

Abstract
ATP-sensitive potassium (KATP) channels are reversibly inhibited by intracellular ATP. Agents that interact with sulfhydryl moieties produce an irreversible inhibition of KATP channel activity when applied to the intracellular membrane surface. ATP appears to protect against this effect, suggesting that the cysteine residue with which thiol reagents interact may either lie within the ATP-binding site or be inaccessible when the channel is closed. We have examined the interaction of the membrane-impermeant thiol-reactive agent p-chloromercuriphenylsulphonate (pCMPS) with the cloned β cell KATP channel. This channel comprises the pore-forming Kir6.2 and regulatory SUR1 subunits. We show that the cysteine residue involved in channel inhibition by pCMPS resides on the Kir6.2 subunit and is located at position 42, which lies within the NH2 terminus of the protein. Although ATP protects against the effects of pCMPS, the ATP sensitivity of the KATP channel was unchanged by mutation of C42 to either valine (V) or alanine (A), suggesting that ATP does not interact directly with this residue. These results are consistent with the idea that C42 is inaccessible to the intracellular solution, and thereby protected from interaction with pCMPS when the channel is closed by ATP. We also observed that the C42A mutation does not affect the ability of SUR1 to endow Kir6.2 with diazoxide sensitivity, and reduces, but does not prevent, the effects of MgADP and tolbutamide, which are mediated via SUR1. The Kir6.2-C42A (or V) mutant channel may provide a suitable background for cysteine-scanning mutagenesis studies.
Keywords: ATP-sensitive K+ channel, Kir6.2, adenosine triphosphate, gating, sulfhydryl reagent
introduction
ATP-sensitive K channels (KATP channels) couple electrical activity to the metabolic state of the cell in a variety of tissues, including muscle, nerve, and endocrine cells (Ashcroft and Ashcroft, 1990; Nichols and Lederer, 1991; Quayle et al., 1997; Trapp and Ashcroft, 1997). These channels are inhibited by ATP and activated by MgADP and it is currently thought that metabolic regulation of the KATP channel is achieved through changes in the cytosolic levels of these nucleotides. KATP channels are also blocked by sulfonylurea drugs, which are used to treat non–insulin-dependent diabetes mellitus, and activated by a group of chemically unrelated drugs collectively known as K channel openers (Ashcroft and Ashcroft, 1992; Edwards and Weston, 1993). Recent studies have demonstrated that the β cell KATP channel is an octameric 4:4 complex of two structurally distinct proteins: an inwardly rectifying K channel subunit, Kir6.2, and a sulfonylurea receptor, SUR1 (Inagaki et al., 1995, 1997; Sakura et al., 1995; Clement et al., 1997; Shyng and Nichols, 1997). Both subunits are required for functional expression. Kir6.2 acts as an ATP-sensitive pore, while SUR1 is a regulatory subunit that endows Kir6.2 with sensitivity to sulfonylureas, K-channel openers, and the potentiatory effects of Mg nucleotides such as MgADP (Nichols et al., 1996; Gribble et al., 1997a ; Shyng et al., 1997; Trapp et al., 1997; Tucker et al., 1997). Two different sulfonylurea receptor genes have been cloned, SUR1 and SUR2, that encode proteins with different pharmacological sensitivity and show different tissue expression (Chutkow et al., 1996; Inagaki et al., 1996; Isomoto et al., 1996). KATP channels in pancreatic β cells comprise Kir6.2 and SUR1 subunits, while those of cardiac muscle appear to be composed of Kir6.2 and a splice variant of SUR2, SUR2A (Aguilar-Bryan et al., 1998).
Agents that interact with sulfhydryl (−SH) moieties produce an irreversible inhibition of both β cell and cardiac muscle KATP channel activity when applied to the intracellular membrane surface (Weik and Neumcke, 1989; Lee et al., 1994; Coetzee et al., 1995). Inhibition can be reversed, however, by subsequent application of the reducing agent dithiothreitol (DTT).1 These results argue that a sulfhydryl group, and thus a cysteine residue, is associated with the normal function of the KATP channel. ATP appears to protect against the effects of thiol reagents, for if ATP is applied before the sulfhydryl-modifying agent is added, and removed after it is washed out, the irreversible inhibition of channel activity is substantially reduced (Weik and Neumcke, 1989; Lee et al., 1994). A similar protective effect was observed with the sulfonylurea tolbutamide, which also blocks the KATP channel (Lee et al., 1994). A possible explanation for the ability of both ATP and tolbutamide to protect against the effects of sulfhydryl agents is that the critical cysteine is not accessible when the channel is closed.
In the present study, we examined the interaction of the membrane-impermeant thiol-reactive agent p-chloromercuriphenylsulphonate (pCMPS) with the KATP channel. We used a truncated isoform of Kir6.2 (Kir6.2ΔC26), which expresses ATP-sensitive channel activity in the absence of a sulfonylurea receptor (Tucker et al., 1997), to demonstrate that the cysteine residue involved in channel inhibition by pCMPS lies on Kir6.2. Using site- directed mutagenesis, we show that this residue is located at position 42, which lies within the intracellular NH2 terminus of the protein. The ATP sensitivity of the channel was unchanged by mutation of C42 to either valine or alanine, suggesting that ATP does not interact directly with this residue. It remains possible, however, that C42 lies within the ATP-binding motif, although it does not contribute to the affinity for ATP. Our results indicate that C42 is inaccessible to the intracellular solution, and thereby protected against inhibition by pCMPS, when the channel is closed by ATP. They also suggest that the inhibitory effect of pCMPS is state dependent.
materials and methods
Molecular Biology
Mouse Kir6.2 (Genbank D50581; Inagaki et al., 1995; Sakura et al., 1995) and rat SUR1 (Genbank L40624; kindly provided by Dr. G. Bell, University of Chicago, Chicago, IL; Aguilar-Bryan et al., 1995) were used in this study. A 26 amino acid COOH-terminal deletion of mouse Kir6.2 (Kir6.2ΔC26) was also used (Tucker et al., 1997). Site-directed mutagenesis of Kir6.2ΔC26 was carried out by subcloning the appropriate fragments into the pALTER vector (Promega, Madison, WI). The mutations are indicated by the single amino acid letter code. Synthesis of capped mRNA was carried out using the mMessage mMachine large scale in vitro transcription kit (Ambion Inc., Austin, TX).
Electrophysiology
Oocyte collection.
Female Xenopus laevis were anaesthetized with MS222 (2 g/liter in water). One ovary was removed via a mini laparotomy, the incision was sutured, and the animal was allowed to recover. Once the wound had completely healed, the second ovary was removed in a similar operation and the animal was then killed by decapitation while under anaesthesia. Immature stage V–VI Xenopus oocytes were incubated for 60 min with 1.0 mg/ml collagenase (type V; Sigma Chemical Co., St. Louis, MO) and manually defolliculated. In most experiments, oocytes were injected with ∼2 ng of mRNA encoding Kir6.2ΔC26 (either wild type or mutant). For coexpression experiments, ∼0.04 ng of full-length Kir6.2 (wild type or mutant) was coinjected with ∼2 ng of SUR1 (giving a 1:50 ratio). The final injection volume was ∼50 nl/oocyte. Control oocytes were injected with water. Isolated oocytes were maintained in tissue culture and studied 1–4 d after injection (Gribble et al., 1997b ).
Macroscopic currents were recorded from giant excised inside-out patches at a holding potential of 0 mV and at 20–24°C (Gribble et al., 1997b ). Currents were evoked by repetitive 3-s voltage ramps from −110 to +100 mV and recorded using an EPC7 patch-clamp amplifier (List Electronik, Darmstadt, Germany). They were filtered at 0.2 kHz, digitized at 0.5 kHz using a Digidata 1200 Interface and analyzed using pClamp software (Axon Instruments, Burlingame, CA).
The pipette solution contained (mM): 140 KCl, 1.2 MgCl2, 2.6 CaCl2, 10 HEPES, pH 7.4 with KOH, and the internal (bath) solution contained (mM): 110 KCl, 2 MgCl2, 1 CaCl2, 30 KOH, 10 EGTA, 10 HEPES, pH 7.2 with KOH, and nucleotides as indicated. pCMPS was dissolved directly in the solution. Tolbutamide was made up as a 0.05 M stock solution in 0.1 M KOH and diazoxide as a 0.02 M stock solution in 0.1 M KOH. Solutions containing nucleotides were made up fresh each day. The pH of all solutions was checked and readjusted, if required, after drug and nucleotide addition. Rapid exchange of solutions was achieved by positioning the patch in the mouth of one of a series of adjacent inflow pipes placed in the bath.
Data Analysis
The slope conductance was measured by fitting a straight line to the current–voltage relation between −20 and −100 mV. The average of five consecutive ramps was calculated in each solution.
ATP dose–response relationships were measured by alternating the control solution with a test ATP concentration. The extent of inhibition by ATP was then expressed as a fraction of the mean of the value obtained in the control solution before and after ATP application. Dose–response curves were fitted to the Hill equation G/G c = 1/{1 + ([ATP]/K i)h}, where [ATP] is the ATP concentration, K i is the ATP concentration at which inhibition is half maximal, and h is the slope factor (Hill coefficient). The time course of the block by pCMPS was fit with a monoexponential function to obtain the time constant of block.
results
Effects of pCMPS on the KATP Wild-Type Channel
We first investigated whether the effect of sulfhydryl-modifying reagents on the cloned β cell KATP channel (Kir6.2/SUR1) was the same as that reported for native KATP channels (Weik and Neumcke, 1989; Lee et al., 1994; Coetzee et al., 1995). Fig.1 A shows that the sulfhydryl reagent pCMPS (50 μM) produced a complete block of Kir6.2/SUR1 currents when applied to the intracellular surface of an inside-out patch. Inhibition was not reversible on removal of pCMPS, but application of the reducing agent DTT partially restored the current. In seven patches, 5 mM DTT restored the current amplitude to 31 ± 4% of its control value. This supports the idea that the inhibitory effect of pCMPS is mediated by sulfhydryl modification of the channel. The fact that the current amplitude did not recover to its original value may be due to an incomplete action of DTT, which acts only slowly at physiological pH. Alternatively, it may reflect the presence of rundown, a time-dependent decline in channel activity that is observed for both native and cloned KATP channels in excised patches.
ATP has been shown to protect against the effects of thiol reagents on native KATP channels (Weik and Neumcke, 1989; Lee et al., 1994). This was also the case for Kir6.2/SUR1 currents (Fig. 1 B). Provided that the patch was exposed to pCMPS in the continuous presence of ATP, the sulfhydryl reagent did not induce an irreversible block and the currents recovered on removal of ATP (to 40 ± 13% of the control value, n = 5). This suggests that the cysteine residue with which the thiol reagent interacts is not accessible from the intracellular solution when the channel is inhibited by ATP, either because it is not exposed when the channel is closed by ATP, or because access to the cysteine residue is occluded by the presence of ATP in its binding site.
Figure 1.

Effects of pCMPS on wild-type K-ATP currents. Macroscopic currents recorded from oocytes coinjected with mRNAs encoding Kir6.2 and SUR1 in response to a series of voltage ramps from −110 to +100 mV. pCMPS, DTT, or ATP were added to the internal solution as indicated by the bars. The dashed line indicates the zero current level.
Effects of pCMPS on Kir6.2ΔC26 Currents
We next explored whether the site at which thiol reagents interact with the KATP channel lies on the pore-forming Kir6.2 subunit, or on SUR1, by examining the effect of pCMPS on a truncated isoform of Kir6.2 (Kir6.2ΔC26) that is able to form functional channels independently of SUR1. Fig. 2 shows that 50 μM pCMPS completely inhibited Kir6.2ΔC26 currents and that DTT was able to partially reverse this inhibition. This result indicates that pCMPS block is not mediated by SUR1, and further suggests that the thiol reagent interacts either directly with Kir6.2ΔC26 or (possibly) with a third protein, endogenously expressed in Xenopus oocytes, that modulates the activity of Kir6.2ΔC26.
Figure 2.

Effects of pCMPS on Kir6.2ΔC26 currents. Macroscopic currents recorded from an inside-out patch excised from an oocyte injected with Kir6.2ΔC26 mRNA in response to voltage ramps from −110 to +100 mV. pCMPS and DTT were added to the internal solution as indicated by the bars.
Cysteine Mutations Identify the Residue Involved in pCMPS Block
To determine whether pCMPS interacts directly with Kir6.2, and to identify the cysteine residue involved, we carried out site-directed mutagenesis of Kir6.2ΔC26. The critical cysteine is accessible to the intracellular solution and must therefore lie either on the NH2 or COOH terminus of Kir6.2ΔC26, or within the inner mouth of the channel pore itself. There are five cysteines within these regions of Kir6.2ΔC26, at positions 42, 81, 166, 197, and 344 (Fig. 3 A). We therefore tested the effects of mutating each of these, in turn, to serine.
Figure 3.
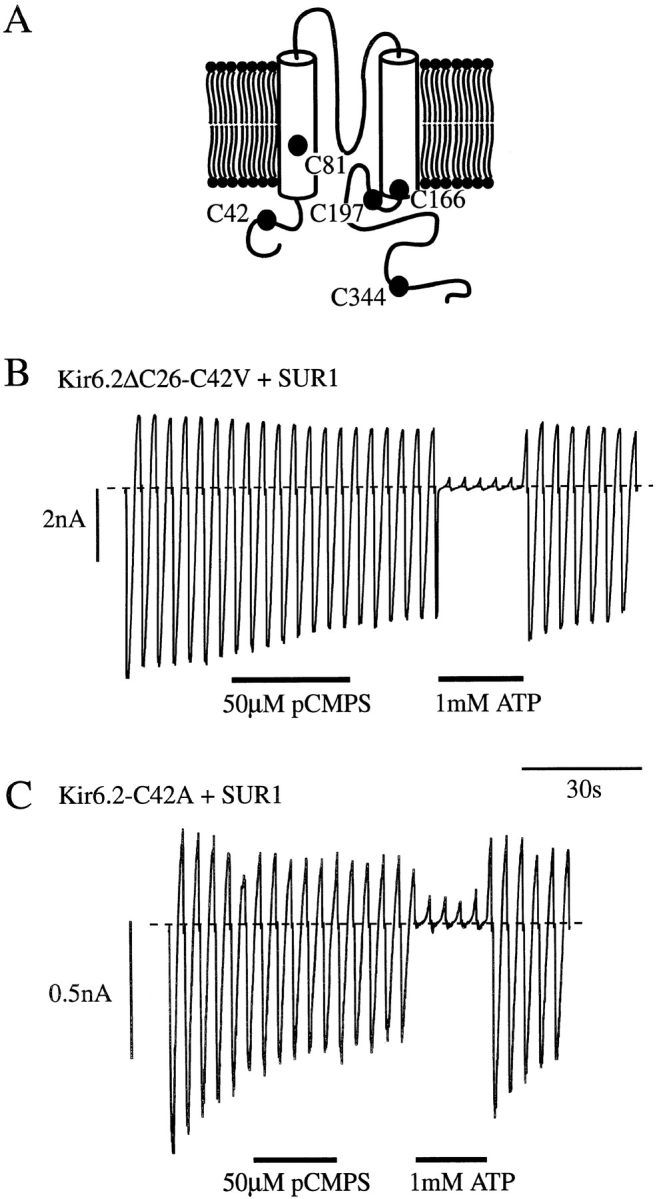
Mutation of C42 abolishes pCMPS but not ATP block of KATP currents. (A) Putative membrane topology of Kir6.2 with the location of intracellular cysteine residues marked. (B and C) Macroscopic currents recorded from inside-out patches excised from oocytes coinjected with either Kir6.2ΔC26-C42V and SUR1 mRNAs (B) or Kir6.2-C42A and SUR1 mRNAs (C). Currents were recorded in response to voltage ramps from −110 to +100 mV. pCMPS and ATP were added to the internal solution as indicated by the bars.
Currents expressed by Kir6.2ΔC26 containing either the C81S or C344S mutations were of similar amplitude to wtKir6.2ΔC26, while those containing the C166S mutation were approximately sevenfold larger (see accompanying paper, Trapp et al., 1998). For all three mutant channels, 50 μM pCMPS caused a complete block of the current. Functional expression of Kir6.2ΔC26-C42S and Kir6.2ΔC26-C197S channels was too low to be able to study: only rare single-channel openings were observed in excised patches containing Kir6.2ΔC26-C42S, and none were detected in patches excised from oocytes injected with Kir6.2ΔC26-C197S mRNA. We therefore tested the effect of both alanine and valine substitutions at positions 42 and 197. When expressed alone, these mutants only produced very small currents but, when coexpressed with SUR1, significant functional expression was observed for Kir6.2ΔC26 containing the C197A, C197V, C42A, or C42V mutations. This suggests that C42 and C197 may be needed for correct targeting of the truncated Kir6.2 isoform to the surface membrane, or for its functional activity.
Mutation of C197 did not prevent inhibition by pCMPS. By contrast, pCMPS was without effect on Kir6.2ΔC26-C42V/SUR1 currents (Fig. 3 B), nor did it affect the KATP currents observed when the full length form of Kir6.2 containing the C42A mutation was coexpressed with SUR1 (Fig. 3 C). These results confirm that pCMPS inhibits the KATP channel by interaction with Kir6.2, rather than with an endogenous oocyte protein. A single cysteine residue in Kir6.2, at position 42, which lies in the cytosolic NH2-terminal part of the protein, appears to be the target for the inhibitory action of pCMPS. Since SUR1 was present in these experiments, they also indicate that pCMPS does not interact with additional sites on SUR1 to inhibit channel activity: in wild-type channels, the presence of such sites would be masked by the inhibitory effect of the thiol agent on Kir6.2.
We next examined the rate of pCMPS block of the mutant channels (with the exception of C42A and C42V, which are not blocked). The rate of block at −100 mV was quantified by fitting a single exponential to the decay of the current after pCMPS application, and the mean time constants for the different cysteine mutations are plotted in Fig. 4 C. The mutant channels were fully blocked by pCMPS and, with two exceptions, the kinetics of the block were similar to those observed for both Kir6.2ΔC26 and Kir6.2/SUR1 currents. The rate of block of Kir6.2ΔC26-C197V currents was slightly increased, while that of Kir6.2ΔC26-C166S was markedly slowed. However, mutation of C166 to valine did not alter the kinetics of inhibition by pCMPS: this may reflect the fact that the C166V mutation, unlike C166S, did not alter the single-channel kinetics (see Trapp et al., 1998). The rate of block of wild-type Kir6.2/SUR1 channels by pCMPS (100 μM) was unaffected by membrane potential, being 1.36 ± 0.32 s (n = 7) at −100 mV and 1.76 ± 0.20 s (n = 7) at +100 mV.
Figure 4.

Effect of cysteine mutations on the rate of pCMPS block. (A and B) Macroscopic currents recorded at −100 mV from inside-out patches excised from oocytes injected with Kir6.2ΔC26 or Kir6.2ΔC26-C166S mRNA. pCMPS was added to the internal solution as indicated by the bars. The dotted line indicates the zero current level and the dashed line is an extrapolated linear fit to the slope of the rundown in control solution and extrapolated. (C) Mean time constant for pCMPS block of wild-type and mutant Kir6.2ΔC26 channels (open bars), and for Kir6.2 or Kir6.2ΔC26-C197V coexpressed with SUR1 (solid bars). The number of oocytes is given above the bars. *P = 0.0005 against wild-type Kir6.2ΔC26 currents (wt). + P < 0.005 against wild-type (Kir6.2/SUR1) currents.
One possible explanation for the ability of ATP to protect against the effects of pCMPS is that the critical cysteine lies within, or close to, the ATP-binding site and is thus inaccessible when ATP is bound. We therefore compared the ATP sensitivity of Kir6.2ΔC26-C42V/SUR1 currents with that of Kir6.2ΔC26/SUR1 currents (Fig. 5, A and B). Application of 1 mM ATP to the inner membrane surface markedly inhibited both currents. There was no significant difference in the ATP sensitivity of the channels, half-maximal inhibition being produced by 13 ± 1 μM ATP (n = 4) for Kir6.2ΔC26-C42V/SUR1 and by 18 ± 3 μM ATP (n = 5) for Kir6.2ΔC26/SUR1 currents. In both cases, the Hill coefficient was close to unity: 0.91 ± 0.07 for Kir6.2ΔC26-C42V/SUR1 and 0.92 ± 0.14 for Kir6.2ΔC26/SUR1 currents. Thus, the C42V mutation does not influence the block by ATP.
Figure 5.
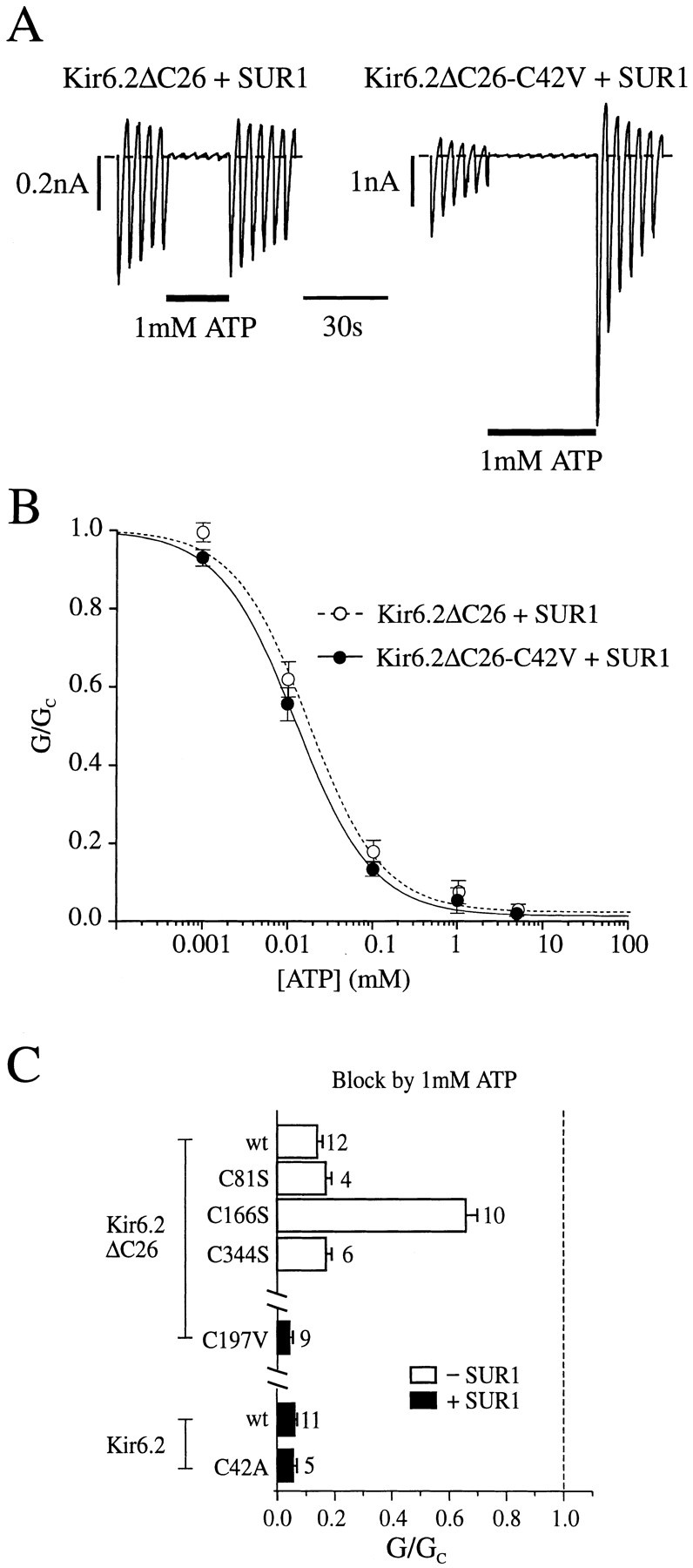
(A) Macroscopic currents recorded from inside-out patches in response to a series of voltage ramps from −110 to +100 mV from oocytes injected with mRNAs encoding SUR1 and either Kir6.2ΔC26 or Kir6.2ΔC26-C42V. ATP was added to the internal solution as indicated by the bar. (B) Mean ATP dose– response relationships for Kir6.2ΔC26/SUR1 currents (○, n = 5) and Kir6.2ΔC26-C42V/SUR1 currents (•, n = 4). Test solutions were alternated with control solutions and the slope conductance (G) is expressed as a percentage of the mean (Gc) of that obtained in control solution before and after exposure to ATP. Conductance was measured between −20 and −100 mV and is the mean of five voltage ramps. The lines are the best fit of the data to the Hill equation using the mean values for K i and h given in the text. (C) Mean current amplitude recorded during exposure to ATP, expressed as a fraction of the mean current amplitude before and after exposure to ATP for patches excised from oocytes injected with wild-type or mutant Kir6.2ΔC26 mRNAs (top), or wild-type or mutant Kir6.2 together with SUR1 (bottom). The dashed line indicates the control current level (in the absence of ATP). The number of oocytes is given above the bars.
No change in ATP sensitivity was observed for the Kir6.2ΔC26 isoform containing either the C81S or C344S mutation, nor for Kir6.2ΔC26-C197V/SUR1 currents or Kir6.2-C42A/SUR1 currents (Fig. 5). When C166 was replaced by serine, however, the ATP sensitivity of the channel was markedly reduced (Fig. 5; see Trapp et al., 1998). It is worth noting that in addition to removing the pCMPS sensitivity, the C42V mutation also induces an increased rate of rundown of the current, and an accompanying increase in the refreshment of the current after MgATP application (Fig. 5 A).
Coupling to SUR1
We next examined the effect of mutating C42 on the ability of SUR1 to couple to Kir6.2, by coexpressing the full length version of Kir6.2 containing the C42A mutation (Kir6.2-C42A) with SUR1. Since Kir6.2 does not express functional channels independently of SUR, this procedure ensures that the KATP current we record only reflects current flow through octameric channels comprising both Kir6.2-C42A and SUR1 subunits. We compared the ability of MgADP, diazoxide, and tolbutamide, all of which interact with SUR1, to influence the activity of wild-type and mutant KATP channels. The stimulatory effect of diazoxide requires the presence of hydrolyzable nucleotides at the inner face of the membrane (Dunne, 1989; Kozlowski et al., 1989) and we therefore tested the ability of this drug to activate channels partially blocked by 100 μM ATP. Fig. 6 shows that the stimulatory effect of diazoxide was not significantly different for Kir6.2-C42A/SUR1 and wild-type KATP currents (unpaired t test). By contrast, both MgADP and tolbutamide were less effective when C42 was mutated. Thus, 100 μM MgADP was without significant effect on Kir6.2-C42A/ SUR1 currents, in contrast to the potentiatory effect of the nucleotide on Kir6.2/SUR1 currents (P = 0.007). In addition to the potentiatory effect of MgADP mediated by the nucleotide-binding domains of SUR1, ADP inhibits the KATP channel by interaction with Kir6.2 (Gribble et al., 1997a ). Complete abolition of the stimulatory effects of MgADP unmasks the inhibitory effect of ADP, reducing the current to ∼50% of its control value (Gribble et al., 1997a ). Since inhibition of Kir6.2-C42A/ SUR1 currents by MgADP was not observed, the stimulatory effect of MgADP must be impaired but not abolished by the mutation. Tolbutamide was also less effective when C42 was mutated (P = 0.017; Fig. 6). Thus, mutation of C42 to alanine reduces, but does not prevent, the ability of SUR1 to transduce binding of MgADP and tolbutamide into changes in Kir6.2 gating.
Figure 6.

Coupling to SUR1 is reduced when C42 in Kir6.2 is mutated to alanine or valine. (A and B) Macroscopic currents recorded from inside-out patches in response to a series of voltage ramps from −110 to +100 mV from oocytes injected with mRNAs encoding SUR1 and either wtKir6.2 or Kir6.2-C42A. MgADP, ATP, diazoxide (DZ), or tolbutamide (TB) were added to the internal solution as indicated by the bars. (C) Mean macroscopic slope conductance recorded in the presence of MgADP, diazoxide, or tolbutamide, expressed as a percentage of the slope conductance in control solution (no additions), for the indicated channels. Diazoxide was added in the presence of MgATP. The dashed line indicates the current level in control solution. The number of oocytes is given above the bars. *P < 0.01; + P < 0.05.
discussion
Both native KATP channels and the cloned wild-type β cell KATP channel (Kir6.2/SUR1) are blocked by the sulfhydryl reagent pCMPS with a time constant of ∼3 s. Our results argue that this effect involves interaction of the sulfhydryl reagent with the cysteine residue at position 42 of Kir6.2. Mutation of C42 to either alanine or valine abolished the inhibitory effect of pCMPS, while mutation of the cysteine residues at positions 81, 166, 197, and 344 had no effect on the magnitude of pCMPS block. The ability of these mutant channels to express functional currents suggests that none of the mutated cysteines participate in disulfide bonds that are critical for channel formation. Although the kinetics of pCMPS block were altered when C166 was mutated to serine, this was not observed for the C166V mutation, indicating that this cysteine is also not involved in inhibition by thiol reagents. The ability of intracellular pCMPS to interact with C42 also implies that the NH2 terminus of Kir6.2 is intracellular, as suggested by hydropathy analysis and by analogy with other Kir channels. The block by pCMPS is not obviously voltage dependent, which implies that C42 lies outside the membrane voltage field and is again consistent with an intracellular location for the NH2 terminus. It is noteworthy that one or more cysteine residues in the NH2 terminus of the voltage-gated K+ channel Kv2.1 must also be targeted by sulfhydryl reagents, since deletion of the NH2 terminus (which contains six cysteines) largely abolishes the effect of these reagents (Pascaul et al., 1997).
Effects of C166 Mutation
Mutation of C166S markedly enhanced the level of current expression. This effect can be entirely explained by the fact that the mutation alters the single-channel kinetics and enhances the channel open probability, rather than affecting the single-channel conductance or channel density (Trapp et al., 1998).
The simplest kinetic model of the KATP channel assumes the presence of a single open state and two closed states, one short and one long (Trapp et al., 1998). The slower time course of pCMPS block of Kir6.2ΔC26-166S currents argues that the thiol reagent may not block the open state of the channel, which is markedly increased in frequency by the C166S mutation (Trapp et al., 1998). Consistent with this idea, the rate of pCMPS block was unaffected in Kir6.2ΔC26-C166V channels, which had single-channel kinetics similar to those of wild-type channels. One possibility, therefore, is that pCMPS reduces the probability of channel reopening from the long closed state. Alternatively, the C166S mutation may independently affect channel gating and slow access to the pCMPS binding site.
Effects of C42 Mutation
Mutation of C42 to either valine or alanine did not alter the ATP sensitivity of Kir6.2ΔC26, indicating that a cysteine at this site is not essential for ATP inhibition. We cannot exclude the possibility, however, that C42 is located close to the ATP-binding site. If access of pCMPS to this residue in wild-type channels is physically prevented by the presence of ATP in its binding site, this would account for the ability of ATP to protect against irreversible pCMPS inhibition. The fact that tolbutamide, which primarily interacts with the SUR rather than the Kir6.2 subunit, also protects against thiol reagent block of native KATP channels (Lee et al., 1994) is less easily explained by this hypothesis. However, we were unable to demonstrate protection against pCMPS block of Kir6.2/SUR1 currents by tolbutamide under our experimental conditions (not shown). Although previous studies have implicated residues in the COOH terminus in ATP inhibition of Kir6.2ΔC26 (Tucker et al., 1997), there is also evidence that residues in the NH2 terminus may be involved: mutation of R50, for example, may also markedly alter the ATP sensitivity (Tucker et al., 1998). It remains unclear, however, whether the ATP-binding site involves residues in both the NH2 and COOH terminus, whether mutations in the NH2 terminus allosterically influence an ATP-binding site in the COOH-terminal part of the molecule or if the NH2 and COOH termini cooperate in channel closure by ATP. A precedent for the latter possibility is provided by the cyclic nucleotide-activated channels, where the cyclic nucleotide binding site is located in the COOH terminus, but channel activation is modified by physical interaction between the NH2 and COOH termini (Gordon and Zagotta, 1995; Varnum and Zagotta, 1997).
The mechanism by which SUR1 enables functional expression of full-length Kir6.2 was unaffected by the C42A mutation. Likewise, the ability of SUR1 to confer diazoxide sensitivity on Kir6.2 was unchanged. By contrast, both MgADP and tolbutamide were significantly less effective than this mutant. This suggests that C42 may be required for full functional coupling of SUR1 to Kir6.2: our data, however, do not allow us to say whether the cysteine residue physically interacts with SUR1, or whether the effect of its mutation on Kir6.2-SUR1 coupling is mediated indirectly.
Conclusion
Cysteine scanning mutagenesis is a well established method for identifying key residues involved in forming the channel pore, in which individual residues are mutated to cysteine and the ability of thiol reagents to block the permeation pathway are investigated. Such studies can only be carried out on a channel that has no intrinsic sensitivity to thiol reagents. This is not the case for either the wild-type KATP channel or the Kir6.2ΔC26 channel. Our results demonstrate that the inhibitory effect of sulfhydryl reagents is mediated by interaction with C42 of Kir6.2, and thus that cysteine scanning mutagenesis could be performed using a Kir6.2 subunit in which C42 has been changed.
Acknowledgments
We thank Dr. G. Yellen for constructive advice, and Drs. F. Gribble and J. Röper for useful discussion.
This study was supported by the Medical Research Council, the Wellcome Trust, and the British Diabetic Association. S. Trapp was supported by a fellowship from the Deutsche Forschungsgemeinschaft. S.J. Tucker is a Wellcome Trust Research Fellow.
Abbreviations used in this paper
- DTT
dithiothreitol
- pCMPS
p-chloromercuriphenylsulphonate
references
- Aguilar-Bryan L, Nichols CG, Wechsler SW, Clement JP, IV, Boyd AE, González G, Herrera-Sosa H, Nguy K, Bryan J, Nelson DA. Cloning of the β-cell high-affinity sulphonylurea receptor: a regulator of insulin secretion. Science. 1995;268:423–425. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- Aguilar-Bryan L, Clement JP, IV, González G, Kunjilwar K, Babenko A, Bryan J. Towards understanding the assembly and structure of KATPchannel. Physiol Rev. 1998;78:227–245. doi: 10.1152/physrev.1998.78.1.227. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM, Ashcroft SJH. Properties and functions of ATP-sensitive K-channels. Cell Signalling. 1990;2:197–214. doi: 10.1016/0898-6568(90)90048-f. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM, Ashcroft SJH. The sulphonylurea receptor. Biochim Biophys Acta. 1992;1175:45–59. doi: 10.1016/0167-4889(92)90008-y. [DOI] [PubMed] [Google Scholar]
- Chutkow WA, Simon MC, Le Beau MM, Burant CF. Cloning, tissue expression, and chromosomal localization of SUR2, the putative drug-binding subunit of cardiac, skeletal muscle, and vascular KATPchannels. Diabetes. 1996;45:1439–1445. doi: 10.2337/diab.45.10.1439. [DOI] [PubMed] [Google Scholar]
- Clement JP, IV, Kunjilwar K, González G, Schwanstecher M, Panten U, Aguilar-Bryan L, Bryan J. Association and stoichiometry of KATPchannel subunits. Neuron. 1997;18:827–838. doi: 10.1016/s0896-6273(00)80321-9. [DOI] [PubMed] [Google Scholar]
- Coetzee WA, Nakamura TY, Faivre J-F. Effects of thiol reagents on KATPchannels in guinea-pig ventricular cells. Am J Physiol. 1995;269:H1625–H1633. doi: 10.1152/ajpheart.1995.269.5.H1625. [DOI] [PubMed] [Google Scholar]
- Dunne MJ. Phosphorylation is required for diazoxide to open ATP-sensitive potassium channels in insulin-secreting cells. FEBS Lett. 1989;250:262–266. doi: 10.1016/0014-5793(89)80734-3. [DOI] [PubMed] [Google Scholar]
- Edwards G, Weston AH. The pharmacology of ATP-sensitive potassium channels. Annu Rev Pharmacol Toxicol. 1993;33:597–637. doi: 10.1146/annurev.pa.33.040193.003121. [DOI] [PubMed] [Google Scholar]
- Gordon SE, Zagotta WN. Localization of regions affecting an allosteric transition in cyclic nucleotide-gated channels. Neuron. 1995;14:857–864. doi: 10.1016/0896-6273(95)90229-5. [DOI] [PubMed] [Google Scholar]
- Gribble FM, Tucker SJ, Ashcroft FM. The essential role of the Walker A motifs of SUR1 in K-ATP channel activation by MgADP and diazoxide. EMBO (Eur Mol Biol Organ) J. 1997a;16:1145–1152. doi: 10.1093/emboj/16.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble FM, Ashfield R, Ämmälä C, Ashcroft FM. Properties of cloned ATP-sensitive K-currents expressed in Xenopusoocytes. J Physiol (Camb) 1997b;498:87–98. doi: 10.1113/jphysiol.1997.sp021843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement JP, IV, Namba N, Inazawa J, Gonzalez G, Aguilar-Bryan L, Seino S, Bryan J. Reconstitution of IKATP: an inward rectifier subunit plus the sulphonylurea receptor. Science. 1995;270:1166–1169. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement JP, IV, Wang CZ, Aguilar-Bryan L, Bryan J, Seino S. A family of sulfonylurea receptors determines the properties of ATP-sensitive K+channels. Neuron. 1996;16:1011–1017. doi: 10.1016/s0896-6273(00)80124-5. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Seino S. Subunit stoichiometry of the pancreatic β-cell ATP-sensitive K+channel. FEBS Lett. 1997;409:232–236. doi: 10.1016/s0014-5793(97)00488-2. [DOI] [PubMed] [Google Scholar]
- Isomoto S, Kondo C, Yamada M, Matsumoto S, Higashiguchi O, Horio Y, Matsuzawa Y, Kurachi Y. A novel sulphonylurea receptor forms with BIR (Kir6.2) a smooth muscle type of ATP-sensitive K+channel. J Biol Chem. 1996;271:24321–24325. doi: 10.1074/jbc.271.40.24321. [DOI] [PubMed] [Google Scholar]
- Kozlowski RJ, Hales CN, Ashford MLJ. Dual effects of diazoxide on ATP-K+currents recorded from an insulin- secreting cell line. Br J Pharmacol. 1989;97:1039–1050. doi: 10.1111/j.1476-5381.1989.tb12560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Ozanne SE, Hales CN, Ashford MLJ. Effects of chemical modification of amino and sulfhydryl groups on KATPchannel function and sulphonylurea binding in CRIG1 insulin secreting cells. J Membr Biol. 1994;139:167–181. doi: 10.1007/BF00232621. [DOI] [PubMed] [Google Scholar]
- Nichols CG, Lederer WJ. Adenosine triphosphate-sensitive potassium channels in the cardiovascular system. Am J Physiol. 1991;261:H1675–H1686. doi: 10.1152/ajpheart.1991.261.6.H1675. [DOI] [PubMed] [Google Scholar]
- Nichols CG, Shyng S-L, Nestorowicz A, Glaser B, Clement JP, IV, Gonzalez G, Aguilar-Bryan L, Permutt MA, Bryan J. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science. 1996;272:1785–1787. doi: 10.1126/science.272.5269.1785. [DOI] [PubMed] [Google Scholar]
- Pascaul JM, Shieh CC, Kirsch GE, Brown AM. Contribution of the NH2terminus of Kv2.1 to channel activation. Am J Physiol. 1997;273:C1849–C1858. doi: 10.1152/ajpcell.1997.273.6.C1849. [DOI] [PubMed] [Google Scholar]
- Quayle JM, Nelson MT, Standen NB. ATP-sensitive and inwardly-rectifying potassium channels in smooth muscle. Physiol Rev. 1997;77:1165–1232. doi: 10.1152/physrev.1997.77.4.1165. [DOI] [PubMed] [Google Scholar]
- Sakura H, Ämmälä C, Smith PA, Gribble FM, Ashcroft FM. Cloning and functional expression of the cDNA encoding a novel ATP-sensitive potassium channel expressed in pancreatic β-cells, brain, heart and skeletal muscle. FEBS Lett. 1995;377:338–344. doi: 10.1016/0014-5793(95)01369-5. [DOI] [PubMed] [Google Scholar]
- Shyng SL, Ferrigni T, Nichols CG. Regulation of KATPchannel activity by diazoxide and MgADP: distinct functions of the two nucleotide binding folds of the sulphonylurea receptor. J Gen Physiol. 1997;110:643–654. doi: 10.1085/jgp.110.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng SL, Nichols CG. Octameric stoichiometry of the KATPchannel complex. J Gen Physiol. 1997;110:655–664. doi: 10.1085/jgp.110.6.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp S, Ashcroft FM. A metabolic sensor in action: news from the ATP sensitive K+channel. News Physiol Sci. 1997;12:255–263. [Google Scholar]
- Trapp S, Tucker SJ, Ashcroft FM. Activation and inhibition of KATPcurrents by guanine nucleotides is mediated by different channel subunits. Proc Natl Acad Sci USA. 1997;94:8872–8877. doi: 10.1073/pnas.94.16.8872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp S, Proks P, Tucker SJ, Ashcroft FM. Molecular analysis of KATP channel gating and implications for channel inhibition by ATP. J Gen Physiol. 1998;112:333–349. doi: 10.1085/jgp.112.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM. Truncation of Kir6.2 produces ATP-sensitive K-channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–183. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- Tucker SJ, Gribble FM, Proks P, Trapp S, Ryder TJ, Haug T, Reimann F, Ashcroft FM. Molecular determinants of KATPchannel inhibition by ATP. EMBO (Eur Mol Biol Organ) J. 1998;17:3290–3296. doi: 10.1093/emboj/17.12.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnum MD, Zagotta WN. Interdomain interactions underlying activation of cyclic nucleotide-gated channels. Science. 1997;278:110–113. doi: 10.1126/science.278.5335.110. [DOI] [PubMed] [Google Scholar]
- Weik R, Neumcke B. ATP-sensitive potassium channels in adult skeletal muscle: characterization of the ATP-binding site. J Membr Biol. 1989;110:217–226. doi: 10.1007/BF01869152. [DOI] [PubMed] [Google Scholar]