth of the channel. Another study reported that C-type inactivation of Kv1.3 is modulated at three putative phosphorylation sites (Kupper et al., 1995) but the mechanism of action was not examined. Other aspects of voltage-dependent gating and expression of K+ channels are modulated by protein kinases (Perozo and Bezanilla, 1991; Jonas and Kaczmareck, 1996). There also, little is known about the underlying molecular mechanisms.
We have previously shown that PKC eliminates N-type inactivation of an A-type K+ channel encoded by Kv3.4 (Covarrubias et al., 1994). This study demonstrated that mutation of two serines (S15A, S21A and S[15,21]A) partially inhibited the effect of PKC on N-type inactivation. However, these mutations were not capable of explaining the totality of the PKC effect. In the present study, we demonstrate that there are two additional sites (S8 and S9) and that the four serines (S8, S9, S15, and S21) are sufficient to account for the PKC-mediated inhibition of inactivation, but each site does not contribute equally to this action. Another goal of this study was to gain insights into the mechanism of PKC action. Because the net positive charge of the inactivation particle of Shaker K+ channels is known to be critical to achieve rapid inactivation (Murrell-Lagnado and Aldrich, 1993a ), the NH2-terminal phosphoserines could simply neutralize the overall positive charge and consequently slow inactivation. In agreement with this possibility, our previous study (Covarrubias et al., 1994) showed that substitution of aspartate (D) for serine at position 15 produced a partial inhibition of inactivation. Here, we have analyzed all possible S → D substitutions at positions 8, 9, 15, and 21 to address two questions. (a) Are all phosphorylation sites functionally equivalent? By mutating one residue at a time or in various combinations, we have tested whether the net charge of the inactivation particle is the sole determinant of the rate of inactivation. (b) Do the phosphorylation sites act independently? Because of the close proximity of the phosphorylation sites, it is conceivable that interactions between these sites are involved in the mechanism of PKC action. By examining the free energy that single S → D mutations contribute to disrupt inactivation, we have determined whether the effects of combined S → D mutations are the result of simple additivity. Finally, aided by the nuclear magnetic resonance–based structure of the inactivation particle of Kv3.4 (Antz et al., 1997), we developed a plausible working hypothesis that can explain the structural changes that the inactivation gate may undergo upon phosphorylation by PKC. This hypothesis represents one of the first opportunities to relate the actual three-dimensional structure of a voltage-gated K+ channel to a particular function and its regulation. Preliminary results were previously reported in abstract form (Covarrubias et al., 1997).
materials and methods
Mutagenesis
A cDNA encoding Kv3.4 was maintained as described previously (Covarrubias et al., 1994). S → A substitutions (except S[8,9,15]A and S[8,9,21]A) were created as described before using the Altered Sites II in vitro Mutagenesis System (Promega Corp., Madison, WI) or an overlap polymerase chain reaction strategy (Covarrubias et al., 1994). All other amino acid substitutions were made using the QuickChange Site-Directed Mutagenesis Kit as described by the manufacturer (Stratagene Inc., La Jolla, CA). Briefly, complementary pairs of oligonucleotides (22mer–29mer) containing the appropriate nucleotide substitutions were prepared at the Nucleic Acid Facility, Kimmel Cancer Center (Thomas Jefferson University, Philadelphia, PA). These oligonucleotides were subsequently used as primers for the complete synthesis of both strands of the Kv3.4 plasmid. We used 20 ng of Kv3.4 plasmid as template, 5 U Pfu DNA polymerase (Stratagene Inc.), primers, and free nucleotides in a total volume of 100 μl. After strand synthesis (12–18 cycles), 10 U of DpnI were added to the reaction mixture to digest the original Kv3.4 methylated plasmid template (37°C, 1–2 h). The restriction endonuclease was heat inactivated (65°C, 15 min), and the mixture used to transform DH5α cells by electroporation. Base substitutions were confirmed by automated DNA sequencing at the Nucleic Acid Facility, Kimmel Cancer Center. It should be noted that QuickChange does not involve a polymerase chain reaction. Pfu DNA polymerase (containing 3′ → 5′ exonuclease activity or proofreading activity) simply catalyzes the extension step of the mutagenesis reaction replicating the template with a mutagenic primer. Nevertheless, to confirm that base misincorporations were unlikely under our reaction conditions, we read 105 DNA sequences (∼500 bp, each) created by QuickChange. Approximately 45 of these sequences correspond to the region that surrounds the S4–S5 loop of three distinct K+ channels (Kv3.4, Kv4.1, and dShaw); the rest correspond to the NH2-terminal region of Kv3.4 (58) or the region surrounding the S6 region of Kv4.1 (2). This analysis did not reveal nucleotide errors introduced by Pfu DNA polymerase activity. In addition, the mutants characterized here did not exhibit any unexpected properties, and subcloning of some mutated sequences (S8D and S[8,15,21]D) back into the wild-type cDNA did not result in different phenotypes. Various studies have investigated the fidelity of this enzyme and other thermostable DNA polymerases (Kunkel, 1988; Lundberg, et al., 1991; Flaman et al., 1994; Cline et al., 1996). They found that Pfu DNA polymerase yields the highest fidelity with an error rate {[mutation frequency]/([base pairs] [effective duplication])} on the order of 1–2 × 10−6. This is at least 10× better than Taq polymerase. By applying this formula, we predicted a mutation frequency of ∼2%. Assuming that all sequences are equally vulnerable to errors and that each analyzed sequence is an independent trial, we expected at least two sequences containing one undesired mutation. Thus, it appears that under our assay conditions, which do not involve PCR, the mutation frequency is overestimated. cRNA for microinjection was produced as described elsewhere (Jerng and Covarrubias, 1997).
Microinjection of Xenopus Oocytes and Electrophysiological Recording
Wild-type and mutant Kv3.4 cRNA was microinjected into defolliculated Xenopus oocytes (∼50 ng/cell) using a Nanoject microinjector (Drummond, Broomall, PA). Whole-oocyte currents were recorded 2–10 d after injection using the two-microelectrode voltage-clamp technique (TEV-200; Dagan Corp., Minneapolis, MN). Microelectrodes were filled with 3 M KCl (tip resistance was <1 MΩ). Bath solution contained: 96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, pH 7.4, adjusted with NaOH. Phorbol 12-myristate-13-acetate (PMA)1 was purchased from Sigma Chemical Co. (St. Louis, MO). Current traces were digitized at 250–500 μs/point after low-pass filtering at 1–2 kHz. The average voltage offset recorded at the end of an experiment was generally small (0.5 ± 2.4 mV, n = 107) and was subtracted from the command voltage when analyzing prepulse inactivation curves. The leak current was subtracted off-line assuming ohmic leak. Capacitive currents were subtracted on-line using a P/4 protocol or off-line using a scaled noise-free template generated from a current trace with no active time-dependent currents (elicited by a depolarization to −80 mV). Experiments were conducted at 23°C using a temperature-controlled microscope stage (PDMI-2; Medical Systems Corp., Greenvale, NY).
Data Acquisition and Analysis
Voltage-clamp protocols and the acquisition of data were controlled by a 486 desktop computer interfaced to a 12-bit A/D converter (Digidata 1200 using pClamp 6.0; Axon Instruments, Foster City, CA). Data analysis was conducted using Clampfit (pClamp 6, Axon Instruments), Sigmaplot (Jandel Scientific, San Rafael, CA), or Origin 4.1 (Microcal, Northampton, MA). The slow inactivation that remained after PMA application exhibited complex and variable kinetics (Covarrubias et al., 1994; see Fig. 1 A). This is probably the result of variable degrees of phosphorylation of multiple serines in four subunits of the channel tetramer. Therefore, to simplify the comparative analysis of wild-type and S → A mutant channels, the normalized current integral was computed (current integral/peak current) before and after PMA. In all cases, this analysis included currents elicited by 900-ms step depolarizations to +50 mV. To analyze inactivation of the S → D mutants, the decaying relaxation of the currents was described assuming a sum of two exponential terms as described previously (Jerng and Covarrubias, 1997). Because the S → D mutations studied here mainly affected the time constant of the fast component and the relative weight of this component correlated with changes in the steady state level of the current (see results), we used the following relation to estimate the rate of inactivation (k i): k i = (1 − I s)/τi, where I s is the sum of the relative weights of the slow and steady state components of the current and τi is the time constant of fast inactivation (Murrell-Lagnado and Aldrich, 1993a ; Panyi et al., 1995). According to the theory of absolute reaction rates (Gutfreund, 1995), the following relation was used to estimate the free energy change that a particular mutation contributes to inhibition of inactivation: ΔG obs = −RT ln(k i h/k B T), where R, T, h, and k B have their usual meaning (Gutfreund, 1995). Then, relative to wild type, the free energy that a single mutation contributes to inhibition of inactivation is: ΔΔG obs = ΔG obs(wild type) − ΔG obs(mutant).
Figure 1.

The effect of NH2-terminal S → A mutations on elimination of rapid inactivation induced by PMA. (A) Wild-type whole-oocyte outward currents recorded in the absence and presence of 20 nM PMA. Currents were evoked by a 900-ms step depolarization to +50 mV from a holding potential of −100 mV. (B and C) Mutant whole-oocyte outward currents recorded in the absence and presence of 20 nM PMA. Currents were evoked as described for A. The solid horizontal line indicates the zero current level. (D) Bar graph representing the normalized current integral (current integral/ peak current) from wild type and several mutants before and after application of 20 nM PMA. Current integral excludes the first millisecond of the trace to eliminate the capacitive current from the calculation. Values are expressed as mean ± SD. The number of independent experiments is indicated by the figures accompanying the bars in the upper part of D. Values for S15A, S21A, and S[15,21]A were extracted from an earlier study (Covarrubias et al., 1994).
If the effect of multiple mutations results from the additive effect of single mutations, the predicted free energy change that a multiple mutation contributes to inhibition of inactivation is: ΔΔG(multiple mutation) = ΔΔG(single mutation 1) + ΔΔG (single mutation 2) + ...; therefore, assuming additivity, the predicted inactivation rate (k pred) associated with a multiple mutation is k pred = (k B T/h) exp(−ΔG pred/RT), where ΔG pred is the predicted free energy change [ΔG pred = ΔG obs(wild type) + ΔΔG(multiple mutation)]. A deviation from this prediction suggests that the mutations do not act independently. A similar procedure was used to examine the free energy that mutations contribute to destabilization of the inactivated state of the channel. In this case, we assumed that the observed rate of recovery from inactivation (k r) = 1/(fast time constant of recovery from inactivation). Although the time course of recovery from inactivation of wild-type and some mutant channels was best described assuming the sum of two exponential terms (the fast component contributed to >65% of the time course; see results), a simple exponential rise was sufficient for several mutants that exhibited faster recoveries from inactivation. For K+ channels that exhibit N-type inactivation, the fast phase of the recovery from inactivation is associated with the exit of the inactivation particle from the pore (Demo and Yellen, 1991).
Synthetic Peptides and Phosphorylation In Vitro
The wild-type peptide corresponding to the first 28 amino acids of Kv3.4 was provided by Dr. R.W. Aldrich (Stanford University, Stanford, CA) and handled as described before (Covarrubias et al., 1994). The mutant peptide S[8,9,15,21]A was purchased from Multiple Peptide Systems (San Diego, CA). All peptides were purified (>95%) by reverse phase chromatography. The peptides were applied to a C8 column attached to the FPLC (Pharmacia LKB Biotechnology Inc., Piscataway, NJ) and the adsorbed material was eluted with a linear acetonitrile gradient (0–30%) in 0.1% trifluoroacetic acid. Recombinant rat PKC isoforms (α, β, and γ) were provided by Dr. C.D. Stubbs (Jefferson Medical College). Phosphorylation in vitro was carried out using a filter assay as described previously (Slater et al., 1993). To evaluate whether a peptide can act as a substrate of PKC, this assay was designed to measure the initial rates of phosphate incorporation. The functionality of the isozymes was always confirmed using the myelin-basic-protein as the control substrate. Phosphorylation data are expressed as molar fraction of phosphorylated peptide in the presence of diacylglycerol.
results
Inhibition of Inactivation by PKC Is Associated with Phosphorylation of Four NH2-Terminal Serines
The first 28 amino acids at the NH2 terminus of the Kv3.4 protein constitute its inactivation gate (Ruppersberg et al., 1991; Rettig et al., 1992; Covarrubias et al., 1994). This sequence includes four putative PKC phosphorylation sites at positions 8, 9, 15, and 21 (underlined; MISSVCVSSYRGRKSGNKPPSKTCLKEE). Previously, we showed that S15 and S21 are important contributors to the action of PKC on N-type inactivation, but are not sufficient to explain the totality of the inhibition of inactivation by PKC (Covarrubias et al., 1994). To identify the remaining residues and further investigate their contribution, we examined a series of new S → A substitutions affecting of S8, S9, S15, and S21 (see also subsequent section). Oocytes expressing the wild-type and mutant currents were exposed to a phorbol ester (PMA, 20 nM) to activate PKC. In sharp contrast to the wild-type currents that exhibit a dramatic inhibition of inactivation induced by PKC activation, currents expressed by the quadruple mutant (S[8,9,15,21]A) were not changed in the presence of PMA (Fig. 1, A and C). Thus, S8, S9, S15, and S21 are the most likely phosphorylation sites associated with inhibition of inactivation by PKC.
To verify this result, we conducted in vitro phosphorylation assays using peptides that correspond to the first 28 amino acids of Kv3.4 (the NH2-terminal inactivation domain of the channel) as substrates. We found that three PKC isoforms (α, β, and γ) phosphorylated the Kv3.4 peptide but, as expected, did not significantly phosphorylate the quadruple mutant peptide S[8,9,15,21]A (Fig. 2). The small signal observed with S[8,9,15,21]A corresponds to unspecific phosphate incorporation observed with unrelated peptides that have no phosphate acceptors (e.g., the ShakerB inactivation domain; data not shown). These results confirmed that the NH2-terminal inactivation domain of Kv3.4 is phosphorylated by PKC and that this modification is associated with elimination of N-type inactivation by PKC in the intact channel.
Figure 2.

Phosphorylation of Kv3.4 NH2-terminal peptides by three PKC isoforms. Diacylglycerol-stimulated phosphorylation of the wild-type peptide and the mutant peptide S[8,9,15,21]A by PKCα, and PKCβ, and PKCγ (materials and methods). Values are expressed as the mean ± SD (n = 3).
Evidence of Nonequivalent and Nonindependent Phosphorylation Sites at the NH2 Terminus of Kv3.4
In the previous section, we showed that PKC-mediated inhibition of inactivation involves multiple phosphorylation sites. Thus, to understand the mechanism that disrupts inactivation, it is necessary to know: (a) whether the phosphorylation sites are functionally equivalent, and (b) whether the phosphorylation sites act independently. To investigate these questions, we examined the effect of PKC activation on single, double, and triple S → A mutants. The current integral (see materials and methods) was computed before and after application of 20 nM PMA. In the absence of PMA, the current integrals exhibited little or no difference between the wild-type and mutant channels (Fig. 1 D, top). Note that the time courses of the currents were similar (e.g., Fig. 1, A–C). By contrast, in the presence of PMA, the current integrals of the mutants differed significantly from that of the wild type (Fig. 1 D, bottom). S9A and S[8,9]A exhibited a greater reduction in their responses to PMA (13 and 10% of the control response, respectively) than S8A, S15A, S21A, and S[15,21]A (59, 64, 50, and 39% of the control response, respectively). Thus, the presence of S9 appears to be more critical than that of S8, S15, and S21. Moreover, the results in Fig. 1 D also showed that the sites do not act independently. Addition of the remaining current integrals of the mutants in the presence of PMA did not account for the results observed with wild-type channels. For instance, the sum of the remaining current integrals from S[8,9]A and S[15,21]A in the presence of PMA was only ∼50% of the normalized current integral from wild type. If the sites were independent, this result would be unexpected because phosphorylation of S8, S9, S15, and S21 accounts for the effect of PMA on current inactivation (Fig. 1, A and C). In addition, the apparent contribution of S15 and S21 to the total effect of PMA on inactivation is considerable (∼50%) when these residues were mutated to alanine (individually or in combination; Fig. 1 D). Yet only ∼10% of the response to PMA remained when both S15 and S21 are available (see S[8,9]A). Even lower responses were observed when S15 or S21 were the only available sites (see S[8,9,21]A and S[8,9,15])A, respectively). Although the pooled data (from different batches of oocytes) did not reveal a statistically significant difference between S[8,9,15,21]A and S[8,9,21]A (P ≤ 0.5), this mutant channel exhibited a small but consistent disruption of inactivation in the presence of PMA (Fig. 1 B). Therefore, the sole contribution of S15 and S21 to the effect of PKC on inactivation is small (when the other sites are not available). Later experiments will confirm this observation more directly. Overall, these results suggest that the ability of a phosphorylated site (or set of sites) to disrupt inactivation depends on whether other sites are phosphorylated. Thus, interactions between NH2-terminal phosphoserines play a critical role in determining the disruption of inactivation by PKC.
Serine to Aspartate Mutations at Positions 8, 9, 15, and 21 Mimic the Effect of PKC on K+ Channel Inactivation
To further investigate the interactions described above and how they may disrupt inactivation of Kv3.4, we studied the biophysical properties of serine → aspartate (S → D) mutants. Like phosphorylation, the S → D mutations incorporate negatively charged side chains. Although there is no sequence similarity between the NH2-terminal regions of ShakerB and Kv3.4, both share a net positive charge within the first 25 amino acids (+2 and +5, respectively). Furthermore, positive charges are clearly clustered in the sequence. Previous studies with ShakerB K+ channels have demonstrated that the net positive charge of the inactivation domain determines long-range electrostatic interactions that facilitate the diffusion of the inactivation domain to the inner mouth of the pore (Murrell-Lagnado and Aldrich, 1993a ). If the inactivation domain in Kv3.4 behaves as a point charge, phosphorylation could impair inactivation by simply reducing or reversing the net positive charge of the inactivation domain (from +5 to −3, if all sites are phosphorylated). To test this hypothesis, we created S → D mutations affecting positions 8, 9, 15, and 21 individually and in all possible combinations. We found that the currents mediated by these mutant channels (in the absence of PMA) mimic the action of PKC on K+ channel inactivation (Figs. 3 and 4). For example, wild-type currents recorded in the presence of PMA resemble the currents expressed by S[8,9,15,21]D. Similarly, S[8,9]A currents in the presence of PMA (S15 and S21 are available for phosphorylation) closely resemble currents expressed by S[15,21]D. The similarity was not as striking when comparing S[15,21]A currents in the presence of PMA (S8 and S9 are available for phosphorylation) and currents expressed by S[8,9]D. This was mainly due to the larger sustained current of S[8,9]D. Such a difference may have occurred because: (a) aspartates at 8 and 9 may destabilize the inactivated state to a greater extent than phosphoserines at the same positions; (b) S[15,21]A per se reduces the ability of phosphoserines at 8 and 9 to disrupt inactivation; and (c) with S[8,9]D, all channels are modified, whereas phosphorylation of S[15,21]A may be limited by the degree of PKC activation by PMA. In spite of this finding, S[15,21]A currents in the presence of PMA exhibited intermediate kinetics between that of wild-type and S[8,9]A channels recorded in the presence of PMA, and S[8,9]D currents exhibited intermediate kinetics between that of S[8,9,15,21]D and S[15,21]D currents (Fig. 3). These observations demonstrated that S → D mutations reproduce a phenotype that is reasonably equivalent to that of constitutively phosphorylated channels and that the presence of negative charges at positions 8, 9, 15, and 21 may be important to inhibit N-type inactivation.
Figure 3.

Comparison of PKC action and the effect of NH2-terminal S → D mutations on Kv3.4 inactivation. From a holding potential of −100 mV, 900-ms depolarizing pulses to +50 mV evoked all the currents. The solid horizontal line indicates the zero current level. (top) Currents expressed by wild type, S[15,21]A, and S[8,9]A in the absence and presence of 20 nM PMA (see Fig. 1). Currents are shown without normalization. (bottom) Currents expressed by S → D mutants (S[8,9,15,21)D, S[8,9]D, and S[15,21]D) and their corresponding wild type (no PMA was applied). For comparison, wild-type and S → D mutant peak currents are shown normalized.
Figure 4.
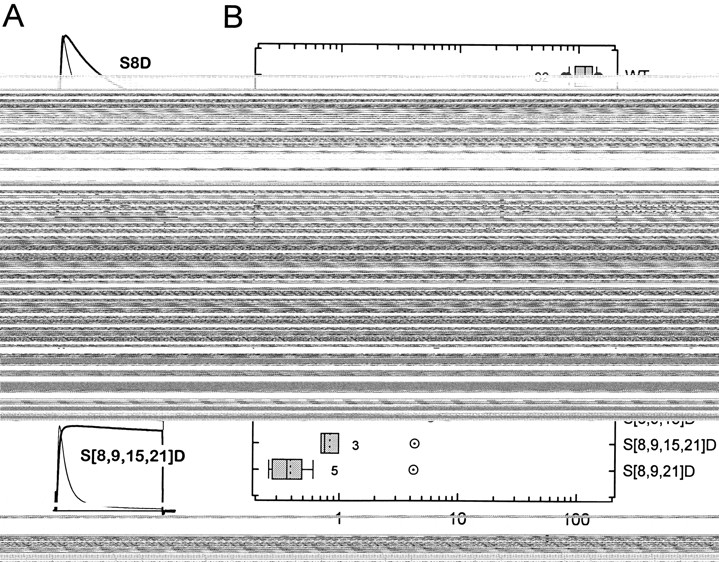
The rates of rapid inactivation of wild-type (WT) Kv3.4 and NH2-terminal S → D mutants. (A) Whole-oocyte currents evoked by 112-ms depolarizing pulses to +50 mV from a holding potential of −100 mV. Capacitive transients were subtracted using a scaled smooth template with no active currents (materials and methods). Each pair compares a mutant (thick trace) current and its corresponding wild type from the same batch of oocytes (thin trace). The solid horizontal line indicates the zero current level. Rates of rapid inactivation were determined as described in materials and methods. For mutant channels that exhibited very slow inactivation, long pulses (900 ms) were used to estimate the time constants of current decay. (B) Tukey box plot summarizing the data from wild type (WT) and mutants. Data were ordered from fast (top) to slow (bottom). Left and right borders of a box represent the 25th and 75th percentiles, respectively. Solid and dashed lines in the box correspond to the 50th percentile and the mean value, respectively. Left and right whiskers represent the 10th and 90th percentiles, respectively. Outliers are shown as •. The number of oocytes examined is indicated by the figure accompanying the boxes. Four oocytes expressing S[8,9,15]D were also examined. However, the time constants of inactivation from this mutant were not determined because it exhibited no inactivation in response to 900-ms pulses to positive membrane potentials. Open symbols with a dot represent the predicted rate of inactivation for multiple mutants assuming a simple additive effect of single mutations as described in materials and methods.
Serine to Aspartate Mutations at Positions 8, 9, 15, and 21 Slow Inactivation and Destabilize the Inactivated State in a Nonadditive Manner
Single S → D mutants exhibited different degrees of impaired inactivation and, as shown above, a quadruple mutation (S[8,9,15,21]D) was necessary to closely mimic the complete effect of PKC (Fig. 4 A). To examine the kinetics of inactivation of the S → D mutants, the decaying relaxation of the currents was described assuming the sum of two exponential terms. Analysis of the wild-type currents gave the following best-fit parameters at +50 mV: τf = 7.5 ± 1.4 ms, τs = 35 ± 9.6 ms, Wf = 0.82 ± 0.05, Ws = 0.14 ± 0.04, and Wss = 0.03 ± 0.02 (mean ± SD, n = 32; τf and τs are the fast and slow time constants, respectively; Wf and Ws are the corresponding relative weights of the fast and slow components of current decay, respectively; and Wss is the relative weight of the steady state level of the current). Notice that the fast component dominates the kinetics of current decay (>80%) and that inactivation is nearly complete (>95%). S → D mutations slowed both τf and τs. However, while Wf was greatly reduced (from 0.84 to 0.02 at the extremes between wild type and S[8,9,21]D), Ws stayed relatively constant (the largest changes were 0.15 and 0.1 above and below the wild-type value, respectively). Parallel to a decrease in Wf, there was an increase in Wss (0.03 and 0.97 at the extremes between wild type and S[8,9,21]D). This result suggested that the inactivated state of the channel has been destabilized by the mutations (see below). We focused the analysis on the fast component of current decay because it was more sensitive to the mutations, and changes that affect the relative weight of this component correlated with changes in the steady state level of the current. First, we estimated the rate constants of inactivation from the decay of the macroscopic currents (materials and methods; Table I). These values were sorted by size and plotted as a Tukey box plot (Tukey, 1977). Then, to test whether the simple additive effect of the single mutants can account for the effects observed with multiple mutants, we calculated the free energies that the mutations contribute to impair inactivation, and plotted the predicted and observed rate constants (materials and methods; Fig. 4 B; Table I). Clearly, the extent of inhibition of inactivation in the single mutants is not equivalent (S8D > S9D > S15D > S21D) and the predicted rates differed significantly from the observed ones. Thus, as suggested earlier, NH2-terminal phosphoserines that regulate N-type inactivation in Kv3.4 K+ channels are not functionally equivalent and interact favorably to eliminate N-type inactivation in Kv3.4. For instance, S21D alone caused no effect on inactivation, but enhanced the effects of S8D or S9D when combined in the double mutants S[8,21]D and S[9,21]D. Similarly, inhibition of inactivation by S[8,15]D and S[9,15]D was enhanced by the presence of S21D in triple mutants S[8,15,21]D and S[9,15,21]D.
Table I.
Inactivation Properties of Wild-Type and Mutant Kv3.4 K+ Channels
| k i | ΔG i | ΔΔG i | k r | ΔG r | ΔΔG r | V0.5 | Slope | INI | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| s−1 | kcal/mol | kcal/mol | s−1 | kcal/mol | kcal/mol | mV | mV/e-fold | |||||||||||
| WT | 111.5 ± 21 | 14.57 | — | 2.4 ± 1.3 | 16.89 | — | −32.5 ± 4.3 | −4.8 ± 0.3 | 0.02 ± 0.02 | |||||||||
| (33) | (23) | (30) | ||||||||||||||||
| S8D | 18.4 ± 2.3 | 15.62 | 1.07 | 6.9 ± 0.5 | 16.19 | 0.65 | −22.2 ± 0.8 | −5 ± 0.2 | 0.1 ± 0.006 | |||||||||
| (4) | (4) | (4) | ||||||||||||||||
| S9D | 35.6 ± 4.6 | 15.22 | 0.68 | 2.7 ± 0.2 | 16.75 | 0.1 | −24.6 ± 2.1 | −5.6 ± 0.3 | 0.02 ± 0.01 | |||||||||
| (10) | (7) | (7) | ||||||||||||||||
| S15D | 82.7 ± 2.8 | 14.73 | 0.18 | 4.4 ± 0.2 | 16.46 | 0.38 | −31.6 ± 3.1 | −4.8 ± 0.1 | 0.02 ± 0.005 | |||||||||
| (4) | (5) | (4) | ||||||||||||||||
| S21D | 115.1 ± 9 | 14.54 | 0.01 | 2.5 ± 0.1 | 16.79 | 0.06 | −31.6 ± 2 | −4.7 ± 0.2 | 0.03 ± 0.02 | |||||||||
| (7) | (7) | (7) | ||||||||||||||||
| S[8,9]D | 2.0 ± 0.4 | 16.92 | 2.37 | 13.8 ± 3.2 | 15.79 | 1.06 | ND | ND | ND | |||||||||
| (6) | (4) | (5) | ||||||||||||||||
| S[8,15]D | 8.4 ± 0.6 | 16.08 | 1.53 | 15.2 ± 0.5 | 15.73 | 1.12 | −15 ± 1.1 | −5.8 ± 0.2 | 0.29 ± 0.02 | |||||||||
| (4) | (4) | (4) | ||||||||||||||||
| S[9,15]D | 11.2 ± 0.3 | 15.91 | 1.36 | 6.23 ± 0.5 | 16.25 | 0.59 | −13 ± 1.4 | −5.6 ± 0.3 | 0.13 ± 0.003 | |||||||||
| (3) | (4) | (4) | ||||||||||||||||
| S[8,21]D | 18.2 ± 0.5 | 15.81 | 1.26 | 11.5 ± 1.8 | 15.89 | 0.95 | −18.4 ± 2 | −5.3 ± 0.2 | 0.18 ± 0.02 | |||||||||
| (4) | (4) | (4) | ||||||||||||||||
| S[9,21]D | 22.8 ± 1 | 15.49 | 0.94 | 4.3 ± 0.3 | 16.47 | 0.38 | −20.3 ± 0.9 | −5.2 ± 0.1 | 0.04 ± 0.003 | |||||||||
| (4) | (3) | (3) | ||||||||||||||||
| S[15,21]D | 60.3 ± 5 | 14.92 | 0.37 | 6.6 ± 0.6 | 16.22 | 0.62 | −24.3 ± 1.4 | −4.8 ± 0.2 | 0.03 ± 0.007 | |||||||||
| (9) | (4) | (8) | ||||||||||||||||
| S[8,9,15]D | ND | ND | ND | ND | ND | ND | ND | ND | ND | |||||||||
| (5) | (5) | (5) | ||||||||||||||||
| S[8,9,21]D | 0.4 ± 0.1 | 17.9 | 3.35 | ND | ND | ND | ND | ND | ND | |||||||||
| (5) | (6) | (6) | ||||||||||||||||
| S[8,15,21]D | 2.6 ± 0.2 | 16.77 | 2.22 | 16.1 ± 2.8 | 15.69 | 1.15 | ND | ND | ND | |||||||||
| (4) | (3) | (5) | ||||||||||||||||
| S[9,15,21]D | 6.5 ± 0.4 | 16.22 | 1.68 | 8.2 ± 0.5 | 16.1 | 0.75 | ND | ND | ND | |||||||||
| (4) | (4) | (4) | ||||||||||||||||
| S[8,9,15,21]D | 0.8 ± 0.2 | 17.44 | 2.89 | ND | ND | ND | ND | ND | ND | |||||||||
| (3) | (4) | (7) | ||||||||||||||||
| S8N | 84.3 ± 4.1 | 14.72 | 0.16 | 4.9 ± 0.3 | 16.39 | 0.5 | −28.7 ± 0.8 | −5.1 ± 0.2 | 0.01 ± 0.005 | |||||||||
| (4) | (4) | (4) | ||||||||||||||||
| S8E | 17.4 ± 0.9 | 15.65 | 1.09 | 10.2 ± 0.5 | 15.97 | 0.93 | −13.3 ± 1.2 | −6.7 ± 0.1 | 0.1 ± 0.01 | |||||||||
| (4) | (4) | (3) |
The rate constants of inactivation (k i) and recovery from inactivation (k r) and the associated free energy changes (ΔG i, ΔΔG i, ΔG r, and ΔΔG r) were calculated as described under Experimental Procedures. The best-fit parameters of the prepulse inactivation curve (V0.5, Slope, and INI) were obtained assuming a Boltzmann distribution. V0.5 and INI represent the midpoint potential and the noninactivating fraction, respectively. The values were expressed as mean ± SD. The number of oocytes examined is indicated in parenthesis. ND indicates that the experiments were not analyzed quantitatively as explained in the text and legends of Figs. 5–7.
Single S → D mutants also exhibited different degrees of accelerated recovery from inactivation at −100 mV (S8D > S9D ≅ S15D > S21D; Fig. 5 A), and the multiple mutations caused greater acceleration of recovery from inactivation than expected from the corresponding single mutations (see below). Kinetic analysis revealed that recovery from inactivation of wild-type Kv3.4 channels was best described assuming the sum of two exponential terms. The fast and slow time constants were 538 ± 198 and 3,304 ± 960 ms, respectively, and their corresponding relative weights were 0.66 ± 0.04 and 0.30 ± 0.04 (mean ± SD, n = 21). S → D mutations decreased both time constants and, in all cases, the weight of the fast component dominated the time course of recovery from inactivation (>65%). In fact, recoveries from inactivation for S[8,9]D, S[8,15]D, S[8,21]D, S[8,15,21]D, and S[9,15,21]D (those channels that exhibited the fastest recoveries) were well described assuming a simple exponential rise. The estimated rates of recovery from inactivation (materials and methods) are summarized in Fig. 5 B and Table I. The recoveries from inactivation of other triple mutants and the quadruple mutant were not examined quantitatively because they exhibited little inactivation during a 900-ms pulse to +50 mV. Resembling the onset of macroscopic inactivation (Fig. 4), the effect of multiple mutations on recovery from inactivation could not be explained assuming additivity of the free energies that single mutations contribute to destabilization of the inactivated state (materials and methods; Table I). The predicted rate constants of recovery were systematically slower than the observed values (Fig. 5 B). Overall, however, deviation from additivity appeared moderate with respect to the results obtained from the analysis of the onset of macroscopic inactivation (Fig. 4 B).
Figure 5.

The rate of recovery from inactivation of wild-type (WT) Kv3.4 and several S → D mutants. (A) Time courses of recovery from inactivation of S8D, S9D, S15D, and S21D (○) and their corresponding wild-type from the same batch of oocytes (•). A two-pulse protocol was used to determine the time course of recovery from inactivation at −100 mV. Whole-oocyte currents were evoked and inactivated by a 150-ms step depolarization to +40 mV (p1) from a holding potential of −100 mV. This pulse inactivated ≥80% of the currents expressed by wild type, S8D, S9D, S15D, and S21D. The degree of inactivation at the end of the pulse decreases for the mutants that disrupted inactivation to a greater extent (∼20% for S[8,15,21]D). To allow recovery from inactivation, the interpulse interval (IPI, at −100 mV) was gradually incremented; and a 75-ms test pulse to +40 mV (p2) evaluated the recovery. The peak of the current evoked by the test pulse was divided by the peak of the current evoked by the first pulse (Ip2/Ip1), and this ratio was plotted against the interpulse interval. The time course of recovery was described assuming the sum of two exponential terms (solid lines, see text). The relative weight of the fast term was >65%. (B) Tukey box plot summarizing the results of the analysis of the fast rate of recovery from inactivation. Definitions of this plot are as described herein. The number of oocytes examined is indicated by the figure accompanying the boxes. We also examined S[8,9,15]D, S[8,9,21]D, and S[8,9,15,21]D. However, they were not analyzed quantitatively because they exhibited little or no inactivation (<20% using a 900-ms pulse) or the shortest interpulse interval used in these experiments (50–100 ms) allowed complete recovery from inactivation. Open symbols with a dot represent the predicted rate of recovery from inactivation for multiple mutants assuming a simple additive effect of single mutations as described in materials and methods.
The results presented above demonstrate that S → D mutations not only reduced the rate of inactivation but also increased the rate of recovery from inactivation. Assuming three interconnected states (closed, open, and inactivated), we expected a depolarizing shift of the prepulse inactivation curve and an increased level of the noninactivating current. Accordingly, most of the S → D mutants that exhibited significantly slower rates of inactivation (Fig. 4) and a destabilized inactivated state (Fig. 5) also showed a rightward shift of the midpoint of prepulse inactivation and an increased current level at the foot of the curve (Fig. 6; Table I). Also, as expected from the analyses of the rates of inactivation and recovery from inactivation, the midpoint potentials of prepulse inactivation for various double S → D mutants did not exhibit linear additivity (Fig. 6, legend). Some mutants that exhibited a small or moderate acceleration of the recovery from inactivation did not show an increased level of the noninactivating current after a 10-s conditioning prepulse to depolarized membrane potentials (Fig. 6; Table I). These mutant channels inactivated almost completely like the wild type because they slowly entered a second more stable inactivated state (possibly from closed and open states). The protocol used to determine the rate of recovery from inactivation did not allow a significant entry into that inactivated state (Fig. 5, legend).
Figure 6.

Prepulse inactivation of wild-type Kv3.4 and several S → D mutants. (A) Graph of the normalized peak current against prepulse potential. During a 10-s period, the membrane was held at the indicated prepulse potentials (abscissa). A 112-ms test pulse to +40 mV was delivered at the end of the prepulse. Between episodes, the membrane potential was held at −100 mV and the interepisode interval was no longer than 6 s. The peak of the current evoked by the test pulse was plotted against the prepulse potential and the resulting curve was described assuming a Boltzmann distribution (solid line) to estimate the midpoint potential (V0.5), the slope, and the fraction of noninactivating current (Table I). For display, data were normalized to the maximum current estimated from the Boltzmann distribution (I/Imax). Prepulse inactivation curves from S[8,9]D, triple mutants, and S[8,9,15,21]D were not analyzed quantitatively because they are significantly biased by current activation (notice that these curves exhibit a reduced steepness and a rebounding effect at the foot of the curves). In these cases, the symbols are connected by a dotted line. (B) Tukey box plot summarizing the midpoint potentials of the prepulse inactivation curves from wild-type and several double S → D mutants (see Fig. 4, legend, for definitions). The number of oocytes examined is indicated by the figure accompanying the boxes. Open symbols with a dot represent the predicted values for the double mutants assuming a simple additive effect of single mutations (relative to wild type, the shifts in the midpoints were added and the result added to the wild-type value).
Electrostatic and Steric Interactions Contribute to the Effects of NH2-Terminal S → D Mutations on Inactivation of Kv3.4
To investigate the relative importance of an electrostatic effect versus steric hindrance, we examined two additional substitutions: S8N (N = Asn), S8E (E = Glu). These mutations allowed us to inquire whether the negative charge (S8E and S8D) and/or the size of the side chain (S8N) altered the inactivation properties of the channel. We chose to study the eighth position because the S8D mutant alone exhibited the largest disruption of inactivation (Figs. 4 and 5). S8E slowed the onset of inactivation to the same extent as S8D, whereas S8N only slightly slowed this process (Fig. 7, A and B). Thus, the slower onset of inactivation of the S8D mutant channels is not likely to be caused by the steric effect of the mutation. As previously suggested, the negative charge appears to be the main factor that explains the slow inactivation of the phosphorylated channels. The presence of steric hindrance was, however, more apparent on the recovery from inactivation (Fig. 7, C and D). S8E increased the rate of recovery from inactivation by approximately fivefold, and S8D and S8N also accelerated the recovery from inactivation but to a more moderate extent (approximately three- and twofold, respectively). This small difference between S8D and S8N indicates that the charge of the side chain plays a minor role on the recovery from inactivation. Thus, by contrast to the onset of inactivation, the stability of the inactivated state is more sensitive to the topography of the inactivation particle, which can be altered by phosphorylation.
Figure 7.

The effects of neutral and negatively charged substitutions affecting S8. (A) Whole-cell currents from oocytes expressing wild-type Kv3.4 (WT) and the mutants S8N, S8D, and S8E. The pulse protocol is shown above the currents. For comparison, currents are shown normalized. The peak currents were (μA): 8.5, 10.4, 3.5, and 10.9 for wild type, S8N, S8D, and S8E, respectively. Capacitive transients were subtracted on-line using a P/4 protocol. The solid horizontal line indicates the zero current level. (B) Time courses of recovery from inactivation of Kv3.4 (WT) and the mutants S8N, S8D, and S8E. The pulse protocol and analysis were as described in Fig. 5, legend. The time course of recovery was described assuming the sum of two exponential terms (solid lines, see text). The relative weight of the fast term was >65%. (C and D) Tukey box plots summarizing the rate constants of inactivation and recovery from inactivation obtained from the experiments described in A and B. Definitions of this plot are as described in Fig. 4, legend. The number of oocytes examined is indicated by the figure accompanying the boxes. Data for wild type and S8D were replotted from Figs. 4 and 5.
discussion
This study investigated the molecular physiology of the NH2-terminal phosphorylation sites that regulate inactivation gating of an A-type K+ channel. The main results show that: (a) PKC acts on four phosphate acceptors (S8, S9, S15, and S21) within the inactivation domain because mutation of these residues to alanine is necessary and sufficient to remove the action of PKC on channel inactivation. Moreover, a peptide that corresponds to the inactivation domain of Kv3.4 (the first 28 amino acid) was phosphorylated by three PKC isoforms, but the mutant peptide S[8,9,15,21]A was a much poorer substrate. (b) Single S → A mutants exhibited partially reduced effects of PKC on inactivation. The remaining effects of PKC were, however, not equal among the single mutants and linear summation of these effects could not explain a nearly complete elimination of inactivation caused by PKC. (c) S → D substitutions at the putative phosphorylation sites mimicked PKC action. The inactivation properties conferred by single S → D mutations were also not equivalent, and multiple S → D mutations appeared to interact in a cooperative manner. Thus, a simple reduction or neutralization of the net positive charge of the inactivation domain of Kv3.4 is not sufficient to explain the data. (d) Analysis of the S → N and S → E mutants revealed that a combination of electrostatic and steric factors contribute to disruption of inactivation by S → D substitutions or phosphorylation at the NH2-terminal domain.
Structural Basis of PKC Action on Kv3.4 Inactivation
To explain how phosphorylation of a set of four serines at positions 8, 9, 15, and 21 by PKC causes elimination of rapid N-type inactivation in Kv3.4, we have considered two possible mechanisms. First, the inactivation domain could behave as a point charge (Murrell-Lagnado and Aldrich, 1993a ). In such a case, the factor that determines the onset of inactivation would be the net charge of the inactivation domain. Accordingly, the addition of negatively charged phosphate groups could simply reduce or neutralize the net positive charge of the inactivation domain. Then, individual phosphoserines should behave equivalently and act independently. Although aspartates at positions 8, 9, 15, and 21 can mimic PKC action (as predicted by this hypothesis), our data showed that these positions are not functionally equivalent and that multiple mutations interact (Figs. 1, and 4–6). Also, the net charge change does not appear to be critical because monoanionic aspartates can closely mimic the action of dianionic phosphates introduced by phosphorylation. Thus, the inactivation domain of Kv3.4 does not behave as a simple point charge.
Second, the topography and electrostatic profile of the inactivation domain may determine binding to its receptor in the channel. In this case, the localization of an active group is more critical. In agreement with this possibility, the effects of mutations at S8 and S9 were, in general, more dramatic than those of mutations at S15 and S21 (Figs. 1–6) and, while the rate of inactivation was mainly sensitive to electrostatic interactions, the rate of recovery from inactivation was mainly sensitive to steric hindrance (Fig. 7). Additional results also suggested the presence of other structural changes that could result from the combination of electrostatic and steric effects. For instance, S → D mutations decreased the rate of inactivation and increased the rate of recovery from inactivation, but the energetic effects of the mutations were not linearly additive (Figs. 4–6). Analysis of multiple mutations in other proteins has shown that deviation from simple free energy additivity may reflect conformational changes induced by the substitutions or functional coupling between the mutated residues (Andersen and Koeppe, 1992). For tyrosyl-tRNA synthetase, crystallographic data have confirmed this interpretation (Carter et al., 1984; Fothergill and Fersht, 1991). Similarly, the contribution of individual deep pore residues to the conductance and tetraethyl ammonium block in certain K+ channels is not additive (Kirsch et al., 1992). This appears to reflect coupling between deep pore residues and the importance of the backbone structure in the pore region. Also, as shown for certain metabolic enzymes of known structure (glycogen phosphorylase and isocitrate dehydrogenase), phosphoserines may alter function by a combination of local and allosteric effects (Johnson and Barford, 1993; Johnson and O'Reilly, 1996).
Inspection of the nuclear magnetic resonance–based tertiary structure of the inactivation domain of Kv3.4 (Antz et al., 1997) allowed us to visualize the topography and electrostatic profile of critical regions and possible structural changes induced by phosphorylation. On one face of the molecule, the side chains of S8 and S15 are located in a region that is partly surrounded by basic side chains (R11, R13, K14, K18, and K26). The local electrostatic potential of this region appeared especially important because single negatively charged substitutions at S8 (S8D and S8E) most effectively slowed the rate of inactivation, and a neutral substitution (S8N) had little or no effect on the onset of inactivation (Fig. 7). Its topography seemed critical too because, independently of the charge, S8D, S8E, and S8N destabilized the bound state of the inactivation particle (i.e., accelerated the recovery from inactivation). On another face, the side chain of S9 is located in a less polar region, and that of S21 (in close spatial relationship with the side chain of K22) is relatively more distant from the other three sites. S9D, S15D, and S21D were significantly less effective in disrupting inactivation than S8D. The effects of the substitutions on inactivation were, however, greatly enhanced when they appeared in double, triple, or quadruple combinations (even when S8 remained). Energetically, this enhancement was greater than that predicted by a simple additive interaction, suggesting the presence of cooperativity (Figs. 4–6). Thus, a combination of electrostatic and steric effects (as a result of phosphorylation or negatively charged substitutions) may serve as an incentive that favors a structural change of the inactivation domain. Phosphoserines could simply shift the structural equilibrium toward a disordered conformation. In support of this model, recent nuclear magnetic resonance experiments have demonstrated that S8D, S15D, and S21D (and their phosphorylated counterparts) differentially disordered the NH2- and COOH-terminal portions of the inactivation domain (Antz et al., 1998).
Regulation of K+ Channel Inactivation by PKC: A Physiological Scenario
Neurotransmitter receptors that activate phospholipase C initiate the second messenger cascade that activates PKC (Nicoll, 1988; Kandel et al., 1995). Thus, under physiological conditions, regulation of rapid K+ channel inactivation by PKC may be controlled by a neurotransmitter. We have previously demonstrated that such a system can be reconstituted in Xenopus oocytes by coexpressing a metabotropic serotonin receptor that activates phospholipase C and Kv3.4 K+ channels (Velasco et al., 1998). This result has important implications because Kv3.4 and metabotropic receptors linked to phospholipase C (e.g., type 2c serotonin receptor, muscarinic acetylcholine receptor, glutamate receptor, etc.) may coexist in the nervous system. In a possible physiological scenario, the neurotransmitter could be released from inhibitory neurons acting on a presynaptic terminal. In fact, the terminal areas of major projection tracts in the brain are rich in Kv3.4 (Roeper and Pongs, 1996). Thus, activation of a metabotropic receptor in the synapse will trigger the second messenger cascade that activates PKC. Both PKCα and PKCβ are found in the nervous system, and PKCβ seems to be especially abundant in certain terminal areas (Nishizuka, 1988). Phosphorylation of the inactivation particle in Kv3.4 by PKC causes slower inactivation of this channel. As a result, the action potential will be shortened and, consequently, synaptic transmission will be depressed. Alternatively, dephosphorylation by phosphatases may broaden action potentials and enhance synaptic activity.
Acknowledgments
We thank Mr. T. Harris for harvesting and injecting oocytes, Drs. A. Wei and A. Matlapudi for helping with site-directed mutagenesis, and Dr. T.B. Vyas for conducting preliminary experiments. We thank Drs. C. Antz, B. Fakler, and R. Horn for insightful discussions and critical comments to an earlier version of the manuscript. We are also grateful to Dr. C.D. Stubbs for providing recombinant PKC isoforms.
This work was supported by National Institutes of Health (NIH) grant NS32337 (M. Covarrubias). E. Beck was supported by NIH Training Grant AA07463.
Abbreviation used in this paper
- PMA
phorbol 12-myristate-13-acetate
references
- Andersen OS, Koeppe RE., II Molecular determinants of channel function. Physiol Rev. 1992;72:S89–S158. doi: 10.1152/physrev.1992.72.suppl_4.S89. [DOI] [PubMed] [Google Scholar]
- Antz C, Geyer M, Fakler B, Schott MK, Guy R, Frank R, Ruppersberg JP, Kalbitzer HR. NMR structure of inactivation gates from mammalian voltage-dependent potassium channels. Nature. 1997;385:272–274. doi: 10.1038/385272a0. [DOI] [PubMed] [Google Scholar]
- Antz C, Bauer T, Kalbacher H, Frank R, Ruppersberg JP, Fakler B. A structural switch in fast inactivation of brain K+channels mediated by protein phosphorylation. Biophys J. 1998;74:A229. . (Abstr.) [Google Scholar]
- Baukrowitz T, Yellen G. Modulation of K+ current by frequency and external [K+]: a tale of two inactivation mechanisms. Neuron. 1995;15:951–960. doi: 10.1016/0896-6273(95)90185-x. [DOI] [PubMed] [Google Scholar]
- Carter PJ, Winter G, Wilkinson AJ, Fersht AR. The use of double mutants to detect structural changes in the active site of the tyrosyl-tRNA synthetase (Bacillus stearothermophilus) Cell. 1984;38:835–840. doi: 10.1016/0092-8674(84)90278-2. [DOI] [PubMed] [Google Scholar]
- Cline J, Braman JC, Hogrefe HH. PCR fidelity of Pfupolymerase and other thermostable DNA polymerases. Nucleic Acids Res. 1996;24:3546–3551. doi: 10.1093/nar/24.18.3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covarrubias M, Wei A, Salkoff L, Vyas TB. Elimination of rapid potassium channel inactivation by phosphorylation of the inactivation gate. Neuron. 1994;13:1403–1412. doi: 10.1016/0896-6273(94)90425-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covarrubias M, Beck EJ, Matlapudi A, Sorensen RG. Phosphorylation at multiple N-terminal sites directly modulates inactivation of Kv3.4. Biophys J. 1997;72:341. . (Abstr.) [Google Scholar]
- Demo SD, Yellen G. The inactivation gate of the Shaker K+channel behaves like an open-channel blocker. Neuron. 1991;7:743–753. doi: 10.1016/0896-6273(91)90277-7. [DOI] [PubMed] [Google Scholar]
- Drain P, Dubin AE, Aldrich RW. Regulation of Shaker K+channel inactivation by cAMP-dependent protein kinase. Neuron. 1994;12:1097–1109. doi: 10.1016/0896-6273(94)90317-4. [DOI] [PubMed] [Google Scholar]
- Flaman J-M, Frebourg T, Moreau V, Charbonnier F, Martin C, Ishioka C, Friend SH, Iggo R. A rapid PCR fidelity assay. Nucleic Acids Res. 1994;22:3259–3260. doi: 10.1093/nar/22.15.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fothergill MD, Fersht AR. Correlation between kinetic and x-ray analyses of engineered enzymes: crystal structure of mutants Cys-Gly-35 and Tyr-Phe-34 of tyrosyl-tRNA synthetase. Biochemistry. 1991;30:5157–5164. doi: 10.1021/bi00235a007. [DOI] [PubMed] [Google Scholar]
- Gomez-Lagunas F, Armstrong CM. Inactivation in ShakerB K+channels: a test for the number of inactivating particles on each channel. Biophys J. 1995;68:89–95. doi: 10.1016/S0006-3495(95)80162-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutfreund, H. 1995. Kinetics for the Life Sciences. 1st ed. Cambridge University Press, Melbourne, Australia. 231–280.
- Hille, B. 1992. Ionic Channels of Excitable Membranes. 2nd ed. Sinauer Associates Inc., Sunderland, MA. 116–121.
- Hoffman DA, Magee JC, Colbert CM, Johnston D. K+channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Science. 1997;387:869–875. doi: 10.1038/43119. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–568. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Two types of inactivation in Shaker K+channels: effects of alterations in the carboxy-terminal region. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- Jerng HH, Covarrubias M. K+channel inactivation mediated by the concerted action of the cytoplasmic N- and C-terminal domains. Biophys J. 1997;72:163–174. doi: 10.1016/S0006-3495(97)78655-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LN, Barford D. The effects of phosphorylation on the structure and function of proteins. Annu Rev Biophys Biomol Struct. 1993;22:199–232. doi: 10.1146/annurev.bb.22.060193.001215. [DOI] [PubMed] [Google Scholar]
- Johnson LN, O'Reilly M. Control by phosphorylation. Curr Opin Struct Biol. 1996;6:762–769. doi: 10.1016/s0959-440x(96)80005-4. [DOI] [PubMed] [Google Scholar]
- Jonas EA, Kaczmarek LK. Regulation of potassium channels by protein kinases. Curr Opin Neurobiol. 1996;6:318–323. doi: 10.1016/s0959-4388(96)80114-0. [DOI] [PubMed] [Google Scholar]
- Kandel, E.R., J.H. Schwartz, and T.M. Jessel. 1995. Essentials of Neural Science and Behavior. Appleton & Lange, Norwalk, CT. 243–692.
- Kirsch GE, Drewe JA, Hartmann HA, Taglialatela M, De Biasi M, Brown AM, Joho RH. Differences between the deep pores of K+channels determined by an interacting pair of nonpolar amino acids. Neuron. 1992;8:499–505. doi: 10.1016/0896-6273(92)90278-l. [DOI] [PubMed] [Google Scholar]
- Kunkel TA. Exonucleolytic proofreading. Cell. 1988;53:837–840. doi: 10.1016/s0092-8674(88)90189-4. [DOI] [PubMed] [Google Scholar]
- Kupper J, Bowlby MR, Marom S, Levitan IB. Intracellular and extracellular aminoacids that influence C-type inactivation and its modulation in a voltage-dependent potassium channel. Pflügers Arch. 1995;430:1–11. doi: 10.1007/BF00373833. [DOI] [PubMed] [Google Scholar]
- Levitan IB. Modulation of ion channels by protein phosphorylation and dephosphorylation. Annu Rev Physiol. 1994;56:193–212. doi: 10.1146/annurev.ph.56.030194.001205. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J, Hoshi T, Heinemann SH, Aldrich RW. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- Lundberg KS, Shoemaker DD, Adams MWW, Short JM, Sorge JA, Mathur EJ. High fidelity amplification using thermostable DNA polymerase isolated from Pyrococcus furiosus. . Gene (Amst) 1991;108:1–6. doi: 10.1016/0378-1119(91)90480-y. [DOI] [PubMed] [Google Scholar]
- Murrell-Lagnado RD, Aldrich RW. Interactions of the amino terminal domains of Shaker K+channels with a pore blocking site studied with synthetic peptides. J Gen Physiol. 1993a;102:949–975. doi: 10.1085/jgp.102.6.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell-Lagnado RD, Aldrich RW. Energetics of Shaker K+channels blocked by inactivation peptides. J Gen Physiol. 1993b;102:977–1003. doi: 10.1085/jgp.102.6.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA. The coupling of neurotransmitter receptors to ion channels in the brain. Science. 1988;241:545–551. doi: 10.1126/science.2456612. [DOI] [PubMed] [Google Scholar]
- Nishizuka Y. The molecular heterogeneity of protein kinase C and its implications in cellular regulation. Nature. 1988;334:661–665. doi: 10.1038/334661a0. [DOI] [PubMed] [Google Scholar]
- Ogielska EM, Zagotta WN, Hoshi T, Heinemann SH, Haab J, Aldrich RW. Cooperative subunit interaction in C-type inactivation of K+channels. Biophys J. 1995;69:2449–2457. doi: 10.1016/S0006-3495(95)80114-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panyi G, Sheng Z, Tu L, Deutsch C. C-type inactivation of a voltage-gated K+channel occurs by a cooperative mechanism. Biophys J. 1995;69:896–903. doi: 10.1016/S0006-3495(95)79963-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perozo E, Bezanilla F. Phosphorylation of K+channels in the squid giant axon. A mechanistic analysis. J Bioenerg Biomembr. 1991;23:599–610. doi: 10.1007/BF00785813. [DOI] [PubMed] [Google Scholar]
- Rettig J, Wunder F, Stocker M, Lichtinghagen R, Mastiaux F, Beckh S, Kues W, Pedarzani P, Schroeter KH, Ruppersberg JP, Veh R, Pongs O. Characterization of a Shaw-related potassium channel family in rat brain. EMBO (Eur Mol Biol Organ) J. 1992;11:2473–2488. doi: 10.1002/j.1460-2075.1992.tb05312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeper J, Pongs O. Presynaptic potassium channels. Curr Opin Neurobiol. 1996;6:338–341. doi: 10.1016/s0959-4388(96)80117-6. [DOI] [PubMed] [Google Scholar]
- Roeper J, Lorra C, Pongs O. Frequency-dependent inactivation of mammalian A-type K+channel Kv1.4 regulated by Ca/calmodulin-dependent protein kinase. J Neurosci. 1997;17:3379–3391. doi: 10.1523/JNEUROSCI.17-10-03379.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruppersberg JP, Frank R, Pongs O, Stocker M. Cloned neuronal Ik(A) channels reopen during recovery from inactivation. Nature. 1991;353:657–660. doi: 10.1038/353657a0. [DOI] [PubMed] [Google Scholar]
- Slater SJ, Cox KJ, Lombardi JV, Ho C, Kelly MB, Rubin E, Stubbs CD. Inhibition of protein kinase C by alcohols and anesthetics. Nature. 1993;364:82–84. doi: 10.1038/364082a0. [DOI] [PubMed] [Google Scholar]
- Starkus JG, Kuschel L, Reyner MD, Heinemann SH. Ion conduction through C-type inactivated Shaker channels. J Gen Physiol. 1997;110:539–550. doi: 10.1085/jgp.110.5.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco, I., E.J. Beck, and M. Covarrubias. 1998. Receptor-coupled regulation of K+-channel N-type inactivation. Neurobiology (Bp). In press. [PubMed]
- Tukey, J.W. 1977. Exploratory Data Analysis. Addison-Wesley Publishing Company, Reading, MA. 27–55.